

Abstract

Chemical cross-linking of proteins combined with mass spectrometry provides an attractive and novel method for the analysis of native protein structures and protein complexes. Analysis of the data however is complex. Only a small number of cross-linked peptides are produced during sample preparation and must be identified against a background of more abundant native peptides. To facilitate the search and identification of cross-linked peptides, we have developed a novel software suite, named Hekate. Hekate is a suite of tools that address the challenges involved in analyzing protein cross-linking experiments when combined with mass spectrometry. The software is an integrated pipeline for the automation of the data analysis workflow and provides a novel scoring system based on principles of linear peptide analysis. In addition, it provides a tool for the visualization of identified cross-links using three-dimensional models, which is particularly useful when combining chemical cross-linking with other structural techniques. Hekate was validated by the comparative analysis of cytochrome c (bovine heart) against previously reported data.1 Further validation was carried out on known structural elements of DNA polymerase III, the catalytic α-subunit of the Escherichia coli DNA replisome along with new insight into the previously uncharacterized C-terminal domain of the protein.
Keywords: proteomics, cross-linking, peptides, structure, proteins, software
Introduction
With the field of structural biology moving toward the analysis of larger macromolecular complexes, there is an increasing need for alternative or combinatorial methods for characterization of these complexes. The analysis of protein–protein interactions, native protein structures, and the structure of protein complexes by mass spectrometry is a rapidly advancing field that can be used alone or in combination with structural approaches such as protein crystallography,2,3 single particle electron microscopy,4,5 and small-angle X-ray scattering.6,7 Mass spectrometric techniques such as hydrogen–deuterium exchange (HDX)8 and the analysis of macromolecular complexes by native mass spectrometry9,10 can provide useful low-resolution information on the interaction between proteins in a complex. Chemical cross-linking has for nearly 40 years been used to investigate the structure of protein complexes with one of the earliest examples being the use of dimethyl suberimidate to locate neighboring proteins within the ribosomes of Escherichia coli.11 More recently, these methods have been combined with mass spectrometry to provide spatial information.12−15
Chemical cross-linking of proteins coupled with mass spectrometry (XL-MS) provides a convenient and complementary method for the analysis of protein interactions. XL-MS provides several advantages over other techniques. It can theoretically work at picomole to femtomole concentrations,12,16 and as with other bottom-up proteomic methodologies it is suited to complex mixtures of proteins.17 XL-MS can be performed under near physiological conditions allowing for a particularly interesting opportunity in the development of XL-MS, the analysis of in vivo species with membrane permeable cross-linking reagents.18 Results from XL-MS experiments may help provide insight into the effects of deviation from a native environment. The effects experimental conditions have on the structures determined by different techniques such as NMR and X-ray crystallography have previously been discussed.19
Cross-linking makes use of chemically reactive groups on the external surfaces of native proteins, to form a covalent bond between the chemical cross-linking reagent and the amino acid. The targeting of lysine residues with N-hydroxysuccinimide-activated esters, for example, bis[sulfosuccinimidyl] glutarate (BS2G) and bis[sulfosuccinimidyl]suberate (BS3),20,21 to produce an epsilon-amide bond is one of the most common examples of this. In a typical study, two or more proteins are combined in the presence of a cross-linking reagent across a range of concentrations and samples collected at set time points. The reaction is usually halted by the introduction of a competing nucleophile; for example, in the case of BS2G and BS3, this can be an amine-containing buffer such as ammonium bicarbonate. The cross-linking of the protein complexes can be easily monitored by denaturing polyacrylamide gel electrophoresis (PAGE). The cross-linked sample is then digested with a protease such as trypsin, and the resulting peptides analyzed by mass spectrometry to obtain specific information on the location of sites of interaction by the fragmentation of cross-linked peptides (Figure 1B).
Figure 1.

(A) Protein cross-linking is complicated by the variety of species produced. These can be divided into two categories: linear peptides, which include mono- and loop-links; and nonlinear peptides, which include intra- and inter-cross-links that provide information on the structure and interactions of proteins respectively. (B) Experimental workflow. The 2D-LC steps, shown in the shaded blue area, were not carried out in the analysis of cytochrome c. The red labeled shaded area defines steps carried out by the Hekate software. Mascot (Matrix Software) was used for the validation of linear peptide data. Proteome Discover (Thermo Scientific) was used for the deisotoping and conversion of the experimental data into Mascot Generic Format (MGF). The protein sequences are supplied in FASTA format via the web interface. An in silico digest of the input proteins is used to produce a database of theoretical masses that are compared to the doublets detected. All matches are then scored and returned to the user. (C) Detection of cross-linked peptides can be aided by the use of an isotopic labeled cross-linking reagent. By combining these reagents in a 1:1 ratio, a characteristic mass doublet is formed. The unlabeled form is highlighted in green, and the labeled in red. This is both easily seen by eye and detected using informatics.
However, there are disadvantages of XL-MS. The enzymatic digestion of the protein complexes produces very complex mixtures of peptides that can impair the identification of cross-linked species. First, the chemical reagents used form a number of different species. Linear monolinked peptides are the most common of these and provide little structural information, though in cases of specifically chosen cross-linking reagents they may be able to provide information about surface accessibility. Loop-links and intra-cross-links form between residues within the same protein and provide information on the internal protein structure (Figure 1A). Interlinks provide information on the interactions between proteins within complexes. Second, the enzymatic digestion of the proteins is hindered by the cross-linking of proteins making it less efficient than digestion of non-cross-linked proteins.22 The possibility of modification at the protease cleavage site by the cross-linking reagent further complicates as this may result in the inhibition of digestion at these positions.
Attempts have been made to reduce the complexity of the analysis by the use of ion-exchange chromatography23 or the use of tagged-affinity labeled cross-linking reactions to enrich for the presence of cross-linked peptides.24 However, this is a partial solution as the number of linear peptides still greatly outnumbers the cross-linked peptides. Additionally, fragmentation spectra of cross-linked peptides are more complex than those of linear peptides as they contain an ion series from both peptide chains and therefore cannot be identified using standard protein database search engines.
To aid in the analysis of these peptide mixtures, the use of isotope labeled cross-linkers mixed in a ratio of 1:1 to produce characteristic “mass doublets” has been widely adopted.5,20,25−27 The resultant spectra (Figure 1C) are both visually and computationally identifiable. This provides a filter to reduce the complexity of data analysis.20 Further development of collision-induced dissociation (CID) cleavable cross-links,28 reporter ions,15 or all of these combined (as with protein interaction reporter (PIR) reagents)24 has demonstrated the breadth of work focused on solving this problem.
