

Abstract
Photo-reactive nanogels with an integrated photoinitiator-based functionality were synthesized via a Reversible Addition-Fragmentation Chain Transfer (RAFT) process. Without additional free initiators, this nanogel is capable of radical generation and initiating polymerization of a secondary monomer (i.e. dimethacrylate) that infiltrates and disperses the nanogel particles. Due to the presence of RAFT functionality and the fact that all initiating sites are initially located within the nanogel structure, gelation can be delayed by sequencing the polymerization from the nanogel to the bulk matrix. During polymerization of a nanogel-filled resin system, a progressive delay of gelation conversion from about 2 % for conventional chain growth polymerization to 18 % for the same monomer containing 20 wt% nanogel additive was achieved. A significant delay of stress development was also observed with much lower final stress achieved with the nanogel-modified systems due to the change of network formation mechanics. Compared with the nanogel-free dimethacrylate control, which contained uniformly distributed free initiator, the flexural modulus and mechanical strength results were maintained for the photopolymers with nanogel contents greater than 10 wt%. There appears to be a critical interparticle spacing of the photo-reactive nanogel that provides effective photopolymerization while providing delayed gelation and substantial stress reduction.
INTRODUCTION
Free radical polymerization has been used extensively to form thermosets from vinyl monomers including styrenics, and (meth)acrylates. With a wide range of materials and versatile reaction conditions, it gives the opportunity to form polymers with quite diverse properties. Photopolymerization is an important mode for the generation of initiating radicals with many advantages such as energy efficiency, accommodation of heat sensitive additives/substrates, as well as temporal and spatial control over the polymerization process. These features make photopolymerization an ideal choice for broad applications including films and coatings1, biomaterials2, and photolithography3, 4. These materials are typically composed of crosslinked, often densely crosslinked, polymeric structures. The utilization of free radical polymerization that involves a chain-growth mechanism in di- or multi-vinyl materials is usually accompanied by gelation at a very low extent of reaction. Features such as cyclization, primary chain length and differential reactivity of free monomer compared with pendant vinyl groups affect the gel point but bulk polymerization of multi-functional (meth)acrylates undergo macrogelation at conversion levels typically below 5 %5. A practical consequence of the early gel point is that polymerization-induced stress starts to accumulate in the system during the transition from the liquid to the solid state. Stress increases exponentially along with storage modulus as conversion increases. For a highly crosslinked material with high storage modulus, stress generated during polymerization results in the formation of internal and interfacial defects, warped structures as well as materials that are less tough and fatigue resistant. Many approaches have been developed to achieve reduced polymerization stress. As an example, multi-phase generation6-8 can create internal phase boundaries that when combined with control of local modulus or sequence of property development within each phase can result in partial volume recovery and stress relaxation during polymer formation. Polymer additives have also shown the ability to reduce stress with the replacement of some monomer with variety of prepolymers including oligomer9, nanogel9, 10 or dendrimer11. Other techniques include design of new monomers12, functionalization of inorganic fillers13, or post-gel relaxation based on covalently adaptable networks14.
Delaying gelation is one efficient method of reducing stress because stress is only generated after the transition from a liquid to a viscoelastic solid during polymerization. Beyond gelation, the extent of relaxation decreases while the relaxation time scale increases dramatically as the reaction proceeds. Chain transfer agents (CTA)15 have shown the capability to suppress gelation by favoring limited chain length and intramolecular cyclization. RAFT agents not only behave like CTAs in controlling chain length16, 17, but also suppress early gelation by uniform growth of primary chains18. Scranton and coworkers19 developed a multistage illumination process for shrinkage stress reduction of acrylate coatings based on illumination with micro/macro-patterned light during an initial stage. This allowed monomer migration from the dark regions to partially compensate for the local shrinkage effect. Since bulk stress buildup requires continuous network structure, only when the entire sample was flood cured to form the final crosslinked network did stress begin to evolve and significant shrinkage stress reduction was achieved through this approach.
Initiator-functionalized nanogels (macroinitiators) are able to initiate polymerization under designated conditions. They have certain advantages compared to free initiators, such as predetermined nanoscale spatial control and the ability to generate complex polymer architectures. They have been used for surface modification20, 21, block copolymer synthesis22, and star-shaped nanogels23. Here we report a novel method to limit stress formation during polymerization by incorporation of a photo-reactive nanogel with RAFT functionality with the aim to create nano-scale (5-10 nm) spatially isolated reaction domains in the initial phase of polymerization. This approach unites both the advantages of localized polymerization effects through initiation in the nanogel phase and RAFT characteristics to control primary chain length and homogeneity towards delaying gelation. A versatile procedure was used to synthesize the nanogel architecture through a solution RAFT polymerization of a monovinyl monomer and a photo-initiator functionalized crosslinker. This nanogel is readily dispersible in resin systems, and upon UV irradiation, is capable of initiating polymerization to form crosslinked materials in the presence of multi-vinyl monomers. This study characterized the reaction kinetics, gelation behavior, polymerization stress, modulus development and dynamic mechanical properties of these nanogel-modified materials.
