

Abstract
Mycobacteria use specialized ESX secretion systems to transport proteins across their cell membranes in order to manipulate their environment. In pathogenic Mycobacterium tuberculosis there are five paralogous ESX secretion systems, named ESX-1 through ESX-5. Each system includes a substilisin-like protease (mycosin or MycP) as a core component essential for secretion. Here we report crystal structures of MycP1 and MycP3, the mycosins expressed by the ESX-1 and ESX-3 systems, respectively. In both mycosins the putative propeptide wraps around the catalytic domain and does not occlude the active site. The extensive contacts between the putative propeptide and catalytic domain, which include a disulfide bond, suggest that the N-terminal extension is an integral part of the active mycosin. The catalytic residues of MycP1 and MycP3 are located in a deep active site groove in contrast with an exposed active site in majority of subtilisins. We show that MycP1 specifically cleaves ESX-1 secretion-associated protein B (EspB) in vitro at residues Ala358 and Ala386. We also systematically characterize the specificity of MycP1 using peptide libraries, and show that it has evolved a narrow specificity relative to other subtilisins. Finally, comparison of the MycP1 and MycP3 structures suggest that both enzymes have stringent and different specificity profiles that result from the structurally distinct active site pockets, which could explain the system specific functioning of these proteases.
Keywords: MycP, subtilisin, M. tuberculosis, type VII secretion system, ESX, EspB
1. Introduction
Tuberculosis (TB) is a devastating infectious disease caused by Mycobacterium tuberculosis that resulted in more than 8.7 million new cases and 1.4 million deaths in 2011 (Floyd, 2012). M. tuberculosis possesses five related secretion pathways designated ESX-1 through ESX-5 that are involved in the transport of multiple proteins across the complex cell envelop (Abdallah et al., 2007; Stoop et al., 2012). Although the ESX secretion pathways mediate secretion of proteins across two membranes, they are unlike the known secretion pathways for Gram-negative bacteria and, as such, are also referred to as type VII secretion (Abdallah et al., 2007). The importance of ESX-1 secretion for virulence has been established through comparison of mycobacterial strains containing the full complement of ESX secretion genes versus avirulent strains with deleted regions, such as the Mycobacterium bovis BCG vaccine strain (Guinn et al., 2004; Majlessi et al., 2005; Pym et al., 2002). The ESX-1 secretion apparatus is involved in early bacterial replication in macrophages and contributes to virulence by allowing escape of mycobacteria from the phagosome into the cytosol of infected macrophages (Houben et al., 2012a; Simeone et al., 2012). In contrast, the ESX-3 system is essential for mycobacteria, possibly because of its role in iron acquisition (Serafini et al., 2009; Siegrist et al., 2009). The ESX-5 system of M. tuberculosis is responsible for secretion of multiple proteins of the PE and PPE families that modulate cell wall integrity and macrophage responses (Abdallah et al., 2009; Bottai et al., 2012; Houben et al., 2012a). The functions of ESX-2 and ESX-4 systems in mycobacteria are still completely unknown.
A comparison of five ESX loci present in mycobacteria and ESX clusters identified in closely related species, such as Nocardia farcinica and Streptomyces sp. allowed identification of a set of genes that likely encode the core components of the ESX secretion systems (Guinn et al., 2004; Houben et al., 2012b). Interestingly, all known ESX clusters encode their own subtilisin-like proteases, named mycosins or MycP1 through MycP5 (Brown et al., 2000). Mycosins are membrane proteins with extracytoplasmic serine protease domains (Dave et al., 2002). The functional role of MycP family proteases in the secretion process is poorly understood, but because MycP3 is essential for in vitro growth of M. tuberculosis H37Rv (Griffin et al., 2011; Sassetti et al., 2003), we can infer that these mycosins can not substitute for each other. A substrate has only been identified for MycP1 protease, which is the ESX-1 secretion-associated protein B (EspB), a secreted substrate of ESX-1, in vitro and in vivo (Ohol et al., 2010). EspB is a glycine-rich protein essential for ESX-1 secretion and required for virulence and growth of bacteria in macrophages (Xu et al., 2007). Importantly, the other ESX systems do not seem to secrete an obvious EspB homologue. Furthermore, mycosins seem to have a dual function in ESX secretion, as deletion of the mycP1 gene results in a lack of secretion, however inactivation of MycP1 catalytic activity leads to increased secretion of ESX-1 substrates (Ohol et al., 2010). Importantly, the catalytically inactivated mutant is attenuated in mice compared to wild-type M. tuberculosis (Ohol et al., 2010), suggesting that MycP1 inhibitors might have efficacy as anti-tuberculosis antibiotics.
To better understand the system-specific role of mycosins in the ESX secretion systems, we determined the crystal structures of two mycosins, Mycobacterium thermoresistibile MycP1mth and Mycobacterium smegmatis MycP3msm. We also carried out a detailed characterization of MycP1mth substrate recognition motifs. In addition, we compared our findings with the recently reported structure of M. smegmatis MycP1msm (Solomonson et al., 2013). Our structural and biochemical analyses of MycP1 and MycP3 provide insights into the molecular recognition mechanisms used by this family of enzymes. These insights are important for understanding the diverse roles of individual ESX secretion systems, and provide a starting point for rational design of mycosin-targeted therapeutics.
2. Materials and methods
2.1. Cloning, expression and purification of MycP1mth, MycP1msm, MycP3msm and EspBmtu
The DNA fragment corresponding to the soluble domain of MycP1mth (residues 24–393) was PCR-amplified from genomic DNA of M. thermoresistibile ATCC 19527 and cloned into pET-22b vector (EMD Millipore) for periplasmic expression with an N-terminal pelB signal sequence and a C-terminal tobacco etch virus (TEV) protease cleavage site followed by His6-tag. Alternatively, the same DNA fragment was cloned into pET-21d vector (EMD Millipore) for cytoplasmic expression with a C-terminal TEV protease cleavage site and His6-tag. Initial crystallization screening and mass-spectrometry analysis of disulfide bonds were performed with MycP1mth produced in the periplasm. The crystal growth optimization and kinetic analysis was performed with MycP1mth produced in the cytoplasm. The gene fragment corresponding to the soluble domain of MycP1msm (residues 24–396) was amplified from genomic DNA of M. smegmatis mc2 155 (ATCC Catalog No. 700084) and cloned into a modified pETDuet-1 vector (EMD Millipore) to encode an N-terminal His6-tag and a TEV protease cleavage site. The DNA fragment encoding the soluble domain of MycP3msm (residues 26–403) was cloned into a modified pET-22b vector to include pelB signal sequence followed by a His6-tag and a TEV protease cleavage site. The DNA fragment encompassing full-length EspBmtu and EspLmtu genes was amplified from genomic DNA of M. tuberculosis H37Rv and cloned into a modified pET-28b vector (EMD Millipore) with an N-terminal His6-tag and a TEV protease cleavage site. The catalytically inactive mycosin variants MycP1mth S334A, and MycP3msm S341A were generated using the QuickChange (Stratagene) site-directed mutagenesis protocol. The plasmid to produce the N-terminal EspBmtu fragment (residues 1–358) was constructed by introducing a stop codon in position 359. All plasmids were sequenced to verify absence of inadvertent mutations.