While several attempts have already been made to aid the data analysis,29−35 these have often evolved for specific applications. In particular, pLink36 and xQuest33 have done a great deal to advance proteome wide interaction studies using XL-MS. Hekate looks to develop in the field of structural mass spectrometry.
As such, the development of Hekate realizes an adaptable platform able to analyze both cross-linking products of a wide range of reagents (both with or without the use of stable-isotope labeling) and proteases (Table S1 in the Supporting Information). It is compatible with a variety of instruments and able to undertake the complete analysis of data produced during an experiment without need for manual or additional processing to identify scans containing cross-linked peptides.
Hetake proves two distinct new capabilities to advance the field of structural proteomics: the exporting for 3D visualization of the cross-links in PyMOL (Schrödinger LLC) and the ability to process and rapidly analyze data without specialist knowledge via an intuitive web interface (Video S1 and Figure S1). It also has several advantages, including an improved and robust scoring algorithm based on a proven linear peptide technology (Figure S2),37 results can be exported in a variety of formats including CSV, it can provide detailed annotated mass spectra in an Adobe Illustrator compatible format, and the software is built using an SQL database interface (Figure S4). This final point is crucial as it provides an interface to a technology that is designed for the manipulation and searching of vast data sets, providing wide scope for future expansion to larger systems, for instance, proteome-wide searches. Finally, the software is provided open source to allow continued development and expansion of the capability of the suite.
In the development of the software, we analyzed two proteins: cytochrome c and DNA polymerase III (DNA Pol III) the catalytic α-subunit of the E. coli DNA replication machinery. Cytochrome c is a well-characterized, commercially available protein, making it suitable for an initial study. Detailed information including both a crystal structure and solution lysine–lysine distances provided a basis for the validation of our methods.1,20
DNA Pol III, is ∼10 times larger than cytochrome c and provided a second target for our studies. A crystal structure of the first ∼900 residues is available, but the last 260 residues that are involved in several protein–protein interactions are not present in this structure.38 This, therefore presented a practical and interesting target as a subset of the protein complex within E. coli. DNA Pol III also provided the starting point for the development of a method for the future analysis of more subunits of the DNA replication machinery within E. coli.2
Materials and Methods
Reagents
All chemicals and reagents were purchased from Sigma-Aldrich (Dorset, U.K.) unless otherwise stated.
Cross-Linking and Digestion of Bovine Cytochrome c
A 1:1 mix of BS3-d0/d4 (ThermoPierce, U.K.) was prepared at a concentration of 2 mM in DMSO. This was added to 95 μL of a solution of 10 μM cytochrome c in 100 mM potassium phosphate pH 7.8, to give a final volume of 100 μL. The reaction was incubated at room temperature for 120 min and then quenched by the addition of 100 mM ammonium bicarbonate, 5 μL. Excess cross-linking reagent was removed by dialysis overnight against 100 mM ammonium bicarbonate, pH 8.0, using a 7000 MWCO dialysis membrane (Slide-A-Lyzer MINI Dialysis Unit, Thermo Scientific). The dialyzed sample was subsequently digested with trypsin (porcine sequencing grade, Promega, U.K.), overnight at 37 °C, using a protein to enzyme ratio of 20:1.
Cross-Linking and Digestion of DNA Pol III
A 1:1 mix of BS2G-d0/d4 or BS3-d0/d4 (ThermoPierce, UK) was prepared at a concentration of 2 mM in DMSO. A volume of 1 μL of this was added to Pol III protein (50 μL) prepared at a concentration of 40 μM in 50 mM Hepes pH 7.5, 150 mM NaCl, 2 mM dithiothreitol (DTT). DNA Pol III was purified using a method adapted from Maki and Kornberg.39 The reaction was incubated at room temperature for 15 min and then quenched by the addition of 100 mM ammonium bicarbonate, 5 μL. Nonspecific cross-linked products (i.e., multimers of Pol III) were removed by gel filtration on a PC3.2/30 (2.4 mL) Superdex 200 gel filtration column (GE healthcare, UK) pre-equilibrated in 50 mM Hepes pH 7.5, 150 mM NaCl, and 2 mM DTT. Then 50 μL fractions were collected and analyzed by SDS-PAGE using 4–12% NuPage Bis-Tris precast gels (Life Technologies, UK). Fractions containing cross-linked protein were reduced with DTT (10 mM) and alkylated with iodoacetamide (55 mM). The alkylated sample was brought to a final concentration of 4 M Urea by the addition of 8 M Urea/100 mM ammonium bicarbonate before digestion with trypsin (porcine sequencing grade, Promega, UK) at a protein to enzyme ratio of 20:1 (w:w).
Strong Cation Exchange Chromatography
The digested sample was fractionated by strong cation exchange (SCX) on a Dionex U3000 HPLC using a Poly SULFOETHYL A column (5 μM, 300 Å, 50 mm × 1.0 mm, PolyLC, USA). Peptides were eluted using a linear gradient from 30% v/v acetonitrile in 5 mM KH2PO4 to 30% v/v acetonitrile in 5 mM KH2PO4/350 mM KCl over 75 min at 80 μL/min. Fractions were subdigested with GluC (Promega, UK) at a protein to enzyme ratio of 20:1 (w:w). The resultant peptides were washed and eluted from a C18 ZipTip column (Millipore, UK) (1% v/v trifluoroacetic acid and 50% v/v acetonitrile). The acetonitrile and trifluoroacetic acid was then removed under reduced pressure before mass spectrometric analysis.
Mass Spectrometric Analysis
Digests were analyzed by nanoscale capillary LC-MS/MS using a Ultimate U3000 HPLC (ThermoScientific Dionex, San Jose, CA) to deliver a flow of approximately 200 nL/min. A C18 Acclaim PepMap100 5 μm, 100 μm x 20 mm nanoViper (ThermoScientific Dionex, San Jose, CA), trapped the peptides prior to separation on a C18 Acclaim PepMap100 3 μm, 75 μm x 250 mm nanoViper (ThermoScientific Dionex, San Jose, CA). Peptides were eluted with a gradient of acetonitrile from 5% v/v acetonitrile in 0.1% v/v formic acid to 40% v/v acetonitrile in 0.1% v/v formic acid over 110 min. The column outlet was directly interfaced via a nanoflow electrospray ionization source, with a hybrid dual pressure linear ion trap mass spectrometer (Orbitrap Velos, ThermoScientific, San Jose, CA). Data dependent analysis was carried out, using a resolution of 60 000 for the full MS spectrum, followed by 10 MS/MS spectra in the linear ion trap. MS spectra were collected over a m/z range of 350–1800. MS/MS scans were collected using a threshold energy of 35 for collision induced dissociation.
