

Abstract
Inflammatory bowel disease (IBD) is a complex genetic disorder of two major phenotypes, Crohn's disease (CD) and ulcerative colitis (UC), with increased risk in Ashkenazi Jews. Twelve genome-wide linkage screens have identified multiple loci, but these screens have been of modest size and have used low-density microsatellite markers. We, therefore, performed a high-density single-nucleotide polymorphism (SNP) genome-wide linkage study of 993 IBD multiply affected pedigrees (25% Jewish ancestry) that contained 1709 IBD-affected relative pairs, including 919 CD–CD pairs and 312 UC–UC pairs. We identified a significant novel CD locus on chromosome 13p13.3 (peak logarithm of the odds (LOD) score = 3.98) in all pedigrees, significant linkage evidence on chromosomes 1p35.1 (peak LOD score = 3.5) and 3q29 (peak LOD score = 3.19) in Jewish CD pedigrees, and suggestive loci for Jewish IBD on chromosome 10q22 (peak LOD score = 2.57) and Jewish UC on chromosome 2q24 (peak LOD score = 2.69). Nominal or greater linkage evidence was present for most previously designated IBD loci (IBD1–9), notably, IBD1 for CD families at chromosome 16q12.1 (peak LOD score = 4.86) and IBD6 in non-Jewish UC families at chromosome 19p12 (peak LOD score = 2.67). This study demonstrates the ability of high information content adequately powered SNP genome-wide linkage studies to identify loci not observed in multiple microsatellite-based studies in smaller cohorts.
Keywords: Crohn's disease, ulcerative colitis, linkage, genetics
Introduction
Inflammatory bowel disease (IBD) (MIM # 266600) is a disorder involving chronic intestinal inflammation and is composed of two major phenotypes, Crohn's disease (CD) and ulcerative colitis (UC). CD is characterized by discontinuous transmural inflammation involving any portion of the gastrointestinal tract, with the ileum and colon most commonly affected.1 In UC, inflammation is limited to the mucosa, and always involves the rectum and, to a variable extent, the colon in a continuous manner.1 In approximately 10% of individuals, confirmed IBD limited to the colon cannot be clearly classified as UC or CD and the IBD is labeled as ‘indeterminate colitis.’2 Phenotypic characteristics vary between both CD- and UC-affected individuals, particularly in the location and extent of involved gut, presence of extraintestinal manifestations (approximately 25% of persons with IBD have arthritis, uveitis, pyoderma gangrenosum, erythema nodosum or primary sclerosing cholangitis) and for CD, development of fistulizing or stricturing complications. Other immune-mediated disorders are more frequent among IBD patients, notably, ankylosing spondylitis, asthma, psoriasis and multiple sclerosis.3 IBD may occur at any age, although the peak age at onset is the third decade of life. Both CD and UC have a prevalence of 100–200 per 100 000 in the United States.
Epidemiological studies have long suggested that IBD is a complex genetic disorder: 10–30% of patients have a family history of IBD, monozygotic (MZ) twin concordance is significantly greater than dizygotic (DZ) twin concordance (20–50% for CD and 6–16% for UC versus 0–4% for CD and 0–5% for UC, respectively), and IBD risk is two- to ninefold greater in persons of Ashkenazi Jewish ancestry.4–7 λs, the sibling recurrence risk ratio, has been estimated as 24.7 for IBD, 36.5 for CD and 16.6 for UC.8 Both CD and UC appear genetically related as the cross-disease relative risk was observed to be 3.85 with a CD proband and 1.72 with a UC proband.9
To date, 12 published genome-wide screens and follow-up studies have identified loci with genome-wide linkage evidence and confirmation in single or combined studies (IBD1–9) on chromosomes 16q21 (CD), 12p13–q24, 6p21–23, 14q11–12 (CD), 5q31 (CD), 19p13–q13, 1p36, 16p12 and 3p26–14, respectively.10–22 An additional six loci have been detected with suggestive evidence of linkage in one study and replication evidence in one or more additional studies on chromosomes 2q23–35 (UC), 3q25–28, 4q22–31, 7p13–q21, 11p15–12 and Xp22–21.22 Notably, defined risk alleles/haplotypes have been identified for the IBD1(NOD2), IBD3 (human leukocyte antigen) and IBD5 (5q cytokine cluster) loci.23–26 Despite the tremendous successes of IBD linkage studies to date, several fundamental challenges remain for IBD genetics: (1) replication evidence for loci is highly inconsistent and candidate regions for the majority of loci without established risk haplotypes span large chromosomal regions; (2) individual genome-wide linkage screens have been relatively small (six with less than 100 pedigrees and only one with over 200 pedigrees) and none have had 80% power to identify loci with λs ≤ 1.5; (3) a screening density range of 9–14 cM, frequently using the same marker panels, has likely left important regions unexamined; (4) with no screen of more than 64 Jewish ancestry IBD pedigrees, no loci explain the disproportionate Ashkenazi Jewish risk and (5) no UC locus has been established, perhaps because even the largest IBD genome screen had less than 120 UC relative pairs.
