

Abstract
We studied a two-generation family presenting with conditions that included progressive permanent weakness, myopathic myopathy, exercise-induced contracture before normokalaemic periodic paralysis or, if localized to the tibial anterior muscle group, transient compartment-like syndrome (painful acute oedema with neuronal compression and drop foot). 23Na and 1H magnetic resonance imaging displayed myoplasmic sodium overload, and oedema. We identified a novel familial Cav1.1 calcium channel mutation, R1242G, localized to the third positive charge of the domain IV voltage sensor. Functional expression of R1242G in the muscular dysgenesis mouse cell line GLT revealed a 28% reduced central pore inward current and a −20 mV shift of the steady-state inactivation curve. Both changes may be at least partially explained by an outward omega (gating pore) current at positive potentials. Moreover, this outward omega current of 27.5 nS/nF may cause the reduction of the overshoot by 13 mV and slowing of the upstroke of action potentials by 36% that are associated with muscle hypoexcitability (permanent weakness and myopathic myopathy). In addition to the outward omega current, we identified an inward omega pore current of 95 nS/nF at negative membrane potentials after long depolarizing pulses that shifts the R1242G residue above the omega pore constriction. A simulation reveals that the inward current might depolarize the fibre sufficiently to trigger calcium release in the absence of an action potential and therefore cause an electrically silent depolarization-induced muscle contracture. Additionally, evidence of the inward current can be found in 23Na magnetic resonance imaging-detected sodium accumulation and 1H magnetic resonance imaging-detected oedema. We hypothesize that the episodes are normokalaemic because of depolarization-induced compensatory outward potassium flux through both delayed rectifiers and omega pore. We conclude that the position of the R1242G residue before elicitation of the omega current is decisive for its conductance: if the residue is located below the gating pore as in the resting state then outward currents are observed; if the residue is above the gating pore because of depolarization, as in the inactivated state, then inward currents are observed. This study shows for the first time that functional characterization of omega pore currents is possible using a cultured cell line expressing mutant Cav1.1 channels. Likewise, it is the first calcium channel mutation for complicated normokalaemic periodic paralysis.
Keywords: periodic paralyses, omega pore, voltage sensor, calcium channel
Introduction
Periodic paralyses are recurrent bouts of flaccid limb weakness traditionally differentiated by accompanying changes in serum potassium levels, as hyperkalaemic, hypokalaemic or normokalaemic episodes (Fontaine, 2008). Each of these variants of periodic paralysis is caused by autosomal dominant mutations in skeletal muscle cation channels; however, the specific mutations elicit differing functional defects. Hyperkalaemic periodic paralysis is caused by destabilized inactivation of the main ion conducting pore, whereas normokalaemic and hypokalaemic periodic paralysis are caused by an accessory current through the so-called omega pore, resulting from a short circuit of intracellular and extracellular compartments produced directly by voltage sensor mutation (Cannon, 2010; Catterall 2010; Jurkat-Rott et al., 2012). Mutations for all three types of periodic paralysis are found in the Nav1.4 sodium channel encoded by SCN4A. To date, mutations in the Cav1.1 calcium channel encoded by CACNA1S have been associated only with hypokalaemic periodic paralysis. Nearly all of these mutations are located in the transmembrane S4 segments, and neutralize positive charges important for voltage sensitivity (Matthews et al., 2009). Similar to the hypokalaemic periodic paralysis mutations in Nav1.4, the two most frequent Cav1.1 mutations (R528H and R1239H) produce inward omega currents that result in membrane depolarization, reduced membrane excitability, and muscle weakness (Jurkat-Rott et al., 2009; Wu et al., 2012).
In Nav1.4, the position of the S4 mutation determines the voltage range in which the omega current occurs. For example, mutations of the first arginine in domain II, II-R1, as well as II-R2 and III-R2 mutations generate omega currents that are activated by hyperpolarization, are active at the usual resting potential, and are only closed by depolarization large enough to activate S4 (Sokolov et al., 2007; Struyk et al., 2008; Sokolov et al., 2010; Francis et al., 2011; Wu et al., 2011). Depending on the substituting residue, the omega current may be carried by protons or cations (Struyk and Cannon, 2007). Omega pores conduct currents that show an above-linear increase in amplitude with hyperpolarization that reflects the stochastic process of a voltage-dependent open probability and follows a Boltzmann distribution. In contrast to the hyperpolarization-induced omega currents from substitutions of the outer two arginine residues R1 or R2, II-R3 mutations in Nav1.4 generate cation-selective currents that are activated by depolarization (Sokolov et al., 2008). Such an omega current is deactivated by hyperpolarization and the associated disease phenotype is normokalaemic (Vicart et al., 2004).
Here we report an American family with normokalaemic periodic paralysis and recurrent cramping, oedema and neuronal compression with additional progressive myopathy caused by a novel Cav1.1-R1242G mutation. Similar to normokalaemic periodic paralysis mutations in the sodium channel domain II, R1242G affects the third arginine of S4. Our results suggest that the pathogenesis associated with R1242G differs from the Cav1.1 hypokalaemic periodic paralysis mutations. This is the first report of an omega pore induced by functional expression of a calcium channel mutation in cultured cells, while previously studied muscle fibres may have been altered by secondary changes (Jurkat-Rott et al., 2009; Wu et al., 2012).