All proteins were expressed in Rosetta (DE3) E. coli cells after induction with 0.5 mM IPTG for 4 h at 18 °C (mycosins) or at 24 °C (EspBmtu). Cells were harvested by centrifugation and resuspended in buffer containing 20 mM Tris-HCl pH 8.4, 300 mM NaCl, and 20 mM imidazole. The resuspended cells were lysed using EmulsiFlex-C5 (Avestin) and proteins were purified via a Ni-NTA column (Qiagen). Following the cleavage of His6-tag by TEV protease, proteins were purified on size-exclusion Superdex200 column (GE Biosciences) in buffer containing 20 mM HEPES pH 7.5 and 100 mM NaCl.
2.2. Identification of MycP1 disulfide bonds by mass-spectrometry
MycP1mth samples were subjected to digestion with trypsin in solution. 6 μL of 10 ng μl−1 trypsin in 40 mM (NH4)HCO3 with 9% acetonitrile solution was added to a 50 μl sample MycP1mth (~10 μg ml−1 in 50 mM (NH4)HCO3) and incubated at 37°C for 16 h. Two sets of digested samples were prepared. One set was pre-treated with dithiothreitol and iodoacetamide before trypsin digest, while the other one was not. The digested solution was subjected to LC-MS/MS analysis as previously described (Kuo et al., 2012). LC-MS/MS analysis was performed using an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled with an Eksigent Nanoflex cHiPLC system (Eksigent) through a nanoelectrospray ionization source. The LC-MS/MS data were subjected to database searches for protein identification using Proteome Discoverer software V. 1.3 (Thermo Fisher Scientific) with a local MASCOT search engine. A custom database containing the protein of interest was used.
2.3. Cloning of MycP1mm-HA and MycP1mth-HA and expression in M. marinum
The DNA fragments corresponding to MycP1mm and MycP1mth were PCR-amplified from M. marinum E11 and M. thermoresistibile ATCC 19527 genomic DNA, respectively, using a 5′ primer containing an NheI restriction site and a 3′ primer that contains a BamHI restriction site and a HA-encoding sequence. After cloning of the PCR products into the pJet1.2 vector (Fermentas), the fragments were digested by NheI and BamHI and ligated into the E. coli-mycobacterial shuttle vector pSMT3::LipYtub (Daleke et al., 2011), which was cut with the same enzymes to remove the lipYtub gene. The resulting vectors, named pSMT3::MycP1mm-HA and pSMT3::MycP1mth-HA, were introduced in M. marinum E11 by electroporation. The various M. marinum strains were grown at 30°C in Middlebrook 7H9-broth (Difco) supplemented with 0.2% glycerol, 10% oleic acid-albumin-dextrose-catalase (OADC) (BBC) and 0.05% Tween 80. Expression and subcellular localization of the HA-tagged proteins in M. marinum were analyzed by subcellular fractionation followed by SDS-PAGE and immunoblotting as previously described (Houben et al., 2012b; van der Woude et al., 2013). Briefly, bacteria were grown until an OD600 of ~1, harvested by centrifugation and resuspended in PBS, 250 mM sucrose. The cells were lysed in a One-Shot cell disruptor and cellular debris was removed by centrifugation 3 times for 5 min at 3,000 × g. Subsequently, the cytosolic fraction was separated from the cell envelope fraction by centrifugation at 100,000 × g.
2.4. Crystallization and data collection
The initial crystal hits were found using JCSG Core Suites I–IV (Qiagen) (Lesley and Wilson, 2005). The thin rod-shaped MycP1mth crystals were obtained by vapor diffusion method mixing protein concentrated to 4.5 mg ml−1 and 0.2 M Zn acetate, 0.1 M imidazole pH 8.0, 25% 1,2-propanediol, 10% glycerol. Crystals were harvested directly from crystallization solution and flash-cooled in liquid N2 prior to data collection. The optimized MycP3msm crystals were produced using protein at concentration 10–14 mg ml−1 and 0.2 M MgCl2, 0.1 M Tris-HCl pH 8.4, 20% PEG8000, 0.01 M NiCl2. Crystals were cryoprotected in crystallization solution supplemented with 10% ethylene glycol and vitrified in liquid N2. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. All data were processed and scaled using XDS and XSCALE (Kabsch, 2010).
2.5. Structure determination and refinement
The crystals of MycP1mth belong to space group P212121 with one molecule in the asymmetric unit. The structure was solved by molecular replacement using BALBES (Long et al., 2008) and thermitase, a subtilisin-fold protease from Thermoactinomyces vulgaris (PDB 1THM), as a search model (Teplyakov et al., 1990). Following extensive manual rebuilding using Coot (Emsley et al., 2010) the model was completed and solvent molecules were added using ARP/wARP and AutoSolve module in PHENIX (Adams et al., 2010; Langer et al., 2008). The structure was refined to 2.0 Å resolution with Rwork 0.169 and Rfree 0.206 using REFMAC5 (Murshudov et al., 2011) and applying twelve translation, libration and screw-rotation displacement (TLS) groups determined by the TLSMD server (Painter and Merritt, 2006). The final model includes residues 24–391 of MycP1mth, seven Zn2+ ions identified by the presence of positive peaks in anomalous difference map, and other solvent molecules.
The crystals of MycP3msm belong to space group P1 with one molecule in the asymmetric unit. The structure was solved by molecular replacement using Phaser (McCoy et al., 2007) and MycP1mth structure without the N-terminal extension (residues 67–391) as a search model. The model was rebuilt using ARP/wARP and Coot. The structure was refined to 1.5 Å resolution with Rwork 0.151 and Rfree 0.185 using REFMAC5 and two TLS groups defined by the TLSMD server. The final model includes residues 26–399 of MycP3msm with the loop residues 241–245 missing due to disorder.
Data collection and refinement statistics are listed in Table 1. The quality of final models was assessed using Coot and the MolProbity server (Chen et al., 2010). The structural figures were generated using PyMol (www.pymol.org) and Chimera (Pettersen et al., 2004). The coordinates and structure factors were deposited to the Protein Data Bank with accession codes 4HVL (MycP1mth) and 4KG7 (MycP3msm).
Table 1.
Data collection and refinement statistics.
| MycP1mth (PDB 4HVL) | MycP3msm (PDB 4KG7) | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P1 |
| Cell dimensions | ||
| a, b, c (Å) | 61.95, 73.08, 75.42 | 42.82, 46.95, 47.54 |
| α β γ(°) | 90, 90, 90 | 77.27, 68.35, 74.47 |
| Resolution (Å) | 47.9–2.00 (2.11–2.00)1 | 43.8–1.50 (1.58–1.50) |
| Rsym | 0.117 (0.684) | 0.048 (0.411) |
| I/Iσ | 14.2 (2.2) | 15.6 (2.3) |
| Completeness (%) | 99.7 (98.0) | 91.4 (65.4) |
| Multiplicity | 6.7 (4.1) | 2.6 (2.5) |
| Refinement | ||
| Resolution (Å) | 47.9–2.00 | 43.8–1.50 |
| No. reflections (total/free) | 23667/1208 | 48069/2432 |
| Rwork/Rfree | 0.169/0.206 | 0.151/0.185 |
| No. atoms | ||
| Protein | 2653 | 2648 |
| Ligand/ion | 31 | 1 |
| Water | 209 | 533 |
| B-factors | ||
| Protein | 23.5 | 13.3 |
| Ligand/ion | 24.0 | 17.8 |
| Water | 31.1 | 27.0 |
| Wilson B | 28.5 | 19.2 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.010 | 0.010 |
| Bond angles (°) | 1.310 | 1.410 |
| Ramachandran distribution (%)2 | ||
| Favored | 97.6 | 96.4 |
| Outliers | 0.0 | 0.0 |
Values in parentheses are for the highest-resolution shell.