The NIDDK (National Institute of Diabetes and Digestive and Kidney Diseases) IBD Genetics Consortium (NIDDK-IBDGC) was established in 2002 in large part to assemble the large number of IBD pedigrees required to identify overlooked genetic loci, identify loci to account for IBD phenotypes and population subgroups, confirm and narrow down candidate regions and make progress toward disease allele identification. Here, we report the results of a new genome-wide linkage scan, the first scan to use less than 1 cM density single-nucleotide polymorphism (SNP)-based markers, on nearly 1000 well-characterized IBD multiply affected families, one-quarter of them being of Jewish ancestry.
Results
The NIDDK IBDGC assembled DNA samples on 1016 independent multiplex IBD pedigrees, containing two or more IBD, CD or UC informative affected relative pairs with diagnoses confirmed, for a whole-genome SNP-based linkage study. The DNA samples were genotyped using a total of 6008 SNP markers, part of the Illumina Linkage IVb Marker Panel, at the SNP Center at the Center for Inherited Disease Research (Baltimore, MD, USA), with 5912 SNPs released by the SNP Center. We dropped an additional 42 SNPs based on mean allele frequency of less than 0.05 and/or Hardy–Weinberg equilibrium P<0.0001. We used the program Merlin to check for Mendelian inconsistencies and to identify genotypes associated with an excessive number of observed recombinations.27 As a result, an additional 120 genotypes were removed from the analysis. Taken together, the Mendelian consistency rate for this project was 97.55%. Following data cleaning, linkage analysis was performed using the program Merlin on 5750 markers. Twenty-three pedigrees were dropped in the analyses by Merlin as the relative pairs were too distantly related (beyond first cousin pairs), leaving 993 IBD pedigrees analyzed containing 1709 total informative affected IBD relative pairs (Table 1), including CD–CD pairs (n = 919), UC–UC pairs (n = 312) and mixed pairs (n = 478; including CD–UC pairs and pairs with one or both affected relatives diagnosed with indeterminate colitis). Separate analyses, as detailed in Table 1, were performed stratified by Jewish ancestry and for CD–CD pairs (‘CD pairs’) and UC–UC pairs (‘UC pairs’).
Table 1. Family characteristics used in this analysis.
| Analytic set | Informative families per Merlin | Affected relative pairs total (sibling, grandparent, avuncular, 1/2 sibling, cousin) | Average number of members with DNA submitted for genotyping (range) |
|---|---|---|---|
| All families | |||
| All IBD pairs | 993 | 1709 (1049, 77, 371, 5, 207) | 4.33 (2–22) |
| CD pairs | 574 | 919 (623, 21, 174, 3, 98) | 4.40 (2–22) |
| UC pairs | 215 | 312 (185, 16, 78, 0, 33) | 4.74 (2–16) |
| Jewish families | |||
| All IBD pairs | 244 | 374 (228, 32, 68, 0, 46) | 4.11 (2–13) |
| CD pairs | 137 | 196 (140, 11, 26, 0, 19) | 4.03 (2–8) |
| UC pairs | 52 | 68 (29, 8, 16, 0, 15) | 4.67 (2–13) |
| Non-Jewish families | |||
| All IBD pairs | 650 | 1160 (699, 36, 274, 4, 147) | 4.45 (2–22) |
| CD pairs | 386 | 638 (418, 7, 137, 3, 73) | 4.56 (2–22) |
| UC pairs | 140 | 210 (131, 7, 56, 0, 16) | 4.83 (2–16) |
Abbreviations: CD, Crohn's disease; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Merlin can automatically define linkage disequilibrium (LD) clusters that take the LD structure into account (data not shown) and then conduct analysis after modeling marker–marker disequilibrium. When using a cutoff R2 of 0.2, the analysis modeling LD clusters eliminated a few initially identified linkage peaks, including a very strong signal (non-parametric linkage (NPL) = 10.2) on chromosome 21. The results with three phenotypes, IBD, CD and UC, with the full cohort and with the stratified data set by Jewish origin are summarized in Figures 1a, b and c. The significant linkage findings in ‘all ethnicities’, Jewish and non-Jewish populations, have been summarized in Table 2.