Materials and methods
The family members gave their written informed consent to the genetic and 3T 23Na-MRI studies, which were approved of by the Ethics Committee of both Ulm and Heidelberg University and were conducted according to the Declaration of Helsinki in its present form. The exons and exon-intron boundaries of CACNA1S (Jurkat-Rott et al., 1994) were amplified from genomic DNA and bidirectionally sequenced using an automated 373A sequencer (Applied Biosystems).
Clinical neurophysiology and muscle biopsies
In the long exercise test (Tengan et al., 2004), the patients performed seven episodes of finger spreading against strong resistance of 35 to 40 s each, before 2 to 3 s relaxation to ensure sufficient blood flow during testing. Compound muscle action potentials were recorded 2 s immediately after cessation of exercise, every minute for 5 min, and every 5 min for 1 h. Compound muscle action potentials were recorded from the left abductor digiti minimi muscle using surface electrodes. They were evoked by supramaximal nerve stimulation (constant current method, 125%) of the ulnar nerve at the wrist, each lasting 0.2 ms.
Triceps muscle sections of Patient II:2 were fixed with formalin and stained with haematoxylin-eosin and embedded in paraffin. Fresh-frozen sections were stained with haematoxylin-eosin, NADH-TR, modified trichrome, non-specific esterase, and alkaline preincubated ATPase. Additional sections were fixed with glutaraldehyde and post-fixed with OsO4. For light microscopy, semi-thin plastic embedded sections of 1-µm thickness were stained with haematoxylin-eosin and toluidine-blue. For electron microscopy, ultra-thin plastic sections were stained with uranyl acetate and lead citrate. The immunocytological reactions included dystrophin 1–3 and sarcoglycans.
Imaging
The MRI scans were performed on a 3 T clinical MR system (MAGNETOM Trio, Siemens AG Medical Solutions). Hardware specific for broadband spectroscopy and a CE-certified double-resonant birdcage coil (32.59 MHz/123.2 MHz, Rapid Biomed Inc.) were used for recording 23Na as well as 1H signals. The imaging protocol of the lower legs comprised axial T1-weighted turbo spin-echo for the detection of fatty muscle degeneration and axial short-tau inversion recovery 1H magnetic resonance sequences for the identification of muscular oedema. The muscular signal intensity on short-tau inversion recovery images was normalized to the background signal as described previously (Jurkat-Rott et al., 2009).
Two reference phantoms were additionally investigated for use as control signal. One was filled with 51.3 mM Na+ in NaCl solution to mimic Na+ with unrestricted mobility as in extracellular fluid, and the other was filled with 51.3 mM Na+ in 5% agarose to mimic Na+ with restricted mobility as in myoplasm. In some recordings, a third reference was present containing 0.6% NaCl in H2O. For normalization of 23Na signals, the values of samples from the soleus muscles were divided by the signal intensity of the agarose phantom in which NaCl was trapped.
Expression of calcium channels
GFP-α1 s (Grabner et al., 1998) was used for site-directed mutagenesis of R1242G. Expression plasmids were verified by sequencing. Myotubes of the homozygous dysgenic cell line GLT were cultured as previously described (Jurkat-Rott et al., 1998) and transfected in a ratio of 4 µl FuGENE® HD reagent to 2 µg complementary DNA, 2 to 3 days after changing the fusion medium.
Central pore and omega pore currents
Whole-cell recordings were performed on 8 to 10-day-old myotubes with GFP fluorescence. Cells were allowed to stabilize for 5 min after establishment of the whole-cell configuration before data acquisition. Currents were recorded at room temperature (20–22°C) after partial (30–60%) series resistance compensation using an Axopatch 200B amplifier (Axon Instruments). The resistance of pipettes was 2.0 to 3.5 MΩ after filling with internal solution. Data were filtered at 1 kHz and sampled at 2 kHz (for calcium current recordings) or filtered at 1 kHz and sampled at 5 kHz (for omega current recordings). All recordings were performed without on-line leak subtraction. For measurements of calcium or omega currents, the linear passive currents at various potentials were calculated from the value at the holding potential of −90 mV, and these currents were subtracted from the current recordings.
For the electrophysiological characterization of central pore calcium current, the external solution contained (in mM): TEA-Cl 132, CaCl2 10, MgCl2 1, glucose 5, 4-aminopyridine 2.5, HEPES 10, pH 7.4; the internal solution contained (in mM): CsCl 128, EGTA 10, Mg-ATP 5, phosphocreatine 5, HEPES 12, pH 7.2. For the measurements of omega currents, the external solution contained (in mM): TEA-methanesulphonate (MS) 80, guanidine sulphate 38, MgSO4 1, Ca-gluconate 5, glucose 5, 4-aminopyridine 2.5, HEPES 10, pH 7.4; the internal solution contained (in mM): TEA-MS 65, Cs-MS 65, EGTA 10, Mg ATP 5, Na2 creatine PO4 5, HEPES 12, pH 7.2. For ionic substitution experiments, internal Cs-MS was replaced by guanidinium whereas external guanidinium was replaced by NMDG+. Sodium currents were blocked by addition of 5 µM tetrodotoxin to all bath solutions.