Hardware and Software
Hekate has been developed in a combination of Perl v5.10.3 and SQL via the Perl DBD::SQLite module. Additional functionality is provided by Chart::Graph::Gnuplot, Twitter Bootstrap, and the Flot jQuery libraries. Hekate is implemented on a desktop computer with an Intel Core 2 6400 processor and 8 GB of RAM. The operating system was Debian Linux (v6.0.5), and the webserver Apache 2.2.16. Using this hardware, it took under 4 min to process the data given in Table S3.
Results
The Hekate Suite
Hekate is a suite of tools to aid the assignment and discovery of cross-linked peptides. The Hekate suite contains four different applications Doublet, Digest, Fragment, and Score that can be used to aid the interpretation of cross-linked data. In addition, the Hekate Search tool combines all four modules.
Hekate Doublet produces a list of scans that contain doublet spectra using the experimental data input file. The input file is formatted as either Mascot Generic Format (MGF) or mzXML, along with details of the isotopic label used and mass accuracy tolerances. The MGF is converted into an SQLite v3 database upon upload to the server. The use of an SQL database has the advantage over direct processing of the MGF data, as it allows for Hekate to build on the already well-developed indexing and searching algorithms of SQLite.40 The index is created on the monoisotopic mass column of the table containg the imported MGF data. This table is then queried utilizing a join of this field to itself with the mass of the isotopic label added. These results are filtered with other constraints as specified by the user, for example, equal charge and elution time. The final output of this query contains the list of “mass doublets” that is then processed and displayed on the Web site.
When collecting MS/MS information under data dependent acquisition conditions, it is common that the instruments are set to acquire the fragment ion spectrum before the peak maximum. This enables the instrument to gather information on a greater number of peptides over a given time. Because of this, the intensity value for a particular spectrum is not reflective of MS peak intensity; therefore, this parameter cannot be relied upon when matching spectral pairs of isotope labeled and unlabeled peptides. Hence, only the mass and timing (scan number) of the peptides are used in identifying spectral pairs.
Hekate Digest provides a list of theoretical cross-linked peptide molecular weights [M]. The protein sequences are imported in FASTA format and digested in silico to produce a list of theoretical peptides including those up to a default value of three missed cleavages. These peptides are then combined to give cross-linked peptide sequences as defined by the parameters of the reagent specified. Each cross-linked peptide sequence is formed of two linear peptide sequences (called the alpha (α) and beta (β) chain), both sequences must contain a residue compatible with the chemistry of the cross-linking reagent and these residues must not be at the enzymatic cleavage site. The scale of the output of this process presents one of the first major problems of protein cross-linking experiment. The list of possible cross-links species for a single protein is a much larger number of possible sequences than would be generated by a search for linear peptides. For example: an in silico tryptic digest of cytochrome c produces 37 possible peptide sequences when using a maximum of one missed cleavage site. Of these peptides, 18 contain a lysine residue within the sequence that is not the terminal residue. The predicted number of cross-linked products, when using a activated-ester based cross-linking reagent like BS3 and assuming that cleavage cannot occur at the modified lysine, for this sample would produce a database of 162 (n2/2) possible species. As the relationship is nonlinear the scale of the effect increases as the number of proteins increase.
As discussed in the introduction to this an additional complicating factor to data-analysis is that cross-linking reactions produce a variety of different species (Figure 1A). Hekate Digest handles all these aspects when generating the database of possible species. The use of a relational database aids this process as the table of cross-linked peptides can be created using the self-join to combine each record in the linear peptide with each other in a single command.
Hekate Fragment takes a supplied sequence of a cross-linked peptide, provided in plain text as two linear amino acid sequences separated by a hyphen, and produces the table of masses for expected a-, b- and y-ions from both the alpha (α) and beta (β) peptide chains formed during collision induced dissociation (CID). When the relevant amino acids are present in a peptide fragment sequence, Hekate Fragment is able to take into account the formation of further ions due to the neutral-loss of either water (D, E, S, or T) or ammonia (K, N, Q, or R).
Hekate Score provides for the scoring of spectral pairs. The scan data is provided as a list of mass/charge ratios and intensity pairs along with the parent ion charge and sequences in FASTA format. The data is scored and a list of any possible matches returned. If multiple peptide sequences are matched, these are all submitted for scoring in turn.
The scoring algorithm (Figure 2) is based on the work of Cox et al.18,37,41 in the development of the Andromeda peptide search engine for integration with MaxQuant. It is noted a similar method was previously described by Maiolica et al.42 based upon a p-score that was introduced for the identification of MS343 spectra and from which Andromeda derives. The software described in that publication is not available as public download and does not, when available, take advantage of the isotopic shift between spectra or the recent advancements in the development of Andromeda.
Figure 2.

To calculate the score of a theoretical peptide sequence against a fragmentation spectrum, we first compare the data from the labeled and unlabeled peptides. Peaks within their respective spectra are then internally annotated if they appear common to both spectra or if they show a characteristic shift for the isotope label used. Algorithms used in the scoring of linear peptide scan data15,37 would not expect to have this information available to them as it is specific to the analysis of cross-linked peptide. Hekate Score; however, takes advantage of this by only matching theoretical ions that are consistent with this extra level of information. Fragment ions that are unique to either spectrum cannot be matched by the algorithm but are included at peak selection for scoring. This results in noisier spectra, with uncorrelated ions scoring lower. The statistical nature of the Andromeda algorithm also allows for the generation of a meaningful combined score from the product of the probabilities, generated for each of the of the two spectra.