Figure 1.
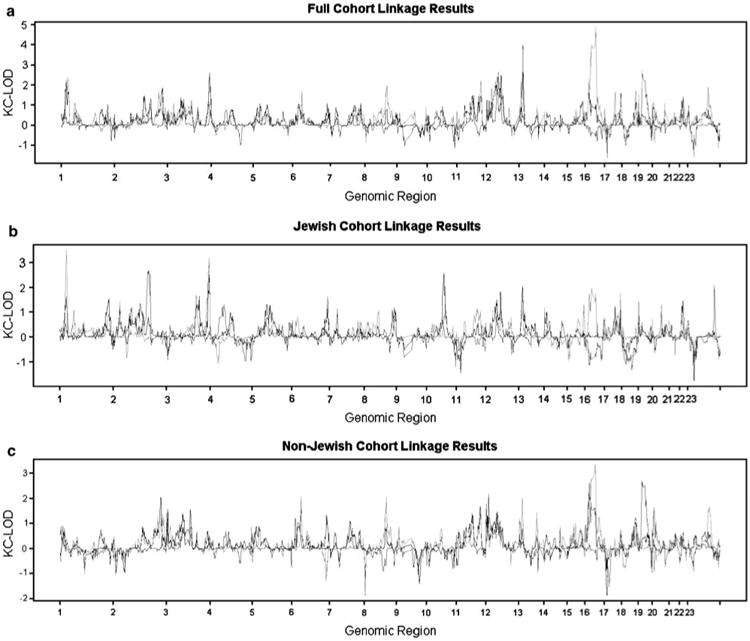
Lod scores by chromosomal regions for the full cohort (a), Jewish pedigrees (b), and Non-Jewish pedigrees (c). KC-LOD, Kong and Cox LOD score; black solid line, all IBD pairs; red solid line, CD–CD pairs; blue solid line, UC–UC pairs. See online version for color Figure.
Table 2. Significant findings by phenotype and ethnicity.
| Band | Cohort | Phenotype | Marker | Positiona | Observed LODb | Sim-Pc | Boundary of 95% | confidence interval |
|---|---|---|---|---|---|---|---|---|
| 16q12.1 | All | CD | rs745230 | 49227683 | 4.86 | 0.002 | rs189378 | rs1123428 |
| 13q13.3 | All | CD | rs20411 | 35129488 | 3.98 | 0.010 | rs1329344 | rs668103 |
| 1p35.2 | Jewish | CD | rs555920 | 30728957 | 3.5 | 0.024 | rs204057 | rs12408030 |
| 3q29 | Jewish | CD | rs2986 | 194013904 | 3.19 | 0.050 | rs2067078 | rs903196 |
Genomic location on chromosome (Build 35.1).
Observed logarithm of the odds score.
P-value determined by using simulation.
Significant linkage findings were detected on chromosomes 16q12.1 (logarithm of the odds (LOD) score = 4.86) and 13q13.3 (LOD score = 3.98) for the CD phenotype for the entire cohort, and on chromosomes 1p35.2 (LOD score = 3.5) and 3q29 (LOD score = 3.19) for Jewish pedigrees. No non-Jewish-only significant linkage findings were detected in this study for the criteria of a genome-wide significance level of 0.05, as determined by permutation testing (Table 2).