EXPERIMENTAL METHODS
Materials
Isobornyl methacrylate (IBMA, technical grade), 2-mercaptoethanol (99 %, ME), 2-cyano-2-propyl dithiobenzoate (97 %, CPBD), tin(II) 2-ethylhexanoate (95 %), azobisisobutyronitrile (98 %, AIBN) and triethylene glycol dimethacrylate (95 %, TEGDMA) were purchased from Sigma-Aldrich. Ethoxylated bisphenol-A dimethacrylate (> 87 %, BisEMA) were from Esstech. 2-Isocyanatoethyl methacrylate (98 %, IEM) was obtained from TCI America. 2-Hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone (97 %, Irgacure 2959 or I2959) was from BASF. AIBN was purified by recrystallization twice in methanol. Inhibitors in IBMA were removed with basic alumina. Dichloromethane (DCM) was dried using molecular sieves (4 Å, Fisher Scientific). All other materials were used without further purification.
Synthesis of I2959-IEM Dimethacrylate Crosslinker (I2959-IEM-DMA)
I2959 (2.89 g, 12.9 mmol) was mixed with IEM (4.00 g, 25.8 mmol) and DCM (16.0 ml) in a sealed 50 ml vial. Two drops of tin (II) 2-ethylhexanoate were added as catalyst. The reaction (Scheme S1) was allowed to continue for 1 day at room temperature. An aliquot of sample was analyzed by Fourier transform infrared spectroscopy (FT-IR) to confirm the disappearance of isocyanate peak (2270 cm−1, stretch) after the reaction. DCM was removed under reduced pressure afterwards and the product (I2959-IEM-DMA) was used without need for further purification as verified spectroscopically: 1H NMR (400 MHz, CDCl3) (Figure S1a) (ppm) 8.04 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.9 Hz, 2H), 6.09 (d, J = 6.2 Hz, 2H), 5.58 (d, J = 3.1 Hz, 2H), 5.10 (dt, J = 27.5, 5.9 Hz, 2H), 4.53 – 4.34 (m, 2H), 4.21 (dt, J = 15.5, 4.9 Hz, 4H), 4.00 (t, J = 5.0 Hz, 2H), 3.51 (q, J = 5.0, 4.5 Hz, 2H), 3.31 (q, J = 5.4 Hz, 2H), 1.93 (dd, J = 1.6, 0.9 Hz, 6H), 1.67 (s, 6H); 13C NMR (400 MHz, CDCl3) (Figure S1b) (ppm) 18.4, 25,7, 40.3, 61.3, 63.7, 66.5, 84.1, 114.1, 126.17, 127.93, 131.1, 136.0, 154.9, 156.2, 161.7, 167.3, 197.7; Mass spec (Figure S1b) m/z: 557.2 [M + Na]+, 535.2 [M + H]+, 517.2, 362.2, 307.1.
Synthesis of Nanogel Materials
The RAFT nanogel was prepared from a mixture of IBMA (13.96 g, 62.8 mmol) and I2959-IEM-DMA (3.73 g, 6.98 mmol) with AIBN initiator (0.11 g, 0.68 mmol) in a magnetically stirred 250 ml round-bottom flask. CPBD (0.77 g, 3.49 mmol) was added as a RAFT agent with ethyl acetate (100 ml) used as solvent (Scheme 1). The reaction was carried out at 80 °C for 20 h in a N2 environment. The resulting nanogel was precipitated in methanol (600 ml) three times and the solid materials were retrieved following centrifugation. The isolated nanogel materials were obtained after residual solvent removal under reduced pressure (nanogel conversion: 85 %; yield: 80 %).
Scheme 1.
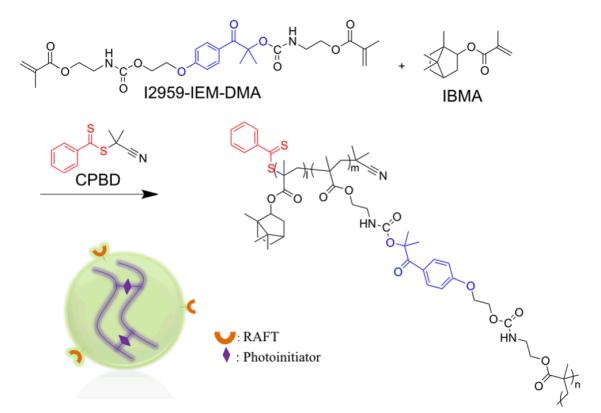
Reaction scheme for nanogel synthesis. Nanogel contains RAFT agent at chain ends and photoinitiators in crosslinking sections.