To calculate the score first, the spectrum being investigated is divided up into sequential 100 Th ranges (one Thompson (Th) is defined as 1 Da divided by the charge on an electron). Within each of these segments, a set number of peaks are selected in descending order of intensity; the number of peaks selected is defined as q. Then these are matched to a theoretical fragmentation of the cross-linked peptide, and the score for each range is probability that that number of ions (or more) is matched by chance. The score for each range is then maximized by optimizing the value of q between 1 and value defined by the user (Figure 2). For this study, the maximum value of q was set to 5. The aim of the process is for the matching of higher intensity peaks within each range to result in a higher score.37 The score of each range is then combined to form an overall probability that the match is by chance for the spectrum. The value returned to the user is then calculated as the minus 10 times the natural logarithm of this value. Further, we provide an option of a threshold value, configured as a percent of maximum intensity, below which fragmentation data is discarded; this was set to 2% within this study. As the algorithm relies on matching peaks, much of this process can be achieved rapidly by utilizing the count function within SQLite. A database table of theoretical ions is generated at the start of the scoring each prospective peptide. By joining these tables of theoretical peaks to the experimental data as part of a database query it is possible directly retrieve the number of peak matches between the two tables.
This method of scoring has a particular advantage in that it is probabilistically based and the reported value tends to zero when no fragment ions are detected. Additionally, we can use the probabilistic nature of the score to combine the underlying probabilities in the cases in which we have multiple spectra for the same species (for example, when using an isotope labeled cross-linking reagent), allowing us to increase the confidence of the assignment.
When available, Hekate Score makes use of the extra information provided from the combination of both the labeled and unlabeled fragmentation spectra. Initially, the spectra are normalized and peaks are categorized into either those that show similar intensity and a mass shift between the two spectra, peaks that show similar intensity but no mass shift, or those that cannot be matched (Figure 3). When matching theoretical peaks to those in the fragmentation data acquired, the algorithm will only score peaks that show the appropriate mass shift between the two spectra. Peaks that cannot be matched in this way are still included in the scoring process but will not be matched as fragment ions. The result is that spectral pairs with a large number of these unpaired ions will score lower. If a stable-isotope-labeled cross-linking reagent is not used and therefore this information is not available, then the scoring is still possible. In these cases, all peaks are considered as potential matches for the theoretical fragmentation ions. The effect on Hekate scores when the information provided by isotope labels is not used is shown in Figure S3.
Figure 3.

The use of an isotope labeled cross-linking reagent in a ratio of 1:1 to the unlabeled reagent at the time of the cross-linking reaction allows for more accurate fragment ion matching. Only fragment ions that still contain the cross-link will show a characteristic shift on comparison of the unlabeled (top) and labeled (bottom) peptide spectrum; this effect is highlighted for βy21+ and αb41+ (inset A). While fragment ions that do not contain the linking regent will not contain the label in either spectra and therefore no shift will be seen, as shown for αy31+ (inset B).
To ascertain the position of variable modifications, including monolinks and cross-links, the fragmentation of all possible positions is calculated and scored separately. The position that returns the highest score is then stored.
Hekate Search combines the four tools into a single workflow (Figure 1B) to provide detection and scoring of possible cross-linked peptides within a data set. The mass spectrometry data are input as a file in Mascot Generic Format (MGF) through a web form along with details of the experiment. Protein sequence data is provided in FASTA format. Sequence data and other search-specific settings can be preconfigured via the Settings web interface for repeat use. No user input is required once the search is submitted.
Hekate Search outputs a list of results for all peptides that were matched, within the user specified tolerance, by accurate mass provided that they have a score greater than zero.
As described in detail below, with respect to false discovery rates (FDR), the analysis of individual cross-links is often important to validating results. The cross-link results are provided as a list, sorted by score. A preview of any scan can be viewed by hovering the mouse over the scan number to aid the rapid validation of data.
A detailed view of the fragmentation ion spectra may be displayed in “Hekate Viewer”, which provides a fully interactive view of spectra. The isotopic labeled fragmentation pattern is shown underneath the unlabeled spectra to help the user visualize the peak shifts between the two spectra. The software provides a table showing all matched and unmatched theoretical fragment ions, to aid the user.
False Discovery
While efforts have been made to develop methods for calculating false discovery rates for application in XL-MS experiments, the data generated provides a unique set of problems. In a traditional experiment, for example, the analysis of bands from an SDS-page gel, the aim is to identify the proteins within a mixture. In these situations, redundancy is achieved by the analysis of multiple peptides from the parent protein within the mixture, and thus, a statistically significant method is available for the effective analysis of the data and the number of independent results in the data set, n, is much greater than one. Whereas in XL-MS, a detected cross-linked peptide may provide the sole representative for a particular structural restraint. The result can lead to an over-reliance on a single fragmentation pattern to confirm an interaction. Additionally, we cannot use technical replicates to provide a solution to this, as misassigned peptides are unlikely to produce different fragment spectra under repeat conditions and thus continue to be reassigned incorrectly. It is not therefore possible to produce a strong statistical method to verify each interaction independently as for this situation, n = 1, thus over-reliance on any score method should be avoided. Instead independent verification is required of any result from such a study.
However, notwithstanding the limitations, a false discovery can still be a useful tool in the analysis of processed data, and for this reason one is provided by Hekate. To calculate the false discovery rate, the fragmentation data is additionally scored against a decoy database.42 The decoy database is initially generated by in silico digestion of both forward and reversed input sequences. At this point, there are multiple options for how the decoy cross-links are generated. We propose that the generation of both standard decoy cross-linked peptides and hybrid-decoy peptides, that is, those created by the combination of decoy peptides with predicted peptides, provides the most satisfactory solution. The alternative is to use a decoy database containing only the direct combination nondecoy and decoy peptides without the formation of hybrid-decoy peptides; however, it was felt that this did not account for the occasions when only a single chain of a cross-link peptide was correctly scored. The false discovery rate is then calculated as the percent of matches at that score or greater that are matched to either a decoy, or to a hybrid decoy peptide out of the total number of scored spectra. Importantly, this additionally includes the scores of peptides to spectra that were not the top scoring spectra. This we believe provided the most stringent method for the calculation of a false discovery rate, but has clear limitations due to the small database size used and the limited amount of data that is used to generate this value. Additionally, it should be noted, because of the inclusion of hybrid decoy peptides, the ratio of forward peptides to reverse and to hybrid peptides tends to a value of 1:1:2 (F:R:H); as both reverse and hybrid peptides represent a decoy peptide, this gives a final result of 1:3. This is in contrast to a usual reverse decoy where the ratio is 1:1 (F:R). This means the base false discovery rate on selection by chance is 75% compared to 50% of a linear peptide database. These difficulties have been independently noted in other methods for the identification of cross-linked peptides36 with a similar conclusion on how to calculate a false discovery rate.33
Exporting to PyMOL for the Three-Dimensional Visualization, Validation, and Measurement of Linkage Distances
The ability to visualize the cross-links within crystallographic models was thought to be a key feature for the interpretation of data. To facilitate this, Hekate is able to export the cross-links and monolinks into a script that can be read by PyMOL, a widely used molecular visualization program.44 Once exported into PyMOL, the distances are automatically measured and displayed to allow rapid comparison of results from the cross-link study and known data.