The strongest linkage evidence was observed at the IBD1 locus on chromosome 16. We observed a very sharp linkage peak at 49.23Mb (rs745230) on chromosome 16 for the CD (peak LOD score = 4.86) compared with the IBD (peak LOD score = 1.00) phenotype. The NOD2 gene maps adjacent to this peak (between 49.29 and 49.32 Mb). The second strongest signal was observed on chromosome 13q13.3 with the CD phenotype with a genome-wide significance level of 0.01 (Table 2). Because it is known that insufficient adjustment for LD correlation between linkage markers can potentially inflate the LOD score,28 we used a more stringent R2 cutoff of 0.1 to conduct the linkage analysis for the CD phenotype (including both Jewish and non-Jewish families) on chromosome 13; the results varied slightly (from 3.98 to 3.87). We compared the LD structure clusters among the Jewish and non-Jewish groups and the clusters appeared to be very similar, particularly in the region where we found the chromosome 13q13.3 peak. To date, this is the first report of an identification of this novel susceptibility locus on chromosome 13q.22 There was no linkage evidence for the UC phenotype at this locus (Figure 1). In addition, signals on chromosomes 1p35.2 and 3q29 both met the criteria of genome-wide significance levels less than 0.05 in Jewish families but not in non-Jewish families (Table 2). Our screen identified two suggestive loci for Jewish families on chromosome 2q24 (maximum LOD score = 2.69 at marker rs1990760 for the UC phenotype) and chromosome 10q22 (maximum LOD score = 2.57 at marker rs585895 for the IBD phenotype). The only novel linkage signal observed in the non-Jewish families was, for the first time in UC families, to the IBD6 locus at chromosome 19p12 (LOD score = 2.67), although the genome-wide significance level for this signal was 0.194.
The peak LOD scores obtained by this linkage scan in previously identified linkage regions of IBD1–9 are shown in Table 3 for all families. Nominal evidence of linkage greater than LOD score of 1.5 was observed in most of these regions, with exceptions of IBD4, IBD5 and IBD9.
Table 3. LOD scores in the regions harboring previously identified IBD loci.
| IBD locus | Band | Phenotype | Peak LOD of this scan |
|---|---|---|---|
| IBD1 | 16p13.1–q12.2 | CD | 4.86 |
| IBD2 | 12p12–q13 | UC | 1.86 |
| IBD3 | 6p23–p21.1 | IBD | 1.62 |
| IBD4 | 14q11–q12 | IBD | 0.21 |
| IBD5 | 5q31–q35 | UC | 0.62 |
| IBD6 | 19p13.2–q13.2 | IBD | 1.46 (CD) |
| 2.67 (UC) | |||
| IBD7 | 1p36.2–p32 | IBD | 2.17 |
| IBD8 | 16p12 | IBD | 1.73 |
| IBD9 | 3p26 | IBD | 0.58 |
Abbreviations: CD, Crohn's disease; IBD, inflammatory bowel disease; LOD, logarithm of the odds; UC, ulcerative colitis.
Discussion
In this study, we identified genomic regions demonstrating significant evidence for linkage to CD on chromosomes 16q12.1 and 13q13.3, and suggestive evidence for linkage to UC on chromosome 19p12. Furthermore, we found significant linkage between Jewish CD and loci on chromosomes 1p35.2 and 3q29. The reliability of this study is, in part, demonstrated by the strong confirmation of the IBD1 locus with a peak LOD score of 4.86 in CD–CD pairs and no evidence in UC–UC pairs. This is consistent with IBD1, and its putative gene, NOD2, established as CD risk factors. The maximal LOD score at rs745230 is only 60.8kb away from NOD2, highlighting the fine resolution achieved in this study. Interestingly, we continue to observe a ‘shoulder’ to the overall linkage peak on the p-arm of chromosome 16 (Figure 1a). This increased linkage score is observed in both Jewish and non-Jewish CD and is not due to decreased information content in the centromere. In support of a NOD2-independent CD signal in this region, Hampe et al. reported a haplotype-based association29 for CD (IBD8), near D16S3068 with a peak significance level of 0.002, with modest enrichment stratified by NOD2 genotype.30 The present linkage findings are consistent with the presence of additional IBD susceptibility alleles residing on the p-arm of chromosome 16, a possibility that will be examined in further studies.