Calculated using the MolProbity server (http://molprobity.biochem.duke.edu).
2.6. In vitro assay of MycP1mth activity
EspB at a concentration of 0.15 mg ml−1 was incubated with 0.015 mg ml−1 MycP1mth or 0.015 MycP3msm at 37°C in reaction buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 2 mM CaCl2) for the indicated time periods. At each time point a 10 μL aliquot of the mixture was mixed with SDS loading buffer and heated at 95 °C for 5 min. The aliquots were analyzed on 15% SDS-PAGE gel. EspBmtu-C was separated on SDS-PAGE and transferred to PVDF membrane (Pierce) using standard protocols. The appropriate bands were excised and submitted to 5 cycles of N-terminal sequencing using the Edman degradation method. The sequencing was performed on an Applied Biosystems 494 Procise HTS sequencer, Tufts University Core Facility.
For quenched fluorescent peptide assays MycP1mth, MycP1msm or MycP3msm were concentrated in the reaction buffer (50 mM HEPES pH 7.5, 100 mM NaCl). The quenched fluorescent substrate Abz-AVKAASLGK(Dnp)-OH was obtained from Genscript Inc. Concentrated substrate stock solutions were made by dissolving the substrate in dimethylformamide (DMF) to a final concentration of 20 mM. Prior to starting the assay 0.5 mM stocks of the substrate were made by adding the reaction buffer. The concentration was confirmed using a Nanodrop spectrophotometer to measure the absorbance of the Dnp quencher at 360 nm (E = 17,400 M−1 cm−1 at 360 nm) (Carsten and Eisen, 1953). 200 μl volumes of substrate solutions in the reaction buffer at the concentrations indicated were warmed to 37 °C in a 96 well opaque black plate. The protease was added to all wells simultaneously to a final concentration of 2 μM. Fluorescence measurements were read starting immediately and every 30 sec for 1 hr in a Spectramax Gemini XPS plate reader with settings: excitation = 319 nm, emission = 419 nm.
The fluorescence increase per cleavage event was measured by reading the fluorescence of control Abz-AVKAASLGK(Dnp)-OH solutions that were completely degraded by trypsin in parallel with MycP digestions. Vmax of MycP1mth or MycP1msm at each substrate concentration was then calculated by measuring the initial slope of the data between 10 min and 60 min. Vmax versus the substrate concentration was then plotted and kinetic constants calculated using Graphpad Prism software.
For immobilized fluorescent peptide library digestions cellulose discs with individual peptides covalently attached were purchased from JPT Peptide Technologies GmbH (Berlin, Germany). The discs contained ~54 nmol of peptide each and were provided in 96-well format. The discs were washed with methanol and reaction buffer (50 mM HEPES pH 7.5, 100 mM NaCl) according to the manufacturer’s instructions. The fluorescence of the final wash was checked to ensure spontaneous desorption from the discs was not significant. After washing the discs were submerged in 150 μL of reaction buffer containing 4 μM MycP1mth and incubated for 2 hr at 37 °C. After 2 hrs the wells were thoroughly mixed by pipetting up and down then 75 μL was aspirated from each well and transferred to a second 96-well plate. The fluorescence of each well was measured using an M5 Spectramax platereader with settings: excitation = 319 nm, emission = 419 nm.
2.7. Circular dichroism (CD) spectroscopy
Full length EspBmtu and EspBmtu-N were concentrated to 20 μM in 50 mM HEPES, 100 mM NaCl. EspBmtu-C was obtained by digesting EspBmtu with MycP1mth overnight at 20 °C. EspBmtu-C was then purified from the digestion mixture by size exclusion chromatography (SEC) using a Superdex 200 column. A single 0.5 mL fraction with two protein fragments migrating at ~10 kDa and ~11 kDa was concentrated to a final concentration of 20 μM. Ellipticity measurements were obtained using a 0.1 cm quartz cuvette maintained at 37 °C in a Jasco J-810 spectropolarimeter.
3. Results
3.1. MycP1 mycosin forms two disulfide bonds
Based on homology with other subtilisins it has been suggested that mycosins are translated as a preproprotein with a secretory signal peptide (residues 1–21 for MycP1mtu), a putative propeptide (residues 22–63) (Dave et al., 2002), a catalytic domain (residues 64–394) and a predicted C-terminal transmembrane region (Brown et al., 2000) (residues 419–439) (Supplementary Fig. S1). TOPCONS analysis (Bernsel et al., 2009) of the mycosin cellular localization predicts that the protease domain is located in the periplasmic space between the inner membrane and outer mycomembrane of mycobacteria. To provide a suitable folding environment and obtain soluble mycosin variants, we substituted the native signal peptide of MycP1mtu with the signal peptide from Erwinia carotovora PelB and truncated the predicted transmembrane region. Our efforts to produce soluble mycosin proteases were initially hampered by poor expression and/or lack of solubility of M. tuberculosis mycosins expressed heterologously in Rosetta(DE3) cells. However, we successfully expressed and purified homologous mycosins from M. thermoresistibile (MycP1mth), and M. smegmatis (MycP1msm), as well as MycP3 from M. smegmatis (MycP3msm), using the same methods.
All mycosins have four conserved Cys residues present in their sequences (Supplementary Fig. S1), which prompted us to investigate the presence of potential disulfide bonds. Using mass spectrometry methods to analyze tryptic digest peptides of reduced and non-reduced MycP1mth samples, we identified two disulfide bonds, one formed between residues Cys51-Cys120 and the other between Cys206-Cys244 (Supplementary Fig. S2). Surprisingly, Cys51 is located in the putative propeptide region, which raised doubts concerning the proteolytic removal of this predicted propeptide.
To investigate whether the N-terminal extensions of mycosins are processed in vivo, a C-terminally HA-tagged version of MycP1 from M. thermoresistibile and M. marinum (MycP1mth and MycP1mm, respectively) were expressed in M. marinum (Fig. 1A). Although the expression of MycP1mth was poor, we could demonstrate that both MycP1 proteins showed an apparent molecular size of ~52 kDa (calculated molecular mass of MycP1mm without signal peptide is ~43.3 kDa and without putative propeptide ~38.9 kDa), indicating that the N-terminal extension remains attached to the protease domains. Furthermore, MycP1mm-HA almost exclusively localized to the cell envelope fraction (Fig. 1B), in accordance with the presence of a predicted transmembrane region at its C-terminus.
Fig. 1. Expression of HA-tagged MycP1mm and MycP1mth in M. marinum.

Immunoblot analysis with anti-HA antibodies of (A) total cell lysates prepared from M. marinum expressing MycP1mm-HA (lane mm) and MycP1mth-HA (lane mth) and (B) cytosol (lane cyt) and cell envelope (lane CE) fractions prepared from M. marinum expressing empty vector (lane −) and MycP1mm-HA (lane +). The HA-tagged proteins are indicated by an asterisk.
In addition, the presence of the N-terminal extension in MycP3mm and MycP5mm was observed by carefully analyzing previously obtained mass spectrometry results of total cell envelope fractions of M. marinum (van der Woude et al., 2013), while no peptide corresponding to the N-terminus of MycP1mm was identified by tandem MS analysis (Supplementary Fig. S3). Moreover the N-terminal extension of MycP3mtu of M. tuberculosis was also previously identified in a proteomics study (Malen et al., 2007). Together, these findings suggest that the N-terminal extensions of these two different mycosins remain attached to the protease domains in vivo.