BisEMA and TEGDMA were combined at a 70:30 mass ratio as the resin system to disperse the photo-reactive nanogels at various concentrations. Free I2959 was added to the control resin (no nanogel).
The nanogel was characterized by GPC, UV-Vis and rheology. The polymerization kinetics was monitored with either near-IR or mid-IR. Near-IR coupling with a rheometer was applied for determination of material modulus development and gelation behavior. Near-IR coupling with a tensometer was used for measurement of stress generation profiles along with conversion. Dynamic mechanical analysis (DMA) was employed to obtain thermomechanical properties. Detailed characterization methodology can be found in the accompanying supporting information.
RESULTS AND DISCUSSION
Photo-reactive Nanogel Synthesis and Characterization
From NMR spectra, the CPBD concentration is 40 % that of the I2959 functionality with the ratio of the incorporated monomers (IBMA and I2959-IEM-DMA) equivalent to the feed ratio ([IBMA]: [I2959-IEM-DMA] = 9:1) in the nanogel synthesis. With the assumption that each primary chain in this nanogel contains one RAFT functionality, the primary chains are estimated to consist of approximately 25 monomer units. From the triple-detection GPC analysis, a weight average molecular weight (Mw) of 35 kDa (polydispersity index of 2.5) and an average hydrodynamic radius of 3.3 nm were obtained for this nanogel, which corresponds to a calculated average of 14 photo-reactive crosslinker groups present per nanogel particle.
Viscosity
Steady shear flow tests (Figure S2) of monomer-dispersed nanogel from 0 to 20 wt% show Newtonian liquid behavior due to lack of extended interparticle entanglement. This indicates that a 20 % nanogel dispersion is below the dense packing critical point. With an increase in nanogel concentration, zero shear viscosity (η) increased accordingly from 0.082 Pa·s for the monomeric resin to 0.78 Pa·s for the 20 % nanogel composition. By fitting of the data, relative viscosity (ηr) is an exponential function of nanogel concentration with an exponent of 7.4 for loading levels below 10 % and an exponent of 13.6 from 10 % to 20 % (Figure 1). This concentration dependent slope change indicates a change of polymer interaction24. Below 10 wt%, the weaker nanogel-solvent interactions dominate the much larger particle-particle interactions. When nanogel concentration is greater than 10 %, the average interparticle spacing is reduced with interparticle effects no longer negligible. The extended interactions between nanogel domains start to contribute to rapid viscosity change with continued loading. For this particular nanogel/monomer combination, we expect a 10 – 20 % nanogel loading is close to the percolation threshold. The Krieger-Dougherty model25 was applied to fit the rheological behavior of hard sphere particles and microgel particles26, 27 in solutions:
where ∅ is the particle concentration and ∅m is the volume fraction of randomly close packing systems (64 %), and [η] is the intrinsic viscosity of hard sphere ([η] = 2.5 based on Einstein equation). A much higher viscosity profile was found for equivalent loading of the nanogel compared with the theoretical values for hard spheres. Nanogel, as a highly internally branched and cyclized polymer formed by solution polymerization may have a lower elastic modulus versus a comparable microgel, is capable of significant swelling, which enlarges the nanogel effective volume fraction with decreased interparticle spacing that potentially increases viscosity behavior.
Figure 1.
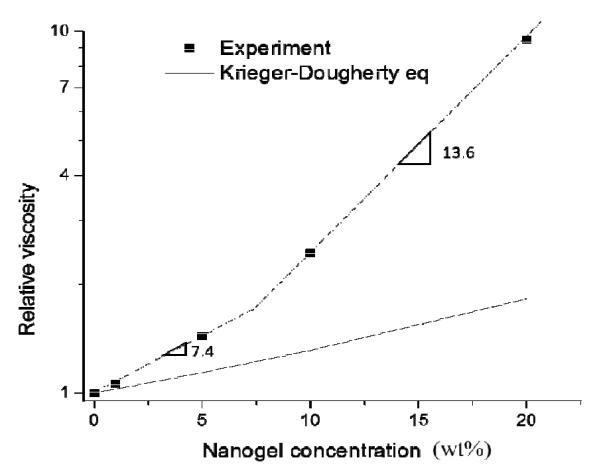
The symbols provide the relative viscosity data as a function of nanogel concentration and the dashed line represents linear fit results. Nanogel was dispersed in TEGDMA from 0 to 20 wt%. The solid line plot was generated from the Krieger-Dougherty equation.