A major finding of the present study is the identification of a novel, significant CD linkage peak on chromosome 13p13.3. In our prior combined analysis of microsatellite-based screens, we observed only modest evidence for linkage (MLOD = 0.5) in this region.31 An increased information content (0.89 in the present screen versus 0.76 in the prior study) for the 1 LOD confidence interval between 31.53 and 37.65Mb combined with a significantly larger number of pedigrees in the CD phenotype analysis (574 families) likely contributed to the novel linkage discovery. For the R2>0.2 analysis, we included all genotyped markers, as none in this region demonstrated an R2>0.2 with each other. Our linkage results were robust to the LD between genotyped markers in the region; limiting markers to R2≤0.1 had minimal effect on the linkage signal. Taken together, this would indicate that as yet unidentified CD-specific susceptibility alleles reside in this region. The most notable gene that maps within the 95% confidence interval is the RFXAP gene, which encodes the 36kb regulatory subunit of regulatory factor X, a nuclear protein complex that is a specific transactivating factor for major histocompatibility complex class II promoters. RFXAP gene mutations are one cause of major histocompatibility complex class II deficiency found in the immunodeficiency syndrome, bare lymphocyte syndrome type II.32 Other genes, listed in the NCBI database, that map within the 95% confidence interval may be found in Supplementary Table 1.
The established IBD associations in NOD2, IBD5, IL23R and ATG16L1 do not account for the several-fold increased IBD prevalence observed in Jewish populations.33 One would anticipate that if directly causative variants are identified, their frequency might be significantly greater in the Jewish population. However, this has not been observed thus far for any of the established risk alleles. Furthermore, association evidence has not been observed for the IBD5 risk haplotype in Jewish CD; however, it has not yet been established if the causal variants at IBD5 have been identified.34 Taken together, this would indicate that major susceptibility alleles of particular importance in the Jewish population have yet to be identified. Toward these ends, our findings of significant linkage of Jewish CD to chromosomes 1p35.2 and 3q29 are of particular importance. Given the allelic architecture of the IBD association within the IL23R gene on chromosome 1p31.3, involving an uncommon, strongly protective allele at Arg381Gln, the IL23R associations are unlikely to generate a significant linkage signal. Consistent with this, the IL23R gene is located outside the 1.5 LOD confidence interval for the observed linkage peak, and therefore the linkage signal at chromosome 1p35 most likely represents an IBD susceptibility allele independent of IL23R.
A comparison of the top linkage findings from the present study (see Table 2) with our recent ileal CD genome-wide association study33 identified (in addition to NOD2-associated SNPs for the IBD1 chromosome 16 peak) only one SNP with association evidence of potential interest (that is, P < 0.0001) within the chromosomes 1p35.2 linkage peak (rs10914850, P = 7.5 × 10−5) and one SNP within the 3q29 linkage peak (rs6787480, P = 9 × 10−5). Conversely, we did not observe any CD linkage peaks among chromosomal segments that correspond to any of the multiple, highly significant novel associations observed among recent CD genome-wide association studies with the exception of NOD2.33,35–38 Notably, for the three most well-replicated associations after NOD2—the IBD5 risk haplotype, IL23R and ATG16L1—the established risk haplotypes or missense variants associated are common in the population and thus the inheritance of risk alleles may frequently come from different founders within a given pedigree weakening potential linkage signals.33,34
The findings of the present study support the concept that genome-wide linkage and association studies represent complementary approaches to gene identification. SNP-based genome-wide linkage studies in large, well-powered cohorts, such as in the present study, provide improved information content and power to identify novel linkage regions, such as those identified presently at chromosome 13p13.3, prioritize the significance of linkage peaks and improve localization information in regions identified in prior studies. The present identification of genome-wide significant linkage signals in regions lacking established IBD associations highlights the complementary information provided by linkage and genome-wide association studies. Genotyping platforms used in genome-wide association studies focus on common SNPs with minor allele frequencies greater than 5%. Assaying of less common, untyped variations can be achieved by multimarker haplotype analyses, but are still dependent on the limitations of the HapMap data set. In contrast, linkage approaches are powered to identify uncommon alleles conferring high genotype relative risks. Association in linkage areas may also not be found should the risk within a gene region be secondary to multiple risk alleles that are not in LD with each other. The presence of significant linkage on chromosomes 1p and 3q in Jewish CD cohorts may reflect the contribution of uncommon variation relatively unique to the Ashkenazi Jewish population. Subsequent studies will involve a careful integration of all sources of information, together with resequencing to identify uncommon genetic variation contributing to IBD.