Action potentials
Action potentials were measured with the whole-cell current clamp technique using an Axopatch 200B amplifier. Hyperpolarizing current was injected into the myotubes to bring the membrane potential to about −110 mV for 6 s to recover sodium channels from inactivation. With anode break of the hyperpolarizing pulse, action potentials were elicited. The bath solution contained (in mM): NaCl 130, KCl 4, CaCl2 1.8, MgCl2 1.2, NaHCO3 18, HEPES 10, glucose 10, pH 7.4; pipette solution contained (in mM): KCl 10, K-gluconate 125, CaCl2 0.6, MgCl2 2, HEPES 5, Na2ATP 4, EGTA 5, sucrose 30, pH 7.2. The cells were continuously perfused with bath solution and maintained at 37°C. Signals were filtered at 5 kHz and sampled at 2 kHz.
Data analysis
The current-voltage dependence of whole cell currents through the central pore was fit according to
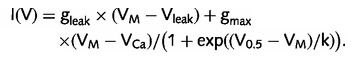 |
Vleak and gleak are reversal potential and conductance of linear leak current, VCa and gmax are reversal potential and maximum conductance of calcium channels, V0.5 is the potential for half-maximal current, VM is the test potential and k is the slope factor. Steady-state inactivation curves were fit to the equation
VM represents the prepulse potential, V0.5 is the potential at which half of channels are inactivated, and C is the fraction of non-inactivated channels, or asymptote. Recovery from inactivation was fitted to I(t)/Imax = −A × exp(−t/τ) + C, with τ as time constant of recovery, A as respective amplitude of time constant and C as asymptote of recovery. Data were analysed by a combination of pClamp (Axon Instruments), Excel (Microsoft), SPSS (IBM) and ORIGIN (Microcal software). Data were presented as mean ± standard error of the mean (SEM). Student’s t-tests were applied for statistical evaluation with the significance levels set to *P < 0.05, **P < 0.01, ***P < 0.001.
Simulation
To compare our results with earlier models of pathogenesis of periodic paralysis, we simulated the membrane current based on the implementation of the inward rectifying potassium current IK by the Boltzmann empirical equation described as
(Hagiwara and Takahashi, 1974). The K+ reversal potential EK = −95.8 mV was given by the Nernst equation with 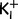 = 145 mM and
= 145 mM and 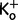 = 4 mM. A maximal conductance gKmax of 48 µS/cm2 for normal human muscle fibres was taken from Kwiecinski et al. (1984). Eh = −80 mV and s = 12 mV are constants that were deduced from measured membrane currents of human muscle (Ruff, 1999; Jurkat-Rott et al., 2009). Similarly, values used to describe the delayed rectifier potassium current were gKmax = 20 µS/cm2, Eh = −80 mV, and s = 12 mV.
= 4 mM. A maximal conductance gKmax of 48 µS/cm2 for normal human muscle fibres was taken from Kwiecinski et al. (1984). Eh = −80 mV and s = 12 mV are constants that were deduced from measured membrane currents of human muscle (Ruff, 1999; Jurkat-Rott et al., 2009). Similarly, values used to describe the delayed rectifier potassium current were gKmax = 20 µS/cm2, Eh = −80 mV, and s = 12 mV.
For the simulation of hypokalaemic periodic paralysis, a corresponding Boltzmann fit using ENa = 61.5 mV given by the Nernst equation with 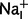 = 15 mM and
= 15 mM and  = 150 mM was performed. This procedure resulted in fit parameters of Eh = −110 mV and s = 10 mV and the maximum conductance of 12 µS/cm2 for R528H (Jurkat-Rott et al., 2009). For the R1242G omega current, attempts to fit the almost constant omega current using Boltzmann fits yielded unrealistic values. Therefore, we took the original data and upscaled them to the maximum conductance of 0.5 × 95 = 47.5 nS/nF to reflect the heterozygous situation in human muscle (95 nS/nF calculated in the results section on omega currents).
= 150 mM was performed. This procedure resulted in fit parameters of Eh = −110 mV and s = 10 mV and the maximum conductance of 12 µS/cm2 for R528H (Jurkat-Rott et al., 2009). For the R1242G omega current, attempts to fit the almost constant omega current using Boltzmann fits yielded unrealistic values. Therefore, we took the original data and upscaled them to the maximum conductance of 0.5 × 95 = 47.5 nS/nF to reflect the heterozygous situation in human muscle (95 nS/nF calculated in the results section on omega currents).
Results
Clinical features
The family over two generations consisted of four affected members (Fig. 1A). The three adults, Patients I:1, II:2 and II:4, had episodes characterized by painful muscle cramping for several hours followed by flaccid weakness lasting up to days. The male child, Patient II:1, was completely paralysed at birth and died from respiratory failure on the seventh post-natal day after the mother received anaesthesia when giving birth.
Figure 1.