3.2. MycP1mth and MycP3msm have a subtilisin-like fold
In order to gain further insight into the basis for MycP specificity, we determined the crystal structures of MycP1mth and MycP3msm (Fig. 2A). Structures were refined to 2.0 Å (MycP1mth) and 1.5 Å (MycP3msm) resolution (see Materials and Methods and Table 1 for structure solution details). Both MycP1mth and MycP3msm contain a typical subtilisin fold, including all the secondary structure elements of the extended core (Siezen et al., 1991). These include a 7-stranded β sheet core surrounded by 8 α helices (Fig. 2A and Supplementary Fig. S4). The structures of MycP1mth and MycP3msm superimpose with RMSD 1.7 Å and 48% sequence identity.
Fig. 2. Crystal structures of MycP1mth and MycP3msm.

A. The structures are shown in the same orientation with α helices and β strands of core subtilisin domain in green, the insertion loops in purple and the N-terminal extension in blue. The catalytic triad residues (yellow) are shown as sticks. Bound ions are omitted for clarity. The disordered amino acid residues in loop 2 of MycP3msm are indicated by a purple dashed line.
B. Structures of MycP1mth and MycP3msm and subtilisin Carlsberg (PDB 3UNX) (Fisher et al., 2012) are shown in surface representation with catalytic triad residues highlighted in yellow.
A Dali search (Holm and Rosenstrom, 2010) using the structure of MycP1mth indicated that subtilisin Carlsberg (PDB 3UNX) of Bacillus licheniformis is the closest structural homolog with an overall RMSD of 2.0 Å (Fisher et al., 2012). However, relative to subtilisin Carlsberg, MycP1mth and MycP3msm have three sequence insertions consisting of residues 160–176 (loop 1, MycP1mth numbering), 241–262 (loop 2), and 319–330 (loop 3) (Figs 2 and 3). These loops pack within the N-terminal extensions of MycP1mth (residues 24–65) and MycP3msm that wrap around the perimeter of the molecule, and all three insertion loops cluster around the active site pocket. These loops and the N-terminal extension together contribute to three 2-stranded anti-parallel β sheet elements in addition to the core subtilisin fold. These new structural elements surround the active site creating a relatively deep active site pocket (Figs 2B and 4). The two disulfide bonds, which are present in both MycP1mth and MycP3msm structures, likely contribute to the stability of these extended loops.
Fig. 3. Structure based sequence alignment of MycP1mth, MycP3msm and subtilisin Carlsberg.

Secondary structure elements of MycP1mth and subtilisin Carlsberg are displayed above and below the sequences, respectively. The signal protease cleavage site is indicated by vertical arrow. The N-terminal extension is indicated by a blue line. The mycosin insertion loops are indicated by purple lines. The catalytic triad residues and conserved Asn residue coordinating oxyanion hole are highlighted in blue. The conserved Cys residues that form disulfide bonds in mycosins are highlighted in yellow.
Fig. 4. The mycosin-specific N-terminal extension and loop insertions.

MycP1mth and MycP3msm structures are displayed as ribbons with semitransparent gray surface around the core subtilsin domain. The catalytic triad residues are highlighted in yellow. Loop insertions are highlighted in purple. The N-terminal extension amino acid residues are shown as blue sticks surrounded by σA-weighted 2FO–FC electron density map contoured at 1 σ.
3.3. A unique N-terminal extension in mycosins does not occlude the active site
Subtilase propeptides normally prevent substrate access to the active site until degradation of the propeptide occurs (Gallagher et al., 1995; Shinde and Inouye, 1995). However, we found that the N-terminal extensions of MycP1mth and MycP3msm follow a meandering path around the outer surface of the protease domain without obscuring the active sites (Fig. 4). Thus the catalytic residues are exposed at the bottom of the active site pocket. Similarly, the structure of a mycosin from M. smegmatis revealed an enzyme with intact propeptide without any obvious obstruction of the active site (Solomonson et al., 2013). Together these data suggest that, in contrast to other subtilisins, the N-terminal extension of MycP1mth and MycP3msm is part of the mature enzyme and not a cleavable propeptide.
Analysis of the N-terminal extension/MycP1mth interface using the PISA server (Krissinel and Henrick, 2007) found 4200 Å2 total buried surface area, 6 salt bridges and 26 hydrogen bonds. Similarly, the interface between the N-terminal extension and protease domain in MycP3msm buries 4100 Å2, and contains 1 salt bridge and 30 hydrogen bonds. These interactions together with the presence of disulfide bonds between residues Cys51-Cys120 (MycP1mth numbering) indicate the integral nature of the N-terminal extension in the mycosin fold and further support a suggested role in fold stabilization (Solomonson et al., 2013).
3.4. Catalytic triad residues are in active conformation
In MycP1mth the catalytic triad consisting of Asp92, His123, and Ser334 adopts the conformation seen in other subtilases with the exception of Ser334 (Fig. 5A). During catalysis the hydroxyl group of the catalytic serine must point upwards toward the substrate, but in MycP1mth it is turned sideways and lies about 1.8 Å out of place. This slightly aberrant positioning was also reported for the catalytic serine of MycP1msm (Solomonson et al., 2013). Conformational rearrangement of the serine side chain probably occurs upon substrate binding. Small side chain rearrangements are common in protein-peptide interactions (London et al., 2010) therefore the observed conformation of Ser334 should not be considered an obstacle to hydrolysis. The catalytic triad of MycP3msm consisting of Asp95, His126 and Ser341 adopts an almost identical configuration (Fig. 5B). Interestingly, Ser341 appears in a double conformation, the first corresponding to the typical subtilisin-like orientation and the second corresponding to the orientation of Ser334 in MycP1mth. Again, we expect that the presence of a substrate at the active site will stabilize the solvent-facing rotamer of Ser341 resulting in the correct orientation for nucleophilic attack on the peptide bond.
Fig. 5. Catalytic triad and binding grooves of MycP1mth, MycP3msm and subtilisin Carlsberg/eglin complex.

A. The surface of MycP1mth is shown in gray with residues of the catalytic triad and Asn239 highlighted in yellow. Zinc ion is shown as a pink sphere. The substrate recognition subsites and important residues are labeled explicitly. Distances between catalytic triad side chains are shown as dashed lines. Residues 242, and 320–329 of MycP1mth (loop 3) were not displayed for clarity.
B. MycP3msm is represented in the same way as (A). Residues 326–335 of MycP3msm (loop 3) were omitted for clarity.
C. Subtilisin Carlsberg/eglin complex (PDB 2SEC) (McPhalen and James, 1988) represented as in (A). Only eglin residues 53–61 are shown as cartoon with side chains at P4–P1′ positions shown as sticks. The glycine/serine rich linker strands (residues 99–103 and 126–130) that make backbone interactions with eglin are shown as cartoons. Subtilisin side chains close enough to interact with eglin are shown as sticks within the surface and labeled explicitly.
The catalytic triad histidine in the MycP1mth structure has an unusual backbone conformation with φ–136.7° ψ–66.5° for His123 in MycP1mth, similar to the conformation in MycP1msm structure (Solomonson et al., 2013). The typical backbone conformation of the catalytic histidine residue is different in MycP3msm (φ–73.5° ψ–50.3° for His126) and other subtilisins, for example φ–61.4° ψ–48.2° for His64 in subtilisin Carlsberg (PDB 3UNX) (Fisher et al., 2012). The functional consequences of these differences in MycP1mth are unclear, because the distances between the catalytic triad side chains in the mycosins are similar relative to other subtilisins. For example Asp → His = 2.6 Å and His → Ser = 3.0 Å in MycP1mth and Asp → His = 2.7 Å and His → Ser = 2.7 Å in subtilisin Carlsberg (PDB ID 2SEC) (Fig. 5C). Most likely there is no functional consequence since all the functional groups participating in the serine protease catalytic mechanism are in place. Furthermore, it is not uncommon for main-chain scaffolds supporting serine protease catalytic triads to vary widely (Dodson and Wlodawer, 1998).