Kinetics Study
From UV-Vis experiments, the modified crosslinker, I2959-IEM-DMA has much lower absorbance than the unmodified I2959 initiator (Figure S3a). Other studies have proved that modification of the tertiary −OH group in this type of photoinitiator through esterification causes low quantum yields28,29 and long triplet state lifetimes28. However, previous reports30, 31 showed functionalization of the primary −OH group in I2959 while leaving the tertiary hydroxyl group intact doesn’t significantly affect initiator efficiency. Here, the divinyl version of the initiator was applied to internally crosslink the nanogel in order to enhance the initial localized polymerization to only inside nanogel domain by avoiding primary radical diffusion into the matrix. On the other hand, the photo-reactive nanogel showed a greater UV absorbance than equivalent concentrations of the free photo-reactive crosslinker monomer apparently due to the light absorption associated with the included RAFT functionality. Upon exposure to 365 nm light, the monomeric crosslinker has only one-tenth the absorbance of I2959 at the same effective molar concentration (Figure S3b) at this single wavelength (365 nm). To normalize the absorptivities, a ten-fold molar excess was applied considering the free, monomeric crosslinker as initiator (4.2 wt%, [I2959] = 88.3 mM) compared with the conventional unmodified initiator control (0.18 wt% I2959, 8.8 mM). From the real-time IR kinetic results of resin photopolymerization (Figure 2), the photo-reactive monomeric crosslinker, even at the higher concentration, has a much longer induction time and slower rate of polymerization compared with the control, which indicates that the modified initiator has a much lower efficiency than free, nonderivatized I2959 under 365 (±10) nm irradiation. This is probably due to the alkyl radical generated from I2959 being more stable than the analogous radical from the crosslinker because of the stronger electron-donating capability of −OH group from I2959 as opposed to the urethane group. In contrast, the nanogel (overall modified I2959 concentration of 44.2 mM) showed almost no induction time (Figure 2). This is due to the potential for RAFT agents to cleave into radicals and initiate polymerization under UV light32, 33. It was confirmed that a variety of RAFT agents, including dithioester (Figure S4) and trithiocarbonate (data not shown) were able to initiate monomer polymerization under the same irradiation conditions even in the absence of photoinitiator. RAFT agents typically restrict polymerization rates due to the reduction of active radical concentration34, 35. In addition, prior investigations have shown that UV irradiation with wavelengths greater than 313 nm may not be suitable for activation of direct initiation via RAFT agents36. However, this experiment demonstrates that some RAFT agents are able to initiate polymerization under relatively long wavelength UV irradiation (i.e. 365 nm or 320-500 nm) without significant O2 inhibition and achieve reasonably high conversion in a limited time during network formation.
Figure 2.
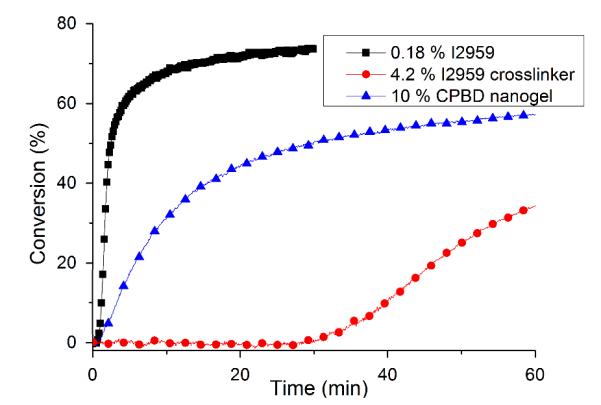
Real-time conversion profiles for 0.18 % I2959, 4.2 % crosslinker (I2959-IEM-DMA) and 10 wt% nanogel in BisEMATEGDMA resin. The C=C stretch (1640 cm−1) was monitored by IR with 2 wavenumber resolution. Continuous irradiation with 365 nm light at 35 mW/cm2 was applied.