Patients and methods
Families with IBD and at least two affected individuals with confirmed IBD diagnoses (CD, UC or indeterminate colitis) were recruited through the IBD genetic research studies at the University of Chicago Genetics Research Center, the Meyerhoff Inflammatory Bowel Disease Genetics Research Center at the Johns Hopkins Hospital, the University of Pittsburgh Genetics Research Center, the University of Montreal Genetics Research Center, the University of Toronto Genetics Research Center and the Cedars-Sinai Genetics Research Center in Los Angeles. Informed consent for a genetic research study for IBD was obtained from all study subjects. All diagnoses of CD, UC or indeterminate colitis were confirmed by physician reviewers from each center from primary chart reviews of endoscopy, radiology and pathology data as meeting standard diagnostic criteria.39–41 Forty-three percent of pedigrees included were previously studied in one of six microsatellite linkage reports from North America,12,14–17,20 and results of a majority of these pedigrees were included in a single genome-wide microsatellite mega-analysis we described previously.31 Study subjects were self-identified as white race. A pedigree was defined as Jewish if two or more affected relatives were of Jewish ancestry (two or more grand-parents Jewish).
Genotyping was performed at the SNP Center at the Center for Inherited Disease Research, Baltimore, MD, USA, using the Illumina Linkage IVb Marker Panel on an Illumina BeadLab system. We computed marker allele frequencies using data from one individual randomly chosen from each pedigree. The NPL method42 implemented in MERLIN, version 0.10.2, was employed.27
Kong and Cox42 propose testing the null hypothesis given δ = 0, where δ is the extent of deviation from IBD sharing. The Kong and Cox LODall statistics were computed since it is known that the score ‘all’ provides more information when relative pairs included in a data set are not independent, and that ‘all’ outperforms ‘pairs’ when the level of heterogeneity underlying a complex trait is relatively high.43 Initially, we computed multipoint NPL scores without taking LD structure into account and then repeated the analysis after modeling marker–marker disequilibrium using Merlin to eliminate the potential for false-positive results; Merlin can automatically define LD clusters using the user-provided marker map and genotype data. We used an R2 cutoff of 0.2 to generate LD clusters for the Jewish and non-Jewish populations separately. The detailed algorithm that clusters tightly linked markers and uses haplotype frequencies to model marker–marker LD was described by Abecasis and Wigginton.28
For the most significant linkage signal detected by this scan (other than the chromosome 16 peak that accounts for the NOD2 locus), we also used a more stringent R2 of 0.1 to conduct the linkage analysis in CD families. All SNP allele frequencies were estimated separately for the Jewish and non-Jewish cohorts.
We estimated empirical P-values by using simulation under the null hypothesis, with 10,000 replicates with Merlin. For each replicate, the original phenotype was used and a new data set, with the same SNP allele frequencies, marker order, genetic distances between markers, as well as missing data pattern was generated. Therefore, the ‘significant signals’ obtained through simulation are chance findings.
Supplementary Material
Acknowledgments
We thank all the participating family members and many colleagues of the NIDDK IBD Genetics Consortium and collaborating centers who took part in the sample collection and data preparation. We also like to acknowledge the Center for Inherited Diseases Research that performed the genotyping. We also thank Mark Daly for his thoughtful review of the paper and Lisa Wu Datta for her assistance in the preparation of the paper. The National Institute of Diabetes and Digestive and Kidney Diseases IBD Genetics Consortium is funded by the following Grants: DK62431 (SRB), DK62422 and DK62429 (JHC), DK62420 (RHD), DK62432 (JDR), DK62423 (MSS) and DK62413 (KDT). This work was also supported by Burroughs Wellcome (JHC), Crohn's and Colitis Foundation of America (RHD, JHC, TMB), Crohn's and Colitis Foundation of Canada (MSS), NIH General Clinical Research Center Grant RR00052 (SRB) and RR00055 (JHC), the Harvey M and Lynn P Meyerh-off Inflammatory Bowel Disease Center (SRB, TMB), the Atran Foundation (SRB), the W Buford Lewis family (SRB) and the Scaife Family Foundation (RHD).