(A) Pedigree of the family. (B) Long exercise tests obtained from the two sisters (normokalaemic periodic paralysis Patients (NPP) II:2 and II:4), a hypokalaemic periodic paralysis patient (HypoPP), and a control subject. (C–E) MRI results of Patient II:2: The T1-weigthed 1H-MRI (C) detected a pronounced fatty degeneration of both triceps surae muscles and the left, paralysed tibialis anterior and the deep posterior compartment, whereas these compartments were spared on the stronger right side. The fat-suppressed T2-weighted 1H-MRI (D) showed an increased water signal of the right tibialis anterior and both deep posterior compartments. The 23Na-MRI (E) identified an elevated sodium signal of the tibialis anterior and the deep posterior compartments on the right side as well as the left peroneal compartment whereas the dystrophic left tibialis anterior showed no increased Na+ signal. Interposed between the lower legs are two references (right tube containing 51.3 mM Na+ in 5% agarose and left tube containing the same Na+ concentration in NaCl-solution). (F–H) MRI results of Patient II:4. Although the T1-weighted 1H-MRI (F) only revealed degeneration of the calf muscles, the fat-suppressed T2-weighted 1H-MRI (G) showed a marked oedema of all other lower leg muscles. The 23Na-MRI (H) identified an elevated sodium signal of the right tibialis anterior.
Of the adults, male Patient I:1 had onset in infancy and delayed motor development. Triggers for the weakness episodes were cold and rest after exercise. Progressive weakness started in adulthood and led to wheelchair dependence at the age of 57. Ictal potassium levels were normal and KCl three times daily did not improve muscle strength. Death occurred at the age of 71 from respiratory failure after anaesthesia.
Female Patient II:2 (63-years-old) had painful muscle cramping in the lower legs elicited by walking and by febrile infections between early teens and adulthood. The cramping muscles were solid for days, could not be relieved by passive stretching, and were paralysed for months or permanently, requiring leg braces for dropped feet. Weakness spells without preceding cramps only occurred twice, once by stress and once following a meal, and both resolved after 1 h. A permanent weakness causing inability to participate in physical education during elementary school increased in late adolescence and adulthood. The overall muscle strength declined, as an inability to climb stairs without railing, to arise from a chair, to get up from a prone position or to arise from bed became the characteristic features. She became wheelchair bound at the age of 50. Progressive weakness led to the latest status at age 62 according to MRC rating scale: diffuse atrophy of extremities, bilateral scapular winging, forearm flexion 2/2 (right/left), wrist extension 5/4, finger flexion 4/4, hip flexion 1/1, knee extension 1/1, knee flexion 1/1, foot dorsiflexion 4/1, and foot plantar flexion 4/4. Tendon reflexes were absent. Ictal and interictal potassium levels always were normal at 4.1 ± 0.3 mM (n = 78), with 500 mg/d acetazolamide administered for decades with equivocal benefit. Diclofenamide was more effective and improved permanent muscle weakness at 25 mg/d but was discontinued because of cognitive impairment including mental fatigue, uneasiness, irritability, depression, drowsiness and confusion. A combination of acetazolamide 125 mg/d, eplerenone 50 mg/d and KCl 40 mmol/d increased serum potassium to 4.8 to 5.2 mM and subjectively improved muscle endurance.
Female Patient II:4 (54-years-old) was born floppy and showed delayed motor development. Cramps leading to solid muscles were triggered by walks and cold environment and were not relieved by passive stretching. During cramping, all muscles innervated by the profound peroneus nerve were weak and exhibited electrical silence in the EMG. Additionally, the corresponding skin of the anteriolateral lower leg and the dorsal foot was hypaesthesic and hypalgesic. After one of these episodes of cramping, serum values for creatine kinase (15 500 U/l), serum glutamic oxaloacetic transaminase (1185 U/l) and potassium (5.4 mM) were markedly increased. Weeks later, EMG recordings of tibialis and peroneus longus muscles revealed fibrillations and positive waves as well as small compound muscle action potentials and long distal latencies. Because of the dropped foot (Supplementary Fig. 1A), a leg brace was required. The only weakness episode without preceding cramps was triggered by local anaesthesia containing epinephrine. The latest status at the age of 53 revealed a positive Trendelenburg sign bilaterally, ubiquitous muscle atrophy especially in the feet, and inability to squat or walk on toes or heels. Foot dorsiflexion was +4 on the right, and +1 on the left side. Tendon reflexes were reduced but not absent. Proximal muscles showed mild myopathic changes in the EMG. Interictal potassium levels were always normal (4.0 ± 0.7 mM, n = 22), also with 500 mg/d acetazolamide administered for decades with equivocal benefit. Similarly, diclofenamide improved permanent muscle weakness at 25 mg/d but was discontinued because of semantic deficit i.e. misusing the names of everyday items, which strikingly reduced her professional practice. A combination of acetazolamide 125 mg/d, eplerenone 50 mg/d and KCl 40 mmol/d increased serum potassium to 4.8 to 5.2 mM and stabilized muscle function. See Supplementary material on ictal potassium, diet and treatment response of the two sisters.
Molecular diagnosis
Sanger sequencing of all coding exons of the CACNA1S and SCN4A genes predicted a novel glycine substitution at arginine 1242 in the Cav1.1 calcium channel of skeletal muscle (Fig. 1A). R1242G is located as the third charged residue in the fourth domain voltage sensor of the channel. SCN4A and KCNJ2 mutations were excluded in all exons by Sanger sequencing. RYR1 was negatively screened by high resolution melting.
Neurophysiology and muscle biopsy
The long exercise tests of the sisters Patients II:2 and II:4 revealed small compound action potentials of unchanging amplitude and shape, but did not reveal the late decrease typically observed in patients with hypokalaemic periodic paralysis (Fig. 1B).