3.5. The MycP1mth and MycP3msm substrate binding sites suggest unique substrate specificity
Subtilase substrate specificity is typically determined by interactions with substrate residues designated P6–P3′ with residues P4–P1 having particular importance (Gron et al., 1992; Siezen et al., 1991). Inhibitor complex structures of subtilisin Carlsberg and subtilisin BPN indicate that residues P4–P1 follow a conserved surface groove and make anti-parallel sheet like interactions between two β sheet elements (Bode et al., 1987; McPhalen and James, 1988; McPhalen et al., 1985; Siezen and Leunissen, 1997). These structural elements correspond to MycP1mth residues 158–162 (β4/β5 linker) and 202–205 (β6/β7 linker) (Fig. 3). In MycP1mth and MycP3msm, as well as MycP1msm (Solomonson et al., 2013), these strands are separated by ~15 Å. This wide groove makes it impossible for a single strand to make sheet-like interactions as expected in classic subtilisin/substrate interactions. Furthermore, in MycP1mth and MycP3msm the canonical binding groove is completely blocked near S4 by loop 1 (residues 160–176 for MycP1mth and 162–175 for MycP3msm), which extends helix α4 by one turn in MycP1mth and by two turns in MycP3msm. This insertion effectively walls off the distal end of the binding groove in both MycP1mth and MycP3msm, albeit more dramatically in MycP3msm (Fig. 5).
Additional differences in MycP1mth and MycP3msm substrate binding surfaces compared to classic subtilisins occur downstream of the active site at substrate positions P1′–P3′. Typically substrates exit abruptly after brief backbone interactions with the β strand corresponding to β13 strand of MycP1mth (residues 325–332). This strand is roughly level with the active site and the rest of the binding groove in subtilisin Carlsberg, but in MycP1mth and MycP3msm it is tilted 90° and overhangs the catalytic triad. Therefore, the substrates must exit at a 90° angle from the active site or else bend to one side of the β13 strand. Either option results in a dramatically bent substrate exit groove that is likely to result in an extended downstream binding groove relative to classic subtilisins. The downstream groove probably mediates interaction with the large aromatic substrate residues that are favored at positions P2′ and P3′ (as discussed in chapter 3.9).
3.6. MycP1mth and MycP3msm structures show system specific differences
The substrate binding groove of MycP1mth is similar to that of MycP1msm (78% sequence identity for full-length MycP1mth and MycP1msm) (Solomonson et al., 2013). All of the residues highlighted in Fig. 5A are identical in the two proteins with the minor exception of His155 (MycP1mth) which is exchanged for Gln155 (MycP1msm). In contrast, the substrate binding grooves of the MycP1mth and MycP3msm structures reveal significant differences that probably result in ESX system specific substrate specificities. First, the overall surface electrostatic potential inside the binding groove of MycP1mth is more negative compared to MycP3msm (Fig. 6). Second, the insertion loop 2, which is located beside the substrate binding groove, is 5 amino acids longer in MycP3msm relative to MycP1mth (residues 241–245 in MycP3msm). In MycP3msm, these residues are disordered, but they appear to occupy the S4 site within the binding groove. Moreover, loop 1 moves 9 Å towards the P4 site in MycP3msm relative to MycP1mth (Fig 2). The combined effect of loop 1 and 2 is to truncate the putative binding groove in MycP3msm, which may decrease the number of residues it recognizes upstream of its cleavage site. Finally, there is a significant difference between MycP1mth and MycP3msm at site S2. This site is relatively deep and polar in MycP1mth. The same site in MycP3msm is shallow, because it is filled by nonpolar Leu202 (Fig. 5B). Thus, the structures of MycP1mth and MycP3msm contain unique architecture that indicates they have evolved to recognize different specific sequences.
Fig. 6. Active site grooves of MycP1mth and MycP3msm have distinct surface properties.
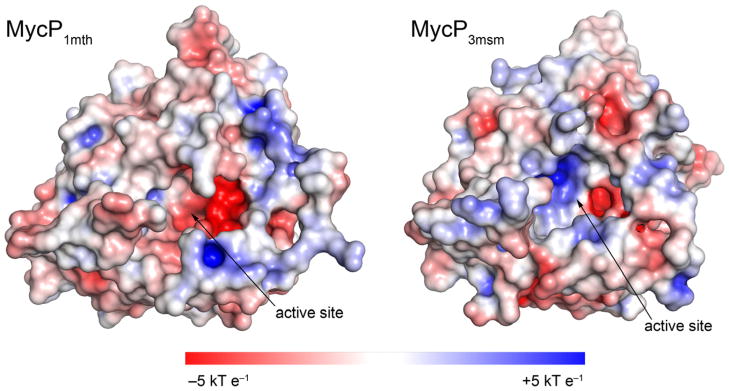
The structures of MycP1mth and MycP3msm are shown as surfaces colored according to the solvent accessible electrostatic potential. The electrostatic surface potential was calculated using the PDB2PQR server, APBS and APBS/PyMol plugin (London et al., 2010; McLaughlin et al., 2007).
3.7. MycP1mth cleaves EspBmtu at Ala358 in vitro
Although most ESX-1 substrates seem to be secreted as full-length proteins, it has been reported that EspB, is processed during secretion in M. tuberculosis, M. smegmatis, and M. marinum (Gao et al., 2004; McLaughlin et al., 2007; Ohol et al., 2010; Xu et al., 2007). However, the exact location of the cleavage site has been the subject of conflicting reports (Ohol et al., 2010; Solomonson et al., 2013). We incubated MycP1mth with EspBmtu then isolated the C-terminal EspBmtu fragments and determined their molecular mass using mass-spectrometry (Fig. 7). The major C-terminal cleavage product had a mass of 9,678 corresponding to residues 359–460, whereas the minor C-terminal fragment had a mass of 7,360 corresponding to residues 387–460. Incubation of EspBmtu with MycP1msm resulted in the same cleavage products, although with a slower kinetics (data not shown). N-terminal sequencing of these fragments further confirmed that Ala358-Ser359 is the primary cleavage site and Ala386-Gly387 is the secondary cleavage site in agreement with the data reported for MycP1msm (Solomonson et al., 2013).
Fig. 7. EspBmtu is processed by MycP1mth in vitro.

A. EspBmtu samples were incubated with MycP1mth or catalytically inactivated variant MycP1mth S334A for indicated time periods and analyzed by SDS-PAGE. The cleavage leads to accumulation of the N-terminal fragment (EspBmtu-N) and two C-terminal fragments (EspBmtu-C) indicated by asterisks.
B. A schematic diagram of the domain structure of EspBmtu with predicted PE and PPE domains. The C-terminal homology region is indicated by an orange bar (Supplementary Fig. S6). The primary and secondary MycP1 cleavage sites are located in the low complexity region of EspBmtu.
C. The sequence conservation of the primary MycP1 cleavage site is displayed as a sequence logo (http://weblogo.berkeley.edu) (Crooks et al., 2004) based on the sequence alignment of EspB homologs shown in (D).