Footnotes
Supplementary Information accompanies the paper on Genes and Immunity website (http://www.nature.com/gene)
References
- 1.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 2.Price AB. Overlap in the spectrum of non-specific inflammatory bowel disease—‘colitis indeterminate’. J Clin Pathol. 1978;31:567–577. doi: 10.1136/jcp.31.6.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein CN, Wajda A, Blanchard JF. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: a population-based study. Gastroenterology. 2005;129:827–836. doi: 10.1053/j.gastro.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 4.Tysk C, Lindberg E, Jarnerot G, Floderus-Myrhed B. Ulcerative colitis and Crohn's disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut. 1988;29:990–996. doi: 10.1136/gut.29.7.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson NP, Driscoll R, Pounder RE, Wakefield AJ. Genetics versus environment in inflammatory bowel disease: results of a British twin study. BMJ. 1996;312:95–96. doi: 10.1136/bmj.312.7023.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orholm M, Binder V, Sorensen TI, Rasmussen LP, Kyvik KO. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand J Gastroenterol. 2000;35:1075–1081. doi: 10.1080/003655200451207. [DOI] [PubMed] [Google Scholar]
- 7.Sandler RS, Targan SR, Shanahan F. Inflammatory Bowel Disease: From Bench To Bedside. Williams & Wilkins; Baltimore, MD: 1994. Epidemiology; pp. 5–30. [Google Scholar]
- 8.Satsangi J, Jewell DP, Bell JI. The genetics of inflammatory bowel disease. Gut. 1997;40:572–574. doi: 10.1136/gut.40.5.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orholm M, Munkholm P, Langholz E, Nielsen OH, Sorensen IA, Binder V. Familial occurrence of inflammatory bowel disease. N Engl J Med. 1991;324:84–88. doi: 10.1056/NEJM199101103240203. [DOI] [PubMed] [Google Scholar]
- 10.Hugot JP, Laurent-Puig P, Gower-Rousseau C, Olson JM, Lee JC, Beaugerie L, et al. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature. 1996;379:821–823. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 11.Satsangi J, Parkes M, Louis E, Hashimoto L, Kato N, Welsh K, et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 12.Cho JH, Nicolae DL, Gold LH, Fields CT, LaBuda MC, Rohal PM, et al. Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q, and 4q: evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci USA. 1998;95:7502–7507. doi: 10.1073/pnas.95.13.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampe J, Shaw SH, Saiz R, Leysens N, Lantermann A, Mascheretti S, et al. Linkage of inflammatory bowel disease to human chromosome 6p. Am J Hum Genet. 1999;65:1647–1655. doi: 10.1086/302677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Y, Ohmen JD, Li Z, Bentley LG, McElree C, Pressman S, et al. A genome-wide search identifies potential new susceptibility loci for Crohn's disease. Inflamm Bowel Dis. 1999;5:271–278. doi: 10.1097/00054725-199911000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, McLeod RS, Griffiths AM, et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66:1863–1870. doi: 10.1086/302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duerr RH, Barmada MM, Zhang L, Pfutzer R, Weeks DE. High-density genome scan in Crohn disease shows confirmed linkage to chromosome 14q11-12. Am J Hum Genet. 2000;66:1857–1862. doi: 10.1086/302947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams CN, Kocher K, Lander ES, Daly MJ, Rioux JD. Using a genome-wide scan and meta-analysis to identify a novel IBD locus and confirm previously identified IBD loci. Inflamm Bowel Dis. 2002;8:375–381. doi: 10.1097/00054725-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Paavola-Sakki P, Ollikainen V, Helio T, Halme L, Turunen U, Lahermo P, et al. Genome-wide search in Finnish families with inflammatory bowel disease provides evidence for novel susceptibility loci. Eur J Hum Genet. 2003;11:112–120. doi: 10.1038/sj.ejhg.5200936. [DOI] [PubMed] [Google Scholar]
- 19.van Heel DA, Dechairo BM, Dawson G, McGovern DP, Negoro K, Carey AH, et al. The IBD6 Crohn's disease locus demonstrates complex interactions with CARD15 and IBD5 disease-associated variants. Hum Mol Genet. 2003;12:2569–2575. doi: 10.1093/hmg/ddg281. [DOI] [PubMed] [Google Scholar]