Several muscle biopsies of Patient I:1 primarily contained fat and connective tissue and led to the diagnosis of atypical muscle dystrophy. In Patient II:2, a triceps brachialis muscle sample taken at the age of 19 showed fibrosis and chronic myopathic changes with internal nucleation, necrotic fibres, increased fibrosis and fatty replacement (Supplementary Fig. 1B). A biopsy of the contralateral triceps at the age of 40 showed mild type I predominance and mild to moderate type I atrophy with diameter variation from 30 to 120 µm. Endomysial fibrosis of severe degree was noted in several areas, and was absent in others. Occasionally myofibres were observed in a state of necrosis, myophagocytosis, or regeneration. Electron microscopy showed neither sarcoplasmic reticulum nor T-tubule dilations. Dystrophins and sarcoglycans were present. In Patient II:4, a deltoid muscle sample biopsied at the age of 17 showed regenerating fibres, a slight increase in fat and perimysial connective tissue, but no vacuoles. Histochemistry was normal.
Imaging
1H-MRI of Patient II:2 at age 58 showed marked symmetrical fatty degeneration and atrophy of all lower leg muscles except the right tibialis anterior, which displayed a normal muscle signal in the T1-weighted sequences (Fig. 1C). However, increased signal intensity was observed in the fat-suppressed T2-weighted (i.e. short-tau inversion recovery) sequences corresponding to muscular oedema (Fig. 1D). The 23Na signal was markedly higher in those muscles with oedema on short-tau inversion recovery images than in the dystrophic muscles displaying distinct fatty changes (Fig. 1E).
The 1H-MRI of Patient II:4 at the age of 47 years of the lower legs revealed a symmetrical fatty degeneration and atrophy more pronounced in the gastrocnemius than in the soleus muscles. The peroneus muscles were affected to a lesser degree (Fig. 1F). The long extensor muscles of the foot such as (bilaterally) the tibialis anterior showed increased signal intensity in the fat-suppressed T2-weighted sequences corresponding to a muscular oedema (Fig. 1G), whereas the intensity of the T1-weighted images was normal (Fig. 1H). When normalized to a 0.3% saline reference solution, the 23Na-MRI of the lower legs showed a muscular sodium signal intensity of 1.32 for the left and 1.26 for the right lower leg. This intensity was higher than in controls (n = 10; mean value ± SD: 0.99 ± 0.12, 95% confidence interval of 0.934–1.053) (Weber et al., 2006). Taking the measurements of the right leg and the left leg of the patient as two measurements and comparing them to the 20 measurements of both legs of the controls, a one-sided t-test yielded a significant difference with P = 0.0007.
Central pore currents
Whole-cell recordings showed that R1242G conducted voltage-dependent calcium currents through the central ion conducting pore (Fig. 2A). Current density of the mutant R1242G, at +30 mV, was significantly reduced to −3.17 ± 0.11 pA/pF compared with wild-type −4.45 ± 0.33 pA/pF (Fig. 2B).
Figure 2.

Electrophysiological characteristics of wild-type (WT, filled circles) or mutant R1242G (open circles) α1 s subunits in GLT cells. (A) Whole-cell currents through wild-type and R1242G calcium channels were recorded at various test potentials between −60 and +55 mV in 5 mV for 600 ms with holding potential of −90 mV. (B) Normalized current-voltage relationships after linear passive current subtraction. (C) Voltage dependence of steady-state inactivation was determined from a holding potential of −90 mV using a series of 60 s prepulses to potentials ranging from −90 mV to +40 mV in 10 mV steps before a test depolarization to +30 mV. (D) Recovery from inactivation was obtained by a two-pulse protocol: a 20-s lasting depolarization prepulse to +30 mV to inactivate the calcium channels, a second 500 ms test pulse to +30 mV followed after an increasing interval (from 0.5 s to 69.7 s) at different holding potentials (−90 mV, −80 mV, and −70 mV). Data are shown as means ± SEM. Solid lines represent fits to corresponding equations given in the text. Fitting parameters are listed in Table 1.
Additionally, the current-voltage relationship (Fig. 2B) was shifted slightly to the left (V0.5 = 21.39 ± 1.51 mV in R1242G versus V0.5 = 25.77 ± 1.26 mV in wild-type). R1242G produced a hyperpolarizing (left) shift of steady-state inactivation of about −20 mV in comparison with wild-type (V0.5 = −5.88 ± 2.19 mV versus V0.5 = 14.68 ± 1.43 mV, Fig. 2C) indicating a loss-of-function for inactivation, in addition to activation. Recovery from inactivation was significantly more complete in R1242G, demonstrating a gain-of-function component (Fig. 2D). Parameter values with significance levels are given in Table 1.
Table 1.