D. Sequence alignment of EspB homologs from the ESX-1 clusters with the primary and secondary MycP1 cleavage sites indicated by vertical arrows. The full-length alignment is shown in Supplementary Fig. S6.
In contrast to the MycP1 proteins, EspB homologs have a lower sequence identity, especially in a low complexity region in their C-terminal parts (Supplementary Fig. S5). However, residues around the identified primary and secondary cleavage sites appear to be conserved in EspB homologs (Figs 8C and 8D). In each EspB protein the conserved residues are consistent with the known specificity of subtilases with an aromatic/hydrophobic residue at position P4, Arg/Lys at P3, Pro/Ala in P2, and Ala or another small residue in P1, (Gron et al., 1992) (using the commonly accepted nomenclature (Schechter and Berger, 1967)).
Fig. 8. Comparison of the relative secondary structure in EspBmtu, EspBmtu-N and EspBmtu-C.

A. Disorder predictions for EspBmtu with location of the predicted PE and PPE domains indicated by rectangles.
B. CD spectra of EspBmtu (black long dashed line), EspBmtu-N (blue short dashed line) and EspBmtu-C (red solid line). Data are expressed as molar ellipticity. EspBmtu-N (residues 1–358) was produced recombinantly and EspBmtu-C was obtained as a mixture of two proteolytic fragments (residues 359–460 and 387–460) as described in Methods.
3.8. The C-terminal EspBmtu fragment lacks secondary structure
According to the HHpred protein homology prediction algorithm the N-terminal fragment of EspB contains two regions that show similarity with PE and PPE domains, respectively (Supplementary Fig. S6) (Soding et al., 2005). Downstream of the identified MycP1mth cleavage site (residues 359–460) the sequence of EspBmtu contains 28% glycine residues with few large hydrophobic or aromatic residues. In silico bioinformatics tools including PONDR-FIT (Xue et al., 2010) predict a disordered C-terminal region (Fig. 8A). We tested the hypothesis that EspBmtu contains a disordered C-terminal region by circular dichroism (CD). CD spectra clearly show that more secondary structure is present in full-length EspBmtu relative to the N-terminal EspBmtu fragment (residues 1–358). However, the CD spectrum of the C-terminal fragments of EspBmtu (residues 359–460 and 387–460) is characteristic of a protein without secondary structure (Fig. 8B). Taken together, these results indicate that the C-terminal domain of EspBmtu is at least partly structured in the context of the full-length protein, but fails to maintain any secondary structure by itself. This lack of secondary structure may indicate the presence of unstructured loop(s) in the C-terminal part of EspBmtu that serve to expose the cleavage site sequence to MycP1mth.
3.9. Specific residues near cleaved bond are necessary for MycP1mth recognition
To further investigate the activity of MycP1mth and MycP1msm we designed a fluorescent resonance energy transfer (FRET) substrate consisting of EspBmtu residues 354–361 with an N-terminal o-aminobenzoic acid (Abz) fluorescent group and an additional C-terminal lysine residue derivatized with 2,4-dinitrophenyl (Dnp) quencher acceptor (Abz-AVKAA|SLGK(Dnp)-OH). Both MycP1mth and MycP1msm hydrolyzed this substrate and the accumulation of fluorescent product was used to follow proteolytic activity (Fig. 9). Mass spectrometry analysis confirmed that the cleavage occurs at the residue corresponding to Ala358. Kinetic analysis using this peptide showed that the substrate associates with MycP1mth with a Km = 60 μM, and a Vmax = 6.0 nmol min−1 mg−1, indicating relatively slow catalysis by MycP1mth. Similarly, kinetic analysis using MycP1msm to degrade the same substrate yielded a Km = 86 μM, and a Vmax = 1.8 nmol min−1 mg−1. These data suggest that the substrate residues at positions P5–P3′ are sufficient for substrate recognition by MycP1 proteases. Notably, MycP3msm protease was completely inactive against this model substrate (data not shown).
Fig. 9. Comparison of the kinetics of MycP1mth and MycP1msm in quenched fluorescent peptide assay.

Rate of substrate (Abz-AVKAA|SLGK(Dnp)-OH peptide) cleavage by MycP1mth and MycP1msm is plotted as a function of substrate concentration. Kinetic parameters were Km = 60 mM, and Vmax = 6.0 nmol min−1 mg−1 (MycP1mth) and Km = 86 mM, and Vmax = 1.78 nmol min−1 mg−1 (MycP1msm).
To determine the sequence specificity requirements for MycP1 in more detail we tested activity of the mycosins against a peptide library composed of 8-residue N-terminal Abz labeled peptides based on the substrate peptide (Abz-AVKAASLG). Peptides were synthesized with positions P5–P3′ systematically changed to every other amino acid and attached to a cellulose matrix. Digestion with MycP1mth revealed that MycP1mth is highly selective in regard to which amino acids it can tolerate at certain positions (Fig. 10). In the P4 position valine is preferred, all other residues significantly abrogate activity. In P3 a basic residue such as lysine or arginine is strictly required consistent with the 100% conservation of Lys356 in EspB (Fig. 7C). Finally, P1 position is selective towards Ala, Met, or Pro residues.
Fig. 10.

Single site substitutional analysis based on the MycP1 substrate Abz-AVKAASLG using ProteaseSpot™ system. Bars represent the fluorescent signal from peptide fragments released from the cellulose membrane over 2 hours. Fluorescence is directly proportional to the activity of MycP1mth against each peptide. The relative activity of MycP1mth against peptides was normalized such that activity versus EspBmtu peptide sequence (Abz-AVKAASLG) = 1.0.
Interestingly, acidic residues were not tolerated at any of the sites tested. This probably reflects electrostatic repulsion from the overall negative charge inside the MycP1mth binding site (Fig. 6). The observed recognition pattern is consistent with the properties of the substrate binding groove of MycP1mth. A model of peptide in the active site of MycP1msm has been proposed (Solomonson et al., 2013), however the preference for a hydrophobic residue in position P4, both in the EspB protein family and in our analysis, is not explained by the model. It should be noted, however, that an altered peptide sequence with an additional Gly residue between P3 and P4 positions of the cleavage site was used for docking (Solomonson et al., 2013).
4. Discussion
Mycobacteria, a group of Gram-positive bacteria, have a very complex cell envelope composed of a typical phospholipid bilayer plasma membrane, followed by a periplasmic space and an outer mycomembrane, containing unique lipids and glycolipids (Brennan, 2003; Hoffmann et al., 2008; Zuber et al., 2008). Therefore, the proteins secreted by mycobacteria have to cross not only the plasma membrane, but also the second highly hydrophobic outer membrane. As a result, mycobacteria developed specialized ESX, or type VII, secretion systems, which transport proteins necessary for virulence in M. tuberculosis. The M. tuberculosis genome encodes five such secretion systems, named ESX-1 through ESX-5. The secreted proteins of the ESX system belong to the EsxAB clan (http://pfam.sanger.ac.uk/clan/EsxAB) including WXG100 and PE/PPE family members. The structures of different members of this clan show that they are usually heterodimers composed of a bundle of four α-helices and one of the monomers has a conserved secretion motif adjacent to the second α-helix (Daleke et al., 2012; Strong et al., 2006). The only known mycosin substrate, EspB, also belongs to the EsxAB clan and contains a similar predicted PE/PPE fold and a secretion motif (Supplementary Fig. S6).