Gelation Measurements and Stress Evaluation
Rheometry is a versatile technique for quantitatively measuring gelation of a crosslinked system where the power law preserves (G’ ~ G” ~ ω1/2)37. The crossover point of the dynamic storage modulus (G’) and loss modulus (G”) evolution during polymerization is one of the generally accepted criteria used to determine the gel point38, 39. An early gelation in terms of conversion (~2 % for dimethacrylates) is normally observed for bulk chain growth network formation. For the nanogel with the photo-initiator integrated as internal crosslinks, multiple initiators and terminal RAFT agents are spatially confined within the discrete nanogel structures with the average distance between initiating sites controlled by the nanogel loading level. Based on the solution polymerization formation of nanogels, they are readily swollen by monomers of appropriate solubility parameter. Initially, localized polymerization would be expected to take place exclusively inside and around the dispersed nanogels. The RAFT functionality can prevent the inter-nanogel coupling at early conversion by control of radical concentration that leads to more uniform chain growth and limiting chain lengths. After the nanogel-based polymerizing domains becoming interconnected through continued network formation, macrogelation occurs and polymerization stress begins to accumulate. Therefore, potentially lower final stress can be achieved with a delayed stress development profile. From the rheology experiments, the crossover point between G’ and G” of the control system took place at very low conversion (~ 2 %, 3a). For the 10 wt% CPBD nanogel composition, the rheological analysis indicated a rise in viscosity, which coincided with the G’/G” crossover that occurred at ~ 12 % conversion due to the localized polymerization effect (Figure 3b). There is a progressive increase in the conversion at gelation with increasing nanogel concentration; the gel point conversion was delayed from 4.7 % for 1 wt% added nanogel to 18.2 % for the 20 wt% nanogel loading level (Table 1). Even if the initiation process is initially confined to in and around the nanogel structure, the reaction still follows a chain growth mechanism that radicals generated from nanogel migrate mainly via propagation into the matrix to eventually interconnect and form a continuous gel. More highly loaded nanogel systems have a higher overall concentration of RAFT groups and greater proportions of internalized monomer that helps to confine the polymerization regime through the formation of shorter chains. Moreover, higher viscosity and faster kinetic rates can also suppress diffusion that may cause inter-nanogel crosslinking and lead to a relatively earlier gelation. This may be why the 20 % nanogel dispersion has a greater conversion at gel point than the other systems. To validate that the gel points of these systems are similar to the crossover points between G’ and G”, polymerization of the 20 % CPBD nanogel system was carried out between salt plates while monitoring conversion using IR. From multiple runs, this material reached physically evident gelation at 14 – 15 % conversion, which is in reasonably good agreement with the value of 18 % conversion determined by the coupled real-time rheometer/NIR experimental technique.
Figure 3.

Storage modulus (G’), loss modulus (G”) and conversion data vs. time for (a) the control resin (BisEMA/TEGDMA with 0.5 wt% I2959) and (b) the resin with 10 wt% nanogel solution. A 320 – 390 nm light was used for irradiation with light intensities of 0.35 mW/cm2 for the free initiator-based control and 10 mW/cm2 for nanogel-filled systems to achieve similar rates of polymerization.
Table 1.
Crossover point conversion for the control and nanogelfilled systems.
| Nanogel content (wt %) | G’/G” crossover conversion (%) |
|---|---|
| 0 (Control) | 2.0 (0.6) |
| 1 | 4.7 (0.9) |
| 5 | 10.1 (1.3) |
| 10 | 12.0 (0.3) |
| 20 | 18.2 (0.5) |
Stress reduction for nanogel-filled resin systems has been reported to be more or less proportional to the nanogel concentration9, 10. This is due to the replacement of monomer by pre-formed nanogel, which contributes limited reactive group concentrations and negligible volume change during network formation. For nanogels containing photo-initiator groups, localized polymerization is another factor that influences stress generation during reaction. From other studies40, light intensities and therefore, reaction rate, appears to have little effect on the stress-conversion correlation profile. Due to the lower efficiency, a higher curing light intensity was used here for the CPBD-based nanogel systems in order to achieve similar final conversion values between the control and experimental materials to facilitate direct comparisons of polymerization stress. In accord with the delayed gelation results, a delay in stress generation was observed compared with the control (final stress value: 1.86 ± 0.05 MPa). The delay in the onset of stress development as well as the point at which rapid stress rise is observed indicates that both macrogelation and bulk vitrification are suppressed as a consequence of the nanogel-based network formation strategy. The photo-active nanogel as initiator also produced much lower final stress for the 1 wt% nanogel system (1.08 ± 0.06 MPa). From Figure 4, the nanogel-based materials reached similar conversion after 30 min reaction compared with the control by adjusting the respective light intensities. The nanogel systems generated much lower stress (Figure 5). Both the 10 % and 20 % nanogel materials reached similar final stress (1.25 ± 0.15 MPa, 1.12 ± 0.07 MPa, respectively) while attaining higher conversion than either the 1 % nanogel composition or the control. By correlating the stress with conversion (Figure 6), it is evident that the nanogel-based photopolymerizations produce a significant delay in the onset of stress compared with the control. The 20 % nanogel had the highest conversion and it only accumulated about a quarter of the stress relative to the control at 80 % conversion. The nanogel concentration played an important role in delaying stress generation and in the final stress value. From Figure 7, the storage modulus for the 1 % nanogel system develops more slowly than the control system in terms of conversion at the early stage. This indicates a more heterogeneous network development for the nanogel system with crosslinking between nanogels taking place at higher conversion leading to a delay of gelation. The 10 % nanogel-modified resin has lower modulus at low conversion than the control or the 1 % nanogel material due to the further delay of gelation.
Figure 4.