If the structure contains a difference in sequence to that used within the cross-linking study it is possible to provide a correction in the PyMOL output. In the case of a preceding sequence, for example, a tag, this is addressed by providing Hetake with a correction value for the resultant shift in sequence numbering. In the cases of multiple subunits all are picked by default for export as PyMOL already then provides the functionality to manipulate the visibility of these cross-links once imported. For more complex variation in structure we propose the method as described in Validation 2.
Validation 1: Cytochrome c
Cytochrome c from bovine heart is a small globular electron carrier protein of molecular weight 12 230 Da (105 amino acids). It is a heme-containing protein and is an essential component of the electron transport chain in mitochondria. The heme group accepts electrons from the b-c1 complex (Complex III) and transfers them to the cytochrome oxidase protein complex (Complex IV). The protein has a compact structure and is well characterized.41 Its small size makes it an attractive test for XL-MS due to the low complexity of the theoretical cross-linked digestion product produced on incubation with trypsin. The primary structure contains 18 lysine residues separated by a range of distances, which provide the required reactivity for an activated carboxylic acid derivative based cross-linking reagent.
Twelve cross-linked peptides were detected by LC-MS/MS analysis of the tryptic digestion product of cytochrome c after incubation with BS3 (Table S2). The locations of the detected interactions were then exported to PyMOL (Figure 4). As predicted, these were consistent with the reported structure.1,30,45
Figure 4.

Cytochrome c. The detected cross-links are shown in red. Three cross-links span the N- and C-terminal α-helices demonstrating cross-linking between two regions that share no proximity within the protein sequence but are topologically near to each other. During the folding of cytochrome c, the N- and C-terminal α-helices form a close tertiary structure between the intermediate I and intermediate II structures. The intermediate II then forms the native structure with the remaining α-helices and coordination of Met 80 to the heme.24,50 The prevalence of cross-linking between these positions within the protein appears to be consistent with this model of cytochrome c folding. The graph shows the distribution of Cα–Cα distances of the detected cross-links.
Validation 2: DNA Pol III
DNA Pol III is the catalytic α subunit of the bacterial DNA replication machinery, a large complex of more than 10 different proteins. DNA Pol III (molecular weight: 130 kDa) is roughly 10 times bigger than cytochrome c and one of the largest single proteins in E. coli. Due to its size (1160 amino acids), we faced a number of challenges. Because of the nature of the generation of peptides from a cross-linking reaction, the predictive capability using accurate mass alone is vastly inferior compared to a linear peptide search. This is because the database produced by the in silico digest of protein is small, even for a reasonably sized protein, and therefore, many of the theoretical peptides will have a uniquely identifiable accurate mass (i.e., not within 2 ppm of another peptide). It is this that forms the basis of peptide mass fingerprinting experiments (PMF) for the identification of proteins without the need for further sequence information.46,47 The result of this increased search space is that any given doublet is much more likely to match multiple different theoretical species by chance. Thus, much like when using a large multiple-proteome wide database, the scoring of the peptide fragmentation data is essential as it serves to resolve which of several potential matches by accurate mass is most likely. The results of our own developed scoring algorithm for all possible species within 2 ppm against a single fragmentation pattern are shown in Table 1. In Figure 5, we show a visual comparison of the scoring process between two of these potential sequences. Using the known part of the E. coli DNA Pol III structure, we could verify what scores of our algorithm reflect a bona fide cross-link and at which number we expect false positives. For this, we measured the distances between cross-linked lysines in PyMOL. The detailed results of the initial study are listed in Table S3, which contains all the doublets detected within 2 ppm of a theoretical peptide mass.
Table 1. Scoring of All Possible Matches to Detected Doublet with Base Peak m/z = 622.66815a.
| score | charge | ppm | residue 1 | residue 2 | sequence |
|---|---|---|---|---|---|
| 776 | 3+ | 0.15 | 621 | 983 | DLIKR-LKDMHPTER |
| 130 | 3+ | 0.15 | 294 | 722 | DYLVKRAK-EmAKQR |
| 94 | 3+ | 1.59 | 743 | 872 | LAMKIFDLVE-TDTKK |
| 61 | 3+ | 0.23 | 796 | 551 | KVVGLVDE-YAGLVKFD |
Oxidized methionine residues are indicated by a lowercase “m”.
Figure 5.

Comparison of fragmentation pattern matching of the highest (A) and second highest (B) matches to detected doublet with bass peak m/z = 622.66815. The lower score represents the lower correlation between the recorded and theoretical fragmentation. Only prominent ions are labeled. A lowercase “m” is used to represent oxidized methionine residues.
The distances given are measured from protein-backbone (Cα atom) to protein-backbone of the specified lysine residues. Most measured distances are equal or shorter than the expected distance for a cross-linked lysine pair: 2 × length of a lysine side chain (2 × 6.4 Å) + length of the cross-linker: 11.4 BS3 or 7.7 Å for BS2G. It is also possible that flexibility within the protein structure will allow for residues separated by greater distances to form cross-links. Analysis of cross-links for two test proteins showed a strong correlation at high scores (>300) between the structure and peptides found (see Table S3 and Figure 6). Below this value, cross-links gradually became less reliable with an increased likelihood that the sequence implies interactions that are not possible due to the distance they span or that contradict the known tertiary structure of the protein. Multiple subsequent studies were carried out in a similar fashion using both BS2G and BS3 as a cross-linking reagent and the combined results are shown in Table 2.
Figure 6.