One of the most intriguing aspects of the ESX secretion system is the presence of membrane associated subtilisin-like proteases, or mycosins, in all ESX clusters. The only functionally characterized mycosin — MycP1 — is essential for ESX-1 secretion and was shown to require its protease activity for efficient infection (Ohol et al., 2010). A link between EspB cleavage and virulence was lacking until it has been recently reported that the N-terminal PE/PPE domain of EspB specifically binds phosphatidic acid (PA) and phosphatidylserine (PS), but only in the absence of the C-terminal region downstream of the MycP1 cleavage site (Chen et al., 2013). Chen et al. show that EspB secretion leads to increased M. tuberculosis virulence in a mouse model of infection, and argue that processed EspB promotes bacterial growth within macrophages by disrupting phospholipid mediated signaling (Chen et al., 2013). Therefore, MycP1 hydrolysis may serve to activate EspB as it is transported into the host cell.
Another ESX secreted protein, LipY, is also known to undergo proteolytic activation upon secretion. LipY promotes virulence by breaking down triacylglycerol in a reaction that may aid in fatty acid acquisition by the bacilli. Hydrolysis of LipY after Gly149 removes the PE domain necessary for secretion via ESX-5 (Daleke et al., 2011) and also activates the enzymatic activity of LipY (Mishra et al., 2008). There is not currently any evidence linking MycP5 to LipY processing, and the sequence around Gly149 does not bear an obvious resemblance to the MycP1 recognition motif described here. However, since there is significant variation in the specificity of mycosins from one system to another it is still possible that MycP5 is responsible. Apart from EspB and LipY, the majority of ESX secreted cargo proteins do not have assigned functions. As more information is learned the mycosins may be appreciated as a means of regulating both the quantity and the functional maturity of secreted proteins.
In this work we determined the crystal structures of M. thermoresistibile MycP1mth and M. smegmatis MycP3msm. We found that MycP1mth does not contain an autoinhibitory propeptide, similar to the recently reported structure of MycP1 from Mycobacterium smegmatis (Solomonson et al., 2013). Indeed, our structures extend this observation to include MycP1 from a second species (MycP1mth) and from a different genetic cluster (MycP3msm). In agreement with these data, our in vivo experiments indicate that the N-terminal extension of MycP1, MycP3 and MycP5 are not cleaved in their homologous host. These analyses further suggest that the mycosins may not require the N-terminal extension removal for activation.
A characteristic feature of the mycosins is a relatively deep active site cleft. In contrast, the majority of subtilisins have a shallow solvent-exposed substrate binding groove, which contributes to low selectivity of many subtilisin-like proteases and dictates the requirement for the propeptide acting an intramolecular inhibitor. Thus, the three loop insertions surrounding the active site may act to increase the specificity of the mycosins toward proteins that meet certain size and conformational requirements. The relatively steep active site pocket of the mycosins suggests that substrates adopt a less extended conformation when inserted into the binding pocket. It is tempting to speculate that substrates may loop back over themselves within the binding pocket. In this scenario the conserved glycine/serine rich regions of EspB (Fig. 7D) would provide a fitting substitute for one of the glycine/serine rich strands seen interacting with substrate in the binding grooves of subtilisin Carlsberg and subtilisin Novo (McPhalen and James, 1988).
We found that MycP1mth and MycP1msm selectively recognize and cleave the C-terminal region of EspBmtu at Ala358 with a secondary recognition site at Ala386. Our experiments with fluorescent peptides based on EspBmtu sequence indicate that eight amino acids are sufficient to be recognized and cleaved by MycP1mth. Moreover, substitution of the amino acids at all eight positions revealed strict specificity at several positions indicating that these enzymes have evolved to hydrolyze only specific sequences. Finally, the MycP1mth peptide substrates identified here are not hydrolyzed by MycP3msm suggesting differences in substrate recognition motifs. This disparate substrate specificity is consistent with the different shape and surface chemistries of MycP1mth and MycP3msm active site grooves (Figs 5 and 7). The effort to identify MycP3 substrates is ongoing.
EspB is the only ESX substrate with a function linked to proteolytic maturation, and it is unique to the ESX-1 system. This raises the question as to what other secreted proteins might be substrates for the remaining mycosin paralogs. CD spectra analysis and in silico predictions suggest that EspB has an α-helical N-terminal domain and a partly disordered C-terminal domain. The cleavage sites of MycP1mth and MycP3msm are located in the intervening domain predicted as disordered. This suggests that mycosins may act to remove autoinhibition of cargo proteins by separating ordered domains with flexible loop regions. In addition, mycosins might promote transport through the ESX machinery by removing PE/PPE domains used as ESX secretory signal sequences.
The results presented here are initial stages in our understanding of the mycosin function in ESX secretion at the molecular level. Information presented here on the sequence recognition motif of MycP1 as well as the unique structural features of MycP3 relative to MycP1, may enable the design of specific competitive inhibitors aimed at disrupting proteolytic activation of ESX secreted proteins and generation of new therapeutics against M. tuberculosis.
5. Accession numbers
The coordinates and structure factors were deposited to the Protein Data Bank with accession codes 4HVL (MycP1mth) and 4KG7 (MycP3msm).
Supplementary Material
Acknowledgments
The authors thank Roy Ummels for technical assistance; Christoph Grundner, Seattle BioMed, for providing M. thermoresistibile genomic DNA; Michael Berne, Tufts University, for N-terminal sequencing; Eun Suk Song, Travis Sexton and K. Martin Chow for advice and assistance with protease assays; Trevor P. Creamer for advice on CD measurements; Natalia Korotkova, Louis B. Hersh and Sidney W. Whiteheart for critical comments on the manuscript. We acknowledge the University of Kentucky Proteomics Core and Protein Analytical Core that are partially supported by grants from the National Center for Research Resources (P20RR020171) and the National Institute of General Medical Sciences (P20GM103486) from the National Institutes of Health. The Orbitrap mass spectrometer was acquired by High-End Instrumentation Grant S10RR029127 (to HZ). We thank staff members of Southeast Regional Collaborative Access Team (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory, for assistance during data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. This study was supported in part by NIH Grant Number P20GM103486 (to KVK) from the National Institute of General Medical Sciences, its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the NIGMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, et al. Type VII secretion — mycobacteria show the way. Nat Rev Microbiol. 2007;5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, et al. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol. 2009;73:329–340. doi: 10.1111/j.1365-2958.2009.06783.x. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernsel A, Viklund H, Hennerdal A, Elofsson A. TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 2009;37:W465–468. doi: 10.1093/nar/gkp363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode W, Papamokos E, Musil D. The high-resolution X-ray crystal structure of the complex formed between subtilisin Carlsberg and eglin c, an elastase inhibitor from the leech Hirudo medicinalis Structural analysis, subtilisin structure and interface geometry. Eur J Biochem. 1987;166:673–692. doi: 10.1111/j.1432-1033.1987.tb13566.x. [DOI] [PubMed] [Google Scholar]
- Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol. 2012;83:1195–1209. doi: 10.1111/j.1365-2958.2012.08001.x. [DOI] [PubMed] [Google Scholar]