Parameters of calcium currents
| Parameter | Wild-type | R1242G |
|---|---|---|
| Current density (pA/pF) | −4.45 ± 0.33 | −3.17 ± 0.11** |
| Capacitance (pF) | 334.7 ± 13.67 | 326.89 ± 16.23 |
| n | 10 | 9 |
| Current activation | ||
| V0.5 (mV) | 25.77 ± 1.26 | 21.39 ± 1.51* |
| k | 6.81 ± 0.36 | 6.50 ± 0.26 |
| n | 10 | 9 |
| Steady state inactivation | ||
| V0.5 (mV) | 14.68 ± 1.43 | −5.88 ± 2.19*** |
| k | −9.65 ± 1.63 | −9.40 ± 1.31 |
| n | 8 | 5 |
| Recovery from inactivation | ||
| −90 mV | ||
| τ (s) | 7.99 ± 0.65 | 7.29 ± 0.37 |
| C | 0.65 ± 0.04 | 0.79 ± 0.02* |
| n | 8 | 10 |
| −80 mV | ||
| τ (s) | 9.76 ± 0.77 | 9.24 ± 0.83 |
| C | 0.61 ± 0.20 | 0.75 ± 0.40** |
| n | 10 | 5 |
| −70 mV | ||
| τ (s) | 10.92 ± 1.19 | 11.48 ± 0.88 |
| C | 0.56 ± 0.21 | 0.71 ± 0.43** |
| n | 7 | 8 |
*P < 0.05, **P < 0.01, ***P < 0.001.
Omega currents
Using solutions to omit effects of the central pore, we were able to detect omega currents in the transfected myotubes when using permeant ions guanidinium and cesium. Steady-state currents elicited by 200 ms step pulses showed that in all myotubes, there was an inward current induced at hyperpolarized potentials with guanidinium in the external solution (Fig. 3A–C). Based on the current-voltage relationship and the fact that the inward current was present in both mutant and wild-type myotubes, we hypothesize that this inward current reflects guanidinium passing through the inwardly rectifying Kir2.1 channels and is not the result of mutation. In contrast, the mutant R1242G showed depolarization-induced outward omega currents carried by caesium that were small or absent in wild-type or in untransfected myotubes which we interpret to be an effect of the mutation (Fig. 3A and B). Ion substitution experiments showed that the large organic ion NMDG+ was not conducted by the omega pore (Fig. 3C).
Figure 3.

Omega currents for R1242G (open circles), but not wild-type (WT, filled circles) or untransfected myotubes (filled triangles). (A) Representative whole-cell currents were measured by a series of 200 ms voltage steps from a −90 mV holding potential to voltages between −140 mV to +40 mV in 5 mV steps without leak subtraction. The R1242G-expressing myotubes showed obvious depolarization-induced outward omega current carried by caesium compared with non-transfected and wild-type-expressing myotubes. (B) Normalized steady-state current-voltage relationships revealed the depolarization-induced outward omega current carried by caesium. (C) Substitution experiments showed that the large organic ion NMDG+ could not pass the omega pore. (D) Current responses of wild-type and R1242G to a ramp pulse (inset). The protocol was: from a holding potential of −90 mV, a 1 min pre-depolarization to +30 mV followed by a ramp from −140 mV to +30 mV over 7 s was performed. The current-voltage curve was constructed by currents versus their corresponding voltages calculated using the known time course of the ramp protocol. The plotted points are not steady-state current values but averages of the corresponding values of the measurements during the ramp. SEM bars were determined at every 5 mV instead of each voltage for clarity. R1242G showed obvious inward current carried by guanidinium responding to ramp depolarization. For consistency, subtraction of linear passive current was done by the values measured at −90 mV for all figure parts. Data are shown as means ± SEM.
From Fig. 3B, the omega outward current showed at 40 mV a mean conductance of about (1.1 pA/pF)/40 mV = 27.5 nS/nF, which is comparable to the 28 nS/nF in the homozygous Cav1.1-R528H mouse myotubes (Wu et al. 2012). Wu et al. (2012) used a membrane capacitance of ∼1 µF/cm2 to estimate their conductance to be 28 µS/cm2 in the homozygous mouse muscle fibres. In the only work measuring omega currents in human muscle fibres (Jurkat-Rott et al. 2009), which are always heterozygous because of the autosomal dominant inheritance of the disease, R528H native muscle fibres had a conductance of 12 µS/cm2, which is indeed about half of the conductance of the homozygous R528H mouse muscle fibres of 28 µS/cm2. Therefore our data in mouse myotube preparations are comparable to all previous data available on Cav1.1 omega currents.
In addition, R1242G showed a hyperpolarization-induced inward omega pore current carried by guanidinium, observed over the entire range of physiological voltages, after 1 min pre-depolarization to +30 mV to inactive calcium channels followed by a ramp from −140 mV to +30 mV over 7 s (Fig. 3D). This current is not because of a shift in the electrode junction potential because pipette offset before and after the recordings remained unchanged. Also, subtraction of the wild-type from R1242G yields a current that is neither constant nor linear (Supplementary Fig. 2A). One explanation could be that the IVS4 segment recovers during the ramp. To test this hypothesis, we recorded the reverse ramp (7 s ramp from +30 mV to −140 mV after 1 min +30 mV depolarization pulse). The resulting inward omega current (Supplementary Fig. 2B) exhibited reduced current values for potentials more negative than −100 mV in agreement with the idea of a recovery of IVS4 after ∼5.5 s. It has a maximal conductance of about (−3.2 + 2.25 pA/pF) / (−140 + 130 mV) = 95 nS/nF in the range of −140 mV to −130 mV. We conclude that the inward omega current is conducted by a IVS4 segment that has moved above the omega pore constriction. As the omega current is visible over the whole voltage range, linear fitting of the values obtained during the ramp was not performed since it would subtract part of the non-linear omega current.