Comparison of cross-links detected in study within the known structure elements of DNA Pol III. (A) Detected cross-links from the analysis of 24 LC-MS/MS experiments involving the alpha subunit that had a score greater 300 are shown in green. All these cross-links are in agreement with the known structural elements. (B) A selection of cross-links whose parent ion matched with 2 ppm of the proposed sequence but where scored less than 300. These cross-links are over a greater than the proposed maximum distance with the exception of 297–410 in this case the cross-link is blocked by the tertiary structure of the protein. (C) Graph showing the combined distribution of Cα–Cα cross-link distances in (A) and (B).
Table 2. Complete List of Intramolecular Cross-Links Detected and Characterized during the Analysis of 24 LC-MS/MS Experiments Involving Polymerase III and Various Cross-Linking Reagentsa.
| m/z | charge | ppm | residue 1 | residue 2 | sequence | reagent |
|---|---|---|---|---|---|---|
| 906.5195 | 2+ | 1.04 | 29 | 714/5 | TAPLVKK-KKPEEMAK | 2 |
| 561.0731 | 4+ | 0.12 | 29/30 | 722 | AMGKKKPEEMAK-TAPLVKK | 3 |
| 677.6235 | 4+ | 0.43 | 229 | 1009 | VAIHDGFTLDDPKRPR-VMVTKR | 3 |
| 580.0756 | 4+ | 1.84 | 316 | 595 | LKR-NGEPPLDIAAIPLDDKK | 3 |
| 705.8922 | 4+ | 0.23 | 439 | 1009 | DAVSQIITFGTMAAKAVIR-VMVTKR | 3 |
| 1052.2451 | 3+ | 1.45 | 461 | 881 | ISKLIPPDPGMTLAK-VLEKLIMSGAFDR | 2 |
| 603.0954 | 4+ | 0.41 | 461 | 1009 | ISKLIPPDPGMTLAK-VMVTKR | 2 |
| 590.5920 | 4+ | 0.33 | 500 | 510 | KLEGVTR-NAGKHAGGVVIAPTK | 3 |
| 418.9995 | 4+ | 1.11 | 500 | 1009 | KLEGVTR-VMVTKR | 3 |
| 764.1073 | 3+ | 0.44 | 510 | 1009 | NAGKHAGGVVIAPTK-VMVTKR | 3 |
| 437.2353 | 5+ | 0.12 | 617 | 983 | GMKDLIKR-LKDMHPTER | 2 |
| 622.6682 | 3+ | 0.15 | 621 | 983 | DLIKR-LKDMHPTER | 2 |
| 510.2637 | 4+ | 0.29 | 855 | 872 | NKGGYFR-TDTKK | 3 |
| 637.6063 | 4+ | 1.69 | 983 | 992 | LKDMHPTER-GKVITAAGLVVAAR | 3 |
Multiple residue positions indicate that more than one cross-link between the peptides was identified. The reagent is represented as either a 2 or 3 for BS2G or BS3, respectively. For clarity where multiple peptides were detected for the same interaction, only one is shown.
Localization of the C-Terminal Domain of DNA Pol III
The structure of the catalytic domain (residues 1:910) of E. coli Pol III has been determined to a resolution of 2.3 Å,38 but the structure of the C-terminal domain (residues 911:1160) still remains elusive. The homologous structure of Thermus aquaticus Pol III was determined for the full-length protein. However, this structure was determined at a considerably lower resolution (3.0 Å) and suffered from poorly defined electron density in the C-terminal domain.48 Therefore, it seemed a reasonable approach to use XL-MS to determine the position of the C-terminal domain in E. coli Pol III, especially because the known part of the protein could be used as a positive control. To visualize the cross-links in the C-terminal domain of E. coli Pol III, we created a model in Modeler45 using the structure of Taq Pol III as a template. The observed cross-links to and within the C-terminal domain fit well with our model, indicating that the C-terminal domain adopts a similar position in both Taq and E. coli Pol III (Figure 7). Furthermore, we have also expanded our search beyond the polymerase alone and characterized by XL-MS the interaction between Pol III and its direct binding partners the sliding clamp β and the proofreading subunit ε, providing a first structural model of the catalytic core of the bacterial DNA replication machinery.2
Figure 7.
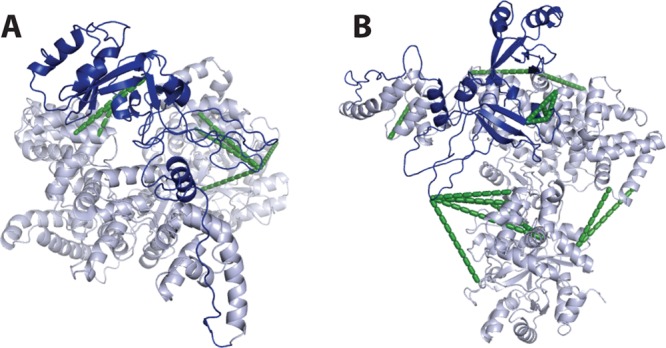
DNA Pol III with modeled C-terminal domain in dark blue. (A) Cross-links from the known structural elements to the previously uncharacterized C-terminal domain are shown in green. (B) An overview of the polymerase showing the modeled C-terminal domain with all cross-links detected in this study.
Conclusions
The two proteins discussed here establish the utility of the Hekate suite for the analysis of data generated in XL-MS experiments to facilitate the modeling and refinement of protein structures. Additionally, our software suite has been used in two more published studies.2,49 This methodology can be of great advantage, either when traditional high-resolution methods have been unsuccessful or provided only limited results on a region of interest. There is additional opportunity to use this technique in parallel to high-resolution techniques to provide an early entry point in the investigation of domain level and protein level structural characteristics.
While protein cross-linking will not displace the clarity provided by high-resolution techniques, as mass spectrometry technologies increase in sensitivity and the computational power increases the ability to investigate larger and more complex data, the method is set to become more commonplace. Combined with other low-resolution techniques including SAXS, hydrogen/deuterium exchange and single particle analysis, it adds to the ever-increasing resources for structural analysis. It is the Hekate suite’s particular focus on providing results in a format to facilitate collaboration, for example, to export crystallographic package to PyMOL, which will drive these fields of research. The Hekate suite and source code are available from either http://evath.net/research/hekate/ or the online code-management tool platform GitHub located at https://github.com/MRC-LMB-MassSpec/Hekate.
Acknowledgments
This work was supported by Medical Research Council.