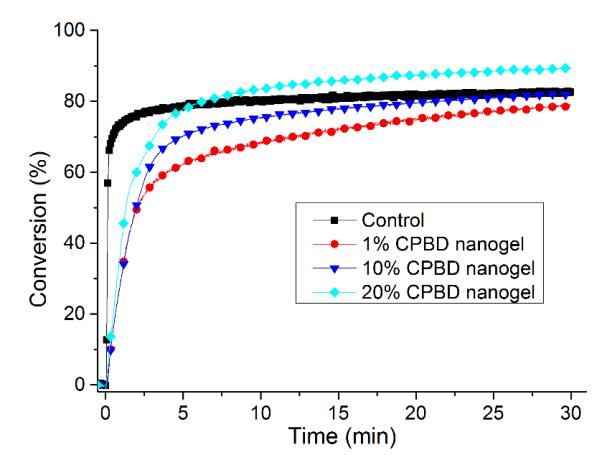
Conversion profiles for control and 1, 10 and 20 wt% nanogel in BisEMA/TEGDMA (70:30 mass ratio). The control system is BisEMA/TEGDMA (70:30) with 0.5 % I2959. A 320 – 500 nm light was used for irradiation with light intensities of 28 mW/cm2 for the control and 85 mW/cm2 for nanogel-filled systems.
Figure 5.
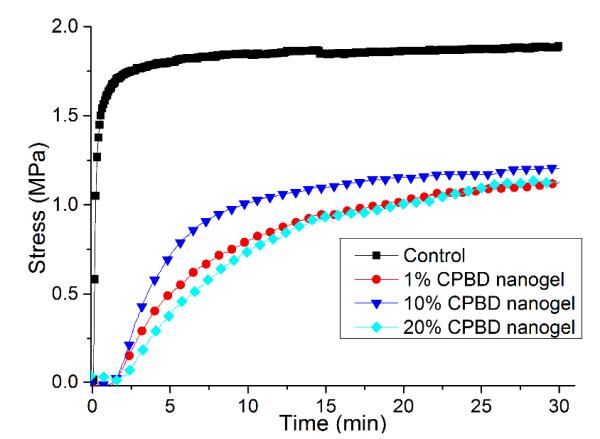
Stress profiles for control as well as 1, 10 and 20 wt% nanogel in BisEMA/TEGDMA (70:30 mass ratio). The control resin includes 0.5 % I2959. A 320 – 500 nm light was used for irradiation with light intensities of 28 mW/cm2 for the control and 85 mW/cm2 for nanogel-filled systems.
Figure 6.
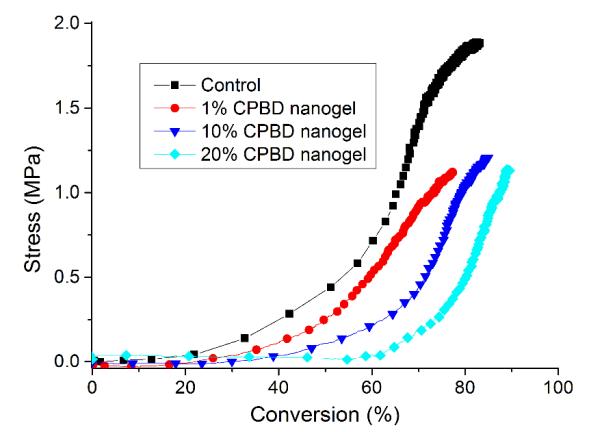
Stress correlation with conversion for control as well as 1, 10 and 20 wt% nanogel in BisEMA/TEGDMA (70:30 mass ratio). The control system is BisEMA/TEGDMA (70:30) with 0.5 % I2959. A 320 – 500 nm light was used for irradiation with light intensities of 28 mW/cm2 for the control and 85 mW/cm2 for nanogel-filled systems.
Figure 7.
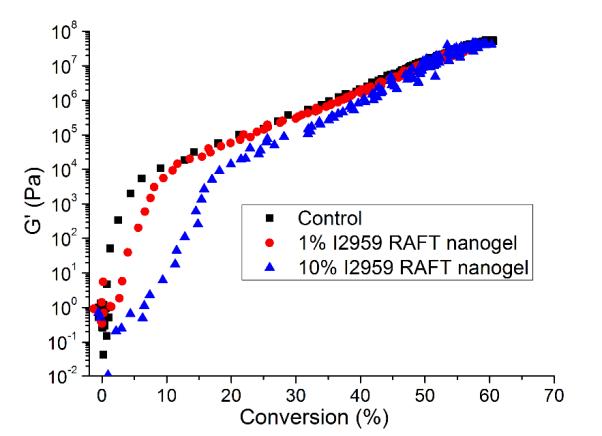
Storage modulus (G’) vs. conversion for the free initiator control (BisEMA/TEGDMA with 0.5 wt% I2959) as well as the 1 % and 10 % nanogel dispersions (in BisEMA/TEGDMA).