Taken together, the decisive element for the omega current seems to be the position of the R1242G residue with respect to the gating pore constriction before testing for the omega currents. If the residue is located below the gating pore as in the resting state then outward currents are observed; if the residue is above the gating pore due to depolarization as in the inactivated state then inward currents are observed. As the inward omega currents are ∼3-fold larger than the outward omega currents, there seems to be some degree of rectification.
Action potentials
We tested the direct effect of the omega current on action potentials using whole-cell current clamp recordings in GLT myotubes. Injection of a negative holding current was followed by an increasing positive current pulse from 0 to 8 nA that did not affect the amplitude or shape of action potentials (Fig. 4A), indicating saturation. The R1242G-expressing myotubes exhibited action potentials with a significantly reduced overshoot by 13 mV, decreased maximal rise slope by 36% and increased threshold compared to wild-type by 4 mV (Fig. 4B, P > 0.05). All measured parameters are shown in Table 2.
Figure 4.

Action potentials generated in myotubes expressing wild-type (WT) and R1242G. (A) Action potential was evoked by a series of 1 ms increasing depolarizing current stimulation (from 0 to 8 nA in 2 nA increments) after 6 s hyperpolarizing current injection to set membrane potential to about −110 mV. (B) Reduced excitability for the R1242G- (n = 11) than wild-type-expressing myotubes (n = 11): reduced overshoot, decreased slope of maximal rise and decreased threshold. Data are shown as means ± SEM. *P < 0.05.
Table 2.
Mean values of action potential parameters generated in the wild-type/R1242G-expressing myotubes
| Parameter | Wild-type (n = 11) | R1242G (n = 11) |
|---|---|---|
| Overshoot (mV) | 34.31 ± 2.30 | 21.25 ± 4.45* |
| Time of overshoot (ms) | 6151.26 ± 4.42 | 6151.75 ± 4.09 |
| 20–80% rise time (ms) | 0.57 ± 0.04 | 0.78 ± 0.11 |
| Half-width (ms) | 4.34 ± 0.74 | 4.93 ± 0.70 |
| Maximal rise slope (mV/ms) | 118.32 ± 6.33 | 84.40 ± 10.56* |
| Time of maximal rise slope (ms) | 6150.81 ± 4.40 | 6151.26 ± 4.07 |
| Maximal decay slope (mV/ms) | −23.10 ± 2.27 | −19.58 ± 2.87 |
| Time of maximal decay slope (ms) | 6151.73 ± 4.42 | 6152.49 ± 4.17 |
| Threshold (mV) | −53.49 ± 1.38 | −49.57 ± 1.18* |
| Time to threshold (ms) | 24.94 ± 3.96 | 25.25 ± 3.83 |
*P < 0.05.
Simulation
We simulated the pathogenesis to compare the situation for the novel phenotype with the situation in hypokalaemic periodic paralysis. By estimating the current–voltage relationship of human control muscle and then adding the omega currents for hypokalaemic periodic paralysis, we simulated the current voltage relationship of hypokalaemic periodic paralysis muscle. Likewise, our omega current was added to the control to mimic current-voltage relationships for normokalaemic periodic paralysis muscle (Fig. 5). The decisive elements for the model are the voltage dependencies and reversal potentials of the omega currents. Although the omega current for hypokalaemic periodic paralysis mutations R528H or R2139H follow a single Boltzmann relationship (Jurkat-Rott et al., 2009; Wu et al., 2012), the omega current of R1242G is almost constant in the range of −120 mV to −20 mV (Supplementary Fig. 2A).
Figure 5.

Simulation of periodic paralysis. On basis of simulated inwardly rectifying potassium current IK, comparison of the steady-state current voltage relationships in controls, hypokalaemic periodic paralysis and our family (normokalaemic periodic paralysis), the latter both obtained by addition of omega currents to the control curve. Note that at normal 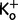 of 4 mM, a depolarizing trigger can depolarize the membrane to −40 mV at which there would be electrically silent cramps. In the inset, a similar depolarization contracture is shown in paramyotonia congenita in which cooling causes a severe depolarization to about −35 mV: after the fifth electrically induced muscle twitch, the excised bundle, that consisted of only few muscle fibre layers, contracted without electrical activity (lower trace; modified from Ricker et al., 1986). The electrical silence during the contracture is verified by wire electrodes that crossed the fibres on both surfaces of the bundle and recorded random action potential spikes; during the time of stimulation the EMG amplifier was overloaded (upper trace). PP = periodic paralysis.
of 4 mM, a depolarizing trigger can depolarize the membrane to −40 mV at which there would be electrically silent cramps. In the inset, a similar depolarization contracture is shown in paramyotonia congenita in which cooling causes a severe depolarization to about −35 mV: after the fifth electrically induced muscle twitch, the excised bundle, that consisted of only few muscle fibre layers, contracted without electrical activity (lower trace; modified from Ricker et al., 1986). The electrical silence during the contracture is verified by wire electrodes that crossed the fibres on both surfaces of the bundle and recorded random action potential spikes; during the time of stimulation the EMG amplifier was overloaded (upper trace). PP = periodic paralysis.