Supporting Information Available
Figure and video showing the Hekate software interface; comparision of Hekate scores against Mascot; analysis of the effect on Hekate scores when the information provided by isotope labels is not used; overview of data structure used by Hekate; This material is available free of charge via the Internet at http://pubs.acs.org
Accession Codes
The accession numbers as listed by UniProt for the proteins discussed in this work are as follows. DNA polymerase III subunit alpha, P10443; and Cytochrome c, P62894.
The authors declare no competing financial interest.
Supplementary Material
References
- Mirkin N.; Jaconcic J.; Stojanoff V.; Moreno A. High resolution X-ray crystallographic structure of bovine heart cytochrome c and its application to the design of an electron transfer biosensor. Proteins 2007, 70, 83–92. [DOI] [PubMed] [Google Scholar]
- Rego A. T.; Holding A. N.; Kent H.; Lamers M. H. Architecture of the Pol III-clamp-exonuclease complex reveals key roles of the exonuclease subunit in processive DNA synthesis and repair. EMBO J. 2013, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybylski M.; Glocker M. O.; Nestel U.; Schnaible V.; Blüggel M.; Diederichs K.; Weckesser J.; Schad M.; Schmid A.; Welte W.; Benz R. X-ray crystallographic and mass spectrometric structure determination and functional characterization of succinylated porin from Rhodobacter capsulatus: implications for ion selectivity and single-channel conductance. Protein Sci. 1996, 5, 1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorca O. Introduction to 3D reconstruction of macromolecules using single particle electron microscopy1. Acta Pharmacol. Sin. 2005, 26, 1153–1164. [DOI] [PubMed] [Google Scholar]
- Leitner A.; Walzthoeni T.; Kahraman A.; Herzog F.; Rinner O.; Beck M.; Aebersold R. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol. Cell. Proteomics 2010, 9, 1634–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti M.; Leggio C.; Federici L.; De Lorenzo G.; Pavel N. V.; Cervone F. Structural Resolution of the Complex between a Fungal Polygalacturonase and a Plant Polygalacturonase-Inhibiting Protein by Small-Angle X-Ray Scattering. Plant Physiol. 2011, 157, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques D. A.; Trewhella J. Small-angle scattering for structural biology-Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Pan J.; Liu Y.-H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224. [DOI] [PubMed] [Google Scholar]
- Hernandez H. Dynamic Protein Complexes: Insights from Mass Spectrometry. J. Biol. Chem. 2001, 276, 46685–46688. [DOI] [PubMed] [Google Scholar]
- Barrera N. P.; Robinson C. V. Advances in the Mass Spectrometry of Membrane Proteins: From Individual Proteins to Intact Complexes. Annu. Rev. Biochem. 2011, 80, 247–271. [DOI] [PubMed] [Google Scholar]
- Clegg C.; Hayes D. Identification of neighbouring proteins in the ribosomes of Escherichia coli. A topographical study with the cross-linking reagent dimethyl suberimidate. Eur. J. Biochem. 1974, 42, 21–28. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Siniossoglou S.; Hurt E. C.; Mann M. A Generic Strategy To Analyze the Spatial Organization of Multi-Protein Complexes by Cross-Linking and Mass Spectrometry. Anal. Chem. 2000, 72, 267–275. [DOI] [PubMed] [Google Scholar]
- Young M. M.; Tang N.; Hempel J. C.; Oshiro C. M.; Taylor E. W.; Kuntz I. D.; Gibson B. W.; Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5802–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley S. C. Building a Replisome Solution Structure by Elucidation of Protein-Protein Interactions in the Bacteriophage T4 DNA Polymerase Holoenzyme. J. Biol. Chem. 2001, 276, 39340–39349. [DOI] [PubMed] [Google Scholar]
- Back J. W.; Hartog A. F.; Dekker H. L.; Muijsers A. O.; de Koning L. J.; de Jong L. A new crosslinker for mass spectrometric analysis of the quaternary structure of protein complexes. J. Am. Soc. Mass Spectrom. 2001, 12, 222–227. [DOI] [PubMed] [Google Scholar]
- Kühn-Hölsken E.; Lenz C.; Sander B.; Lührmann R.; Urlaub H. Complete MALDI-ToF MS analysis of cross-linked peptide-RNA oligonucleotides derived from nonlabeled UV-irradiated ribonucleoprotein particles. RNA 2005, 11, 1915–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J. R.; Ruse C. I.; Nakorchevsky A. Proteomics by Mass Spectrometry: Approaches, Advances, and Applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [DOI] [PubMed] [Google Scholar]
- Guerrero C. An Integrated Mass Spectrometry-based Proteomic Approach: Quantitative Analysis of Tandem Affinity-purified in vivo Cross-linked Protein Complexes (qtax) to Decipher the 26 s Proteasome-interacting Network. Mol. Cell. Proteomics 2005, 5, 366–378. [DOI] [PubMed] [Google Scholar]
- Wagner G.; Hyberts S. G.; Havel T. F. NMR structure determination in solution: a critique and comparison with X-ray crystallography. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 167–198. [DOI] [PubMed] [Google Scholar]
- Pearson K. M.; Pannell L. K.; Fales H. M. Intramolecular cross-linking experiments on cytochrome c and ribonuclease A using an isotope multiplet method. Rapid Commun. Mass Spectrom. 2002, 16, 149–159. [DOI] [PubMed] [Google Scholar]
- Staros J. V. N-hydroxysulfosuccinimide active esters: bis(N-hydroxysulfosuccinimide) esters of two dicarboxylic acids are hydrophilic, membrane-impermeant, protein cross-linkers. Biochemistry 1982, 21, 3950–3955. [DOI] [PubMed] [Google Scholar]
- Hanai R.; Wang J. C. Protein footprinting by the combined use of reversible and irreversible lysine modifications. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 11904–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. A.; Jawhari A.; Fischer L.; Buchen C.; Tahir S.; Kamenski T.; Rasmussen M.; Lariviere L.; Bukowski-Wills J.-C.; Nilges M.; Cramer P.; Rappsilber J. Architecture of the RNA polymerase II – TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010, 29, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Munske G. R.; Siems W. F.; Bruce J. E. Mass Spectrometry Identifiable Cross-Linking Strategy for Studying Protein–Protein Interactions. Anal. Chem. 2005, 77, 311–318. [DOI] [PubMed] [Google Scholar]