Mechanical Properties
Nanogel-initiated polymerization is prone to a more heterogeneous material since the nanogel-surrounded regions are polymerized at the early stage and therefore, achieves a higher conversion and crosslinking density; while regions between nanogels may have a lower crosslinking density due to more limited conversion. From the flexural tests (Figure 8), the 1 % nanogel system has much lower flexural strength and modulus compared with the control system; most likely due to the lower level of conversion achieved within the interparticle polymer matrix region. With the increase in nanogel content, both the flexural strength and modulus increased to values similar to the control while conversion increased. At a nanogel loading greater than 10 %, nanogel interaction starts to increase that helps to enhance both the physical and chemical crosslinking. Higher initiator concentration also leads to a higher final conversion for 10 % and 20 % systems. Moreover, RAFT functionality in nanogel structure helps to reduce termination reaction and promote relatively homogeneous network formation. From DMA tests, nanogel systems are of similar glassy modulus (G’) at room temperature compared with the control resin at around 3 GPa (Figure S5). However, the nanogel systems have a lower rubbery modulus (~ 58 MPa) at 200 °C as compared with the control (113 MPa). From the correlation between rubbery modulus and crosslinking density:
the control system has a crosslinking density of 9.57 mol/L while the values of nanogel systems are around 4.83 – 4.86 mol/L. The lower averaged crosslinking density of nanogel systems comes from the heterogeneous nature of the polymerization reaction mechanism that forms the final materials with potentially lower crosslinking density between nanogel particles. From the tan δ profiles (Figure S5), the 20 wt% nanogel-based polymer has a glass transition temperature (Tg) approximately 10 °C below that of the control with the other nanogel contents yielding polymers with slightly lower Tg.
Figure 8.

(a) Flexural strength and (b) flexural modulus results for the control and systems at 1, 5, 10 and 20 wt% nanogel concentrations.
CONCLUSION
A photo-reactive nanogel with RAFT functionality was synthesized with UV photo-initiator groups integrated within the structure. This nanogel was added to a dimethacrylate resin at various concentrations. It was shown that this nanogel has the ability to initiate polymerization with reasonable (although reduced relative to the control) reaction rate and final conversion without addition of any free initiators. It was demonstrated that the RAFT functionality is capable of assisting in the initiation of polymerization under longer wavelength UV irradiation. By this approach, the desired localized polymerization effect was achieved with a delay in gelation up to 18 % conversion with the 20 wt% nanogel loading. These experimental resins have delayed stress development profiles with much lower final stress values. At appropriate nanogel loading levels, mechanical properties are similar to the control photopolymer obtained with free initiator. RAFT functionality in the nanogel structure is important due to its ability to initiate polymerization, extend the gel point conversion and moderate stress generation while contributing to the formation of a more homogenous network.
Supplementary Material
Acknowledgement
This study was supported by NIH/NIDCR R01 DE022348.
Footnotes
† Electronic Supplementary Information (ESI) available: Detailed characterization, reaction scheme, 1H NMR, 13C NMR and Mass Spec information of crosslinker (I2959-IEM-DMA); UV/Vis spectra and DMA scans.. See DOI: 10.1039/b000000x/
Notes and references
- 1.Decker C. Macromolecular Rapid Communications. 2002;23:1067–1093. [Google Scholar]
- 2.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 3.Bratton D, Yang D, Dai JY, Ober CK. Polym Advan Technol. 2006;17:94–103. [Google Scholar]
- 4.Schanze KS, Bergstedt TS, Hauser BT, Cavalaheiro CSP. Langmuir. 2000;16:795–810. [Google Scholar]
- 5.Natarajan LV, Shepherd CK, Brandelik DM, Sutherland RL, Chandra S, Tondiglia VP, Tomlin D, Bunning TJ. Chem Mater. 2003;15:2477–2484. [Google Scholar]