- Brennan PJ. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis. 2003;83:91–97. doi: 10.1016/s1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- Brown GD, Dave JA, Gey van Pittius N, Stevens L, Ehlers MRW, et al. The mycosins of Mycobacterium tuberculosis H37Rv: a family of subtilisin-like serine proteases. Gene. 2000;254:147–155. doi: 10.1016/s0378-1119(00)00277-8. [DOI] [PubMed] [Google Scholar]
- Carsten ME, Eisen HN. The Interaction of dinitrobenzene derivatives with bovine serum albumin. J Am Chem Soc. 1953;75:4451–4456. [Google Scholar]
- Chen JM, Zhang M, Rybniker J, Boy-Rottger S, Dhar N, et al. Mycobacterium tuberculosis EspB binds phospholipids and mediates EsxA-independent virulence. Mol Microbiol. 2013;89:1154–1166. doi: 10.1111/mmi.12336. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CM, et al. General secretion signal for the mycobacterial type VII secretion pathway. Proc Natl Acad Sci USA. 2012;109:11342–11347. doi: 10.1073/pnas.1119453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daleke MH, Cascioferro A, de Punder K, Ummels R, Abdallah AM, et al. Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. J Biol Chem. 2011;286:19024–19034. doi: 10.1074/jbc.M110.204966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave JA, van Pittius NCG, Beyers AD, Ehlers MRW, Brown GD. Mycosin-1, a subtilisin-like serine protease of Mycobacterium tuberculosis, is cell wall-associated and expressed during infection of macrophages. BMC Microbiol. 2002;2:30. doi: 10.1186/1471-2180-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem Sci. 1998;23:347–352. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SJ, Blakeley MP, Cianci M, McSweeney S, Helliwell JR. Protonation-state determination in proteins using high-resolution X-ray crystallography: effects of resolution and completeness. Acta Crystallogr D Biol Crystallogr. 2012;68:800–809. doi: 10.1107/S0907444912012589. [DOI] [PubMed] [Google Scholar]
- Floyd K. Global Tuberculosis Report. World Health Organization; France: 2012. [Google Scholar]
- Gallagher T, Gilliland G, Wang L, Bryan P. The prosegment-subtilisin BPN′ complex: crystal structure of a specific ‘foldase’. Structure. 1995;3:907–914. doi: 10.1016/S0969-2126(01)00225-8. [DOI] [PubMed] [Google Scholar]
- Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, et al. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol. 2004;53:1677–1693. doi: 10.1111/j.1365-2958.2004.04261.x. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gron H, Meldal M, Breddam K. Extensive comparison of the substrate preferences of two subtilisins as determined with peptide substrates which are based on the principle of intramolecular quenching. Biochemistry. 1992;31:6011–6018. doi: 10.1021/bi00141a008. [DOI] [PubMed] [Google Scholar]
- Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, et al. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004;51:359–370. doi: 10.1046/j.1365-2958.2003.03844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci USA. 2008;105:3963–3967. doi: 10.1073/pnas.0709530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012a;14:1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- Houben EN, Bestebroer J, Ummels R, Wilson L, Piersma SR, et al. Composition of the type VII secretion system membrane complex. Mol Microbiol. 2012b;86:472–484. doi: 10.1111/j.1365-2958.2012.08206.x. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kuo KL, Zhu H, McNamara PJ, Leggas M. Localization and functional characterization of the rat Oatp4c1 transporter in an in vitro cell system and rat tissues. PLoS One. 2012;7:e39641. doi: 10.1371/journal.pone.0039641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley SA, Wilson IA. Protein production and crystallization at the Joint Center for Structural Genomics. J Struct Funct Genomics. 2005;6:71–79. doi: 10.1007/s10969-005-2897-2. [DOI] [PubMed] [Google Scholar]
- London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure. 2010;18:188–199. doi: 10.1016/j.str.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, et al. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J Immunol. 2005;174:3570–3579. doi: 10.4049/jimmunol.174.6.3570. [DOI] [PubMed] [Google Scholar]
- Malen H, Berven FS, Fladmark KE, Wiker HG. Comprehensive analysis of exported proteins from Mycobacterium tuberculosis H37Rv. Proteomics. 2007;7:1702–1718. doi: 10.1002/pmic.200600853. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Chon JS, MacGurn JA, Carlsson F, Cheng TL, et al. A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 2007;3:e105. doi: 10.1371/journal.ppat.0030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhalen CA, James MN. Structural comparison of two serine proteinase-protein inhibitor complexes: eglin-c-subtilisin Carlsberg and CI-2-subtilisin Novo. Biochemistry. 1988;27:6582–6598. [PubMed] [Google Scholar]
- McPhalen CA, Svendsen I, Jonassen I, James MN. Crystal and molecular structure of chymotrypsin inhibitor 2 from barley seeds in complex with subtilisin Novo. Proc Natl Acad Sci USA. 1985;82:7242–7246. doi: 10.1073/pnas.82.21.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra KC, de Chastellier C, Narayana Y, Bifani P, Brown AK, et al. Functional role of the PE domain and immunogenicity of the Mycobacterium tuberculosis triacylglycerol hydrolase LipY. Infect Immun. 2008;76:127–140. doi: 10.1128/IAI.00410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohol YM, Goetz DH, Chan K, Shiloh MU, Craik CS, et al. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe. 2010;7:210–220. doi: 10.1016/j.chom.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. UCSF Chimera — a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Schechter I, Berger A. On the size of the active site in proteases. I Papain Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- Serafini A, Boldrin F, Palu G, Manganelli R. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: essentiality and rescue by iron and zinc. J Bacteriol. 2009;191:6340–6344. doi: 10.1128/JB.00756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde U, Inouye M. Folding mediated by an intramolecular chaperone: autoprocessing pathway of the precursor resolved via a substrate assisted catalysis mechanism. J Mol Biol. 1995;247:390–395. doi: 10.1006/jmbi.1994.0147. [DOI] [PubMed] [Google Scholar]
- Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng TY, et al. Mycobacterial ESX-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA. 2009;106:18792–18797. doi: 10.1073/pnas.0900589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siezen RJ, Leunissen JA. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 1997;6:501–523. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siezen RJ, de Vos WM, Leunissen JA, Dijkstra BW. Homology modelling and protein engineering strategy of subtilases, the family of subtilisin-like serine proteinases. Protein Eng. 1991;4:719–737. doi: 10.1093/protein/4.7.719. [DOI] [PubMed] [Google Scholar]
- Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 2012;8:e1002507. doi: 10.1371/journal.ppat.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonson M, Huesgen PF, Wasney GA, Watanabe N, Gruninger RJ, et al. Structure of the mycosin-1 protease from the mycobacterial ESX-1 protein type VII secretion system. J Biol Chem. 2013;288:17782–17790. doi: 10.1074/jbc.M113.462036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop EJ, Bitter W, van der Sar AM. Tubercle bacilli rely on a type VII army for pathogenicity. Trends Microbiol. 2012;20:477–484. doi: 10.1016/j.tim.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, et al. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplyakov AV, Kuranova IP, Harutyunyan EH, Vainshtein BK, Frommel C, et al. Crystal structure of thermitase at 1.4 Å resolution. J Mol Biol. 1990;214:261–279. doi: 10.1016/0022-2836(90)90160-n. [DOI] [PubMed] [Google Scholar]
- van der Woude AD, Mahendran KR, Ummels R, Piersma SR, Pham TV, et al. Differential detergent extraction of Mycobacterium marinum cell envelope proteins identifies an extensively modified threonine-rich outer membrane protein with channel activity. J Bacteriol. 2013;195:2050–2059. doi: 10.1128/JB.02236-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Laine O, Masciocchi M, Manoranjan J, Smith J, et al. A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol Microbiol. 2007;66:787–800. doi: 10.1111/j.1365-2958.2007.05959.x. [DOI] [PubMed] [Google Scholar]
- Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber B, Chami M, Houssin C, Dubochet J, Griffiths G, et al. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J Bacteriol. 2008;190:5672–5680. doi: 10.1128/JB.01919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.