- Rinner O.; Seebacher J.; Walzthoeni T.; Mueller L.; Beck M.; Schmidt A.; Mueller M.; Aebersold R. Identification of cross-linked peptides from large sequence databases. Nat. Methods 2008, 5, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihling C.; Schmidt A.; Kalkhof S.; Schulz D. M.; Stingl C.; Mechtler K.; Haack M.; Beck-Sickinger A. G.; Cooper D. M. F.; Sinz A. Isotope-labeled cross-linkers and fourier transform ion cyclotron resonance mass spectrometry for structural analysis of a protein/peptide complex. J. Am. Soc. Mass Spectrom. 2006, 17, 1100–1113. [DOI] [PubMed] [Google Scholar]
- Müller D. R.; Schindler P.; Towbin H.; Wirth U.; Voshol H.; Hoving S.; Steinmetz M. O. Isotope-Tagged Cross-Linking Reagents. A New Tool in Mass Spectrometric Protein Interaction Analysis. Anal. Chem. 2001, 73, 1927–1934. [DOI] [PubMed] [Google Scholar]
- Liu F.; Wu C.; Sweedler J. V.; Goshe M. B. An enhanced protein crosslink identification strategy using CID-cleavable chemical crosslinkers and LC/MSn analysis. Proteomics 2012, 12, 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu E. T.; Hawkins A.; Eaton J.; Fabris D. MS3D structural elucidation of the HIV-1 packaging signal. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 12248–12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P.; Panchaud A.; Goodlett D. R. Chemical Cross-Linking and Mass Spectrometry As a Low-Resolution Protein Structure Determination Technique. Anal. Chem. 2010, 82, 2636–2642. [DOI] [PubMed] [Google Scholar]
- Götze M.; Pettelkau J.; Schaks S.; Bosse K.; Ihling C. H.; Krauth F.; Fritzsche R.; Kühn U.; Sinz A. StavroX—A Software for Analyzing Crosslinked Products in Protein Interaction Studies. J. Am. Soc. Mass Spectrom. 2011, 23, 76–87. [DOI] [PubMed] [Google Scholar]
- Rasmussen M. I.; Refsgaard J. C.; Peng L.; Houen G.; Højrup P. CrossWork: Software-assisted identification of cross-linked peptides. J. Proteomics 2011, 74, 1871–1883. [DOI] [PubMed] [Google Scholar]
- Walzthoeni T.; Claassen M.; Leitner A.; Herzog F.; Bohn S.; Förster F.; Beck M.; Aebersold R. False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 2012, 9, 901–903. [DOI] [PubMed] [Google Scholar]
- Gao Q.; Xue S.; Doneanu C. E.; Shaffer S. A.; Goodlett D. R.; Nelson S. D. Pro-CrossLink. Software Tool for Protein Cross-Linking and Mass Spectrometry. Anal. Chem. 2006, 78, 2145–2149. [DOI] [PubMed] [Google Scholar]
- Du X.; Chowdhury S. M.; Manes N. P.; Wu S.; Mayer M. U.; Adkins J. N.; Anderson G. A.; Smith R. D. Xlink-Identifier: An Automated Data Analysis Platform for Confident Identifications of Chemically Cross-Linked Peptides Using Tandem Mass Spectrometry. J. Proteome Res. 2011, 10, 923–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Wu Y.-J.; Zhu M.; Fan S.-B.; Lin J.; Zhang K.; Li S.; Chi H.; Li Y.-X.; Chen H.-F.; Luo S.-K.; Ding Y.-H.; Wang L.-H.; Hao Z.; Xiu L.-Y.; Chen S.; Ye K.; He S.-M.; Dong M.-Q. Identification of cross-linked peptides from complex samples. Nat. Methods 2012, 9, 904–906. [DOI] [PubMed] [Google Scholar]
- Cox J.; Neuhauser N.; Michalski A.; Scheltema R. A.; Olsen J. V.; Mann M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Lamers M. H.; Georgescu R. E.; Lee S.-G.; O’Donnell M.; Kuriyan J. Crystal Structure of the Catalytic α Subunit of E. coli Replicative DNA Polymerase III. Cell 2006, 126, 881–892. [DOI] [PubMed] [Google Scholar]
- Maki H.; Kornberg A. The polymerase subunit of DNA polymerase III of Escherichia coli. II. Purification of the alpha subunit, devoid of nuclease activities. J. Biol. Chem. 1985, 260, 12987–12992. [PubMed] [Google Scholar]
- Allen G.; Owens M.. The definitive guide to SQLite; Springer Science+Business Media, LLC: New York, 2010 [Google Scholar]
- Fisher W.; Taniuchi H.; Anfinsen C. On the Role of Heme in the Formation of the Structure of Cytochrome c. J. Biol. Chem. 1973, 248, 3188–3195. [PubMed] [Google Scholar]
- Maiolica A.; Cittaro D.; Borsotti D.; Sennels L.; Ciferri C.; Tarricone C.; Musacchio A.; Rappsilber J. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol. Cell Proteomics 2007, 6, 2200–2211. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13417–13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.6, Schrödinger LLC.
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M.-Y.; Pieper U.; Sali A.. Comparative protein structure modeling using Modeller. Curr. Protoc Bioinf. 2006, Chapter 5, Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J. R.; Speicher S.; Griffin P. R.; Hunkapiller T. Peptide mass maps: a highly informative approach to protein identification. Anal. Biochem. 1993, 214, 397–408. [DOI] [PubMed] [Google Scholar]
- James P.; Quadroni M.; Carafoli E.; Gonnet G. Protein identification by mass profile fingerprinting. Biochem. Biophys. Res. Commun. 1993, 195, 58–64. [DOI] [PubMed] [Google Scholar]
- Bailey S.; Wing R. A.; Steitz T. A. The Structure of T. aquaticus DNA Polymerase III Is Distinct from Eukaryotic Replicative DNA Polymerases. Cell 2006, 126, 893–904. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Salter H. K.; Holding A. N.; Johnson C. M.; Stephens E.; Lukavsky P. J.; Walshaw J.; Bullock S. L. Bicaudal-D uses a parallel, homodimeric coiled coil with heterotypic registry to coordinate recruitment of cargos to dynein. Genes Dev. 2013, 27, 1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama S.; Takahashi S.; Ishimori K.; Morishima I. Stepwise formation of alpha-helices during cytochrome c folding. Nat. Struct. Biol. 2000, 514–520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.