- 20.Barmada MM, Brant SR, Nicolae DL, Achkar JP, Panhuysen CI, Bayless TM, et al. A genome scan in 260 inflammatory bowel disease-affected relative pairs. Inflamm Bowel Dis. 2004;10:513–520. doi: 10.1097/00054725-200409000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Vermeire S, Vlietinck R, Joossens S, Esters N, Peeters M, Rutgeerts P. Genome-wide scanning in a Belgian IBD population reveals novel linkages and validates previous linkages. Gastroenterology. 2001;120:A456. [Google Scholar]
- 22.Brant SR, Shugart YY. Inflammatory bowel disease gene hunting by linkage analysis: rationale, methodology, and present status of the field. Inflamm Bowel Dis. 2004;10:300–311. doi: 10.1097/00054725-200405000-00019. [DOI] [PubMed] [Google Scholar]
- 23.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 24.Stokkers PC, Reitsma PH, Tytgat GN, van Deventer SJ. HLA-DR and -DQ phenotypes in inflammatory bowel disease: a meta-analysis. Gut. 1999;45:395–401. doi: 10.1136/gut.45.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 26.Rioux JD, Daly MJ, Silverberg MS, Lindblad K, Steinhart H, Cohen Z, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29:223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- 27.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 28.Abecasis GR, Wigginton JE. Handling marker–marker linkage disequilibrium: pedigree analysis with clustered markers. Am J Hum Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clayton D, Jones H. Transmission/disequilibrium tests for extended marker haplotypes. Am J Hum Genet. 1999;65:1161–1169. doi: 10.1086/302566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hampe J, Frenzel H, Mirza MM, Croucher PJ, Cuthbert A, Mascheretti S, et al. Evidence for a NOD2-independent susceptibility locus for inflammatory bowel disease on chromosome 16p. Proc Natl Acad Sci USA. 2002;99:321–326. doi: 10.1073/pnas.261567999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Achkar JP, Dassopoulos T, Silverberg MS, Tuvlin JA, Duerr RH, Brant SR, et al. Phenotype-stratified genetic linkage study demonstrates that IBD2 is an extensive ulcerative colitis locus. Am J Gastroenterol. 2006;101:572–580. doi: 10.1111/j.1572-0241.2006.00451.x. [DOI] [PubMed] [Google Scholar]
- 32.Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, et al. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J. 1997;16:1045–1055. doi: 10.1093/emboj/16.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverberg MS, Duerr RH, Brant SR, Bromfield G, Datta LW, Jani N, et al. Refined genomic localization and ethnic differences observed for the IBD5 association with Crohn's disease. Eur J Hum Genet. 2007;15:328–335. doi: 10.1038/sj.ejhg.5201756. [DOI] [PubMed] [Google Scholar]
- 35.Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn's disease. Hum Mol Genet. 2005;14:3499–3506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 36.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franke A, Hampe J, Rosenstiel P, Becker C, Wagner F, Hasler R, et al. Systematic association mapping identifies NELL1 as a novel IBD disease gene. PLoS ONE. 2007;2:e691. doi: 10.1371/journal.pone.0000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Podolsky DK. Inflammatory bowel disease (1) N Engl J Med. 1991;325:928–937. doi: 10.1056/NEJM199109263251306. [DOI] [PubMed] [Google Scholar]
- 40.Podolsky DK. Inflammatory bowel disease (2) N Engl J Med. 1991;325:1008–1016. doi: 10.1056/NEJM199110033251406. [DOI] [PubMed] [Google Scholar]
- 41.Gower-Rousseau C, Salomez JL, Dupas JL, Marti R, Nuttens MC, Votte A, et al. Incidence of inflammatory bowel disease in northern France (1988–1990) Gut. 1994;35:1433–1438. doi: 10.1136/gut.35.10.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61:1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shugart YY, Feng BJ, Collins A. The power and statistical behaviour of allele-sharing statistics when applied to models with two disease loci. J Genet. 2002;81:99–103. doi: 10.1007/BF02715906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.