- 6.Francis LF, McCormick AV, Vaessen DM, Payne JA. J Mater Sci. 2002;37:4717–4731. [Google Scholar]
- 7.Cheng YJ, Antonucci JM, Hudson SD, Lin NJ, Zhang XR, Lin-Gibson S. Adv Mater. 2011;23:409–413. doi: 10.1002/adma.201001755. [DOI] [PubMed] [Google Scholar]
- 8.Szczepanski CR, Pfeifer CS, Stansbury JW. Polymer. 2012;53:4694–4701. doi: 10.1016/j.polymer.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu JC, Howard GD, Lewis SH, Barros MD, Stansbury JW. Eur Polym J. 2012;48:1819–1828. doi: 10.1016/j.eurpolymj.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moraes RR, Garcia JW, Barros MD, Lewis SH, Pfeifer CS, Liu JC, Stansbury JW. Dent. Mater. 2011;27:509–519. doi: 10.1016/j.dental.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mezzenga R, Boogh L, Manson JAE. Compos Sci Technol. 2001;61:787–795. [Google Scholar]
- 12.Ge JH, Trujillo-Lemon M, Stansbury JW. Macromolecules. 2006;39:8968–8976. doi: 10.1021/ma061284w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye S, Cramer NB, Smith IR, Voigt KR, Bowman CN. Macromolecules. 2011;44:9084–9090. doi: 10.1021/ma2018809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kloxin CJ, Scott TF, Bowman CN. Macromolecules. 2009;42:2551–2556. doi: 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfeifer CS, Wilson ND, Shelton ZR, Stansbury JW. Polymer. 2011;52:3295–3303. doi: 10.1016/j.polymer.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, Newland B, Tai HY, Pandit A, Wang WX. Chem Commun. 2012;48:3085–3087. doi: 10.1039/c2cc17780c. [DOI] [PubMed] [Google Scholar]
- 17.Liu BL, Kazlauciunas A, Guthrie JT, Perrier S. Macromolecules. 2005;38:2131–2136. [Google Scholar]
- 18.Yu Q, Xu SH, Zhang HW, Ding YH, Zhu SP. Polymer. 2009;50:3488–3494. [Google Scholar]
- 19.Scranton AB, Ganahl PD, Smith CM, Coretsopoulos CN. Abstr Pap Am Chem S. 2009:238. [Google Scholar]
- 20.Liu Y, Klep V, Zdyrko B, Luzinov I. Langmuir. 2004;20:6710–6718. doi: 10.1021/la049465j. [DOI] [PubMed] [Google Scholar]
- 21.Nakayama Y, Matsuda T. Macromolecules. 1996;29:8622–8630. [Google Scholar]
- 22.Fan YJ, Chen GP, Tanaka J, Tateishi T. Biomacromolecules. 2005;6:3051–3056. doi: 10.1021/bm050368u. [DOI] [PubMed] [Google Scholar]
- 23.Amamoto Y, Kikuchi M, Masunaga H, Sasaki S, Otsuka H, Takahara A. Macromolecules. 2010;43:1785–1791. [Google Scholar]
- 24.Gupta P, Elkins C, Long TE, Wilkes GL. Polymer. 2005;46:4799–4810. [Google Scholar]
- 25.Krieger IM, Dougherty TJ. T Soc Rheol. 1959;3:137–152. [Google Scholar]
- 26.Meeker SP, Poon WCK, Pusey PN. Phys Rev E. 1997;55:5718–5722. [Google Scholar]
- 27.Borrega R, Cloitre M, Betremieux I, Ernst B, Leibler L. Europhys Lett. 1999;47:729–735. [Google Scholar]
- 28.Jockusch S, Landis MS, Freiermuth B, Turro NJ. Macromolecules. 2001;34:1619–1626. [Google Scholar]
- 29.Lalevee J, Allonas X, Jradi S, Fouassier JP. Macromolecules. 2006;39:1872–1879. [Google Scholar]
- 30.Ruhlmann D, Zahouily K, Fouassier JP. Eur Polym J. 1992;28:1063–1067. [Google Scholar]
- 31.Allen NS, Marin MC, Edge M, Davies DW, Garrett J, Jones F, Navaratnam S, Parsons BJ. J Photoch Photobio A. 1999;126:135–149. [Google Scholar]
- 32.Quinn JF, Barner L, Barner-Kowollik C, Rizzardo E, Davis TP. Macromolecules. 2002;35:7620–7627. [Google Scholar]
- 33.Wang H, Li QB, Dai JW, Du FF, Zheng HT, Bai RK. Macromolecules. 2013;46:2576–2582. [Google Scholar]
- 34.Yu Q, Zhu YS, Ding YH, Zhu SP. Macromol Chem Phys. 2008;209:551–556. [Google Scholar]
- 35.Barner-Kowollik C, Buback M, Charleux B, Coote ML, Drache M, Fukuda T, Goto A, Klumperman B, Lowe AB, Mcleary JB, Moad G, Monteiro MJ, Sanderson RD, Tonge MP, Vana P. J Polym Sci Pol Chem. 2006;44:5809–5831. [Google Scholar]
- 36.Lu LC, Yang NF, Cai YL. Chem Commun. 2005:5287–5288. doi: 10.1039/b512057h. [DOI] [PubMed] [Google Scholar]
- 37.Winter HH, Chambon F. J Rheol. 1986;30:367–382. [Google Scholar]
- 38.Winter HH. Polym Eng Sci. 1987;27:1698–1702. [Google Scholar]
- 39.Chiou BS, Khan SA. Macromolecules. 1997;30:7322–7328. [Google Scholar]
- 40.Lu H, Stansbury JW, Bowman CN. J Dent Res. 2005;84:822–826. doi: 10.1177/154405910508400908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.