At normal extracellular  of 4 mM, there is only stable membrane potential (where current is zero). Typically paralysis is achieved by hypokalaemia, which reduces the inwardly rectifying potassium conductance by collapsing the pore thereby reducing the steepness of the curves so much that a second stable resting potential at −60 mV appears (Jurkat-Rott et al., 2009). In contrast, at
of 4 mM, there is only stable membrane potential (where current is zero). Typically paralysis is achieved by hypokalaemia, which reduces the inwardly rectifying potassium conductance by collapsing the pore thereby reducing the steepness of the curves so much that a second stable resting potential at −60 mV appears (Jurkat-Rott et al., 2009). In contrast, at  of 4 mM a depolarizing trigger (i.e. exertional exercise) can depolarize R1242G muscle to −40 mV at which there would be electrically silent contractures (Fig. 5). The contracture results because −40 mV is the mechanical threshold at which Cav1.1 can interact with the ryanodine receptor to initiate intracellular calcium release (this threshold is below the threshold of the Cav1.1 central pore current of about −20 mV). The electrical silence observed in the cramping patients is because at −40 mV sodium channels are inactivated as well. Depolarization to this value has been shown to produce electrically silent cramps followed by weakness previously, such as in paramyotonia congenita (Fig. 5).
of 4 mM a depolarizing trigger (i.e. exertional exercise) can depolarize R1242G muscle to −40 mV at which there would be electrically silent contractures (Fig. 5). The contracture results because −40 mV is the mechanical threshold at which Cav1.1 can interact with the ryanodine receptor to initiate intracellular calcium release (this threshold is below the threshold of the Cav1.1 central pore current of about −20 mV). The electrical silence observed in the cramping patients is because at −40 mV sodium channels are inactivated as well. Depolarization to this value has been shown to produce electrically silent cramps followed by weakness previously, such as in paramyotonia congenita (Fig. 5).
Discussion
In the periodic paralyses, the level of serum potassium during a paralytic attack determines the diagnosis (Table 3). While hyperkalaemic and hypokalaemic periodic paralysis are accepted as separate entities, ictal normokalaemia has been a matter of debate. The term normokalaemic periodic paralysis was coined in the 1960s. The disorder resembled hyperkalaemic periodic paralysis and its existence as a nosological entity was questioned when the identification of the most frequent hyperkalaemic periodic paralysis mutation, Nav1.4-M1592V, in one of the two original families was identified (Chinnery et al., 2002). Later a potassium-sensitive periodic paralysis with normokalaemia caused by Nav1.4 mutations at the third arginine of the voltage sensor of domain II at codon 675 also was diagnosed as normokalaemic periodic paralysis (Vicart et al., 2004) and again interpreted as hyperkalaemic periodic paralysis because of the triggering effect of potassium (Song et al., 2012).
Table 3.
Clinical features
| Features | Hypokalaemic periodic paralysis | Hyperkalaemic periodic paralysis | Normokalaemic periodic paralysis | Patients II:1/II:2/II:4 |
|---|---|---|---|---|
| Onset age | First or second decade | First decade | First decade | Post-natally |
| Duration | Hours to days | Minutes to hours | Hours to days | Hours to days |
| Dietary triggers | Carbohydrates | K-rich food | − | − |
| Exercise-related triggers | Rest after exercise | Rest after exercise | Rest after exercise | Exercise |
| Other triggers | Stress, cold | Stress, cold | Stress, cold | Stress, cold |
| Myotonia | − | + | − | − |
| Permanent weakness | + | + | + | + |
| Cramping | − | − | − | Pre-ictally |
| Compartment-like syndrome | − | − | − | Pre-ictally |
| Ictal K | Decrease | Increase | Normal/decrease/increase | Normal/decrease/increase |
| Ictal oedema | + | + | + | + |
| Mutations | CACNA1S/SCN4A | SCN4A | SCN4A | CACNA1S |
| Medication | Acetazolamide/K | Acetazolamide | Acetazolamide | Acetazolamide/K |
| Response to K | Improves strength | Triggers weakness | Depends on ictal k | Maintains strength |
| Long exercise test | Late decline | Late decline | Late decline | No late decline |
Because almost all ictal potassium levels of our patients were in the normal range (100 values altogether; Supplementary material), we diagnosed our family as normokalaemic periodic paralysis. In agreement with this, our family shows a number of symptoms that may be found in all types of periodic paralysis such as post-natal hypotonia, periodic paralysis triggered by stress and cold, and response to carboanhydrase inhibitors (Table 3). Further, myoplasmic sodium and water accumulation as an indicator for the permanent weakness was present in our family as in both hypokalaemic and hyperkalaemic periodic paralysis (Jurkat-Rott et al., 2009; Amarteifio et al., 2012). However, our family additionally displays features not found in other forms of periodic paralysis such as: (i) triggering by exercise rather than by rest after exercise; (ii) absence of the periodic paralysis typical late decline in the long exercise test; and (iii) pre-ictal painful muscle swelling and cramping rather than post-ictal muscle soreness (Table 3). The episodes resulted in rhabdomyolysis and compression of nerves, leading up to the development of drop feet and muscle degeneration in the tibialis anterior group. This is in contrast to the previously reported 25 patients with hypokalaemic periodic paralysis in whom mainly the triceps surae muscles, but not the tibilias anterior groups, were affected (Jurkat-Rott et al. 2009). As in the acute phase the patients showed an albeit reduced pulse of the arteria dorsalis pedis and as no operative pressure release was required, our patients did not experience complete compartment syndrome. Therefore, we used the term ‘transient compartment-like syndrome’ to describe the phenomenon (in analogy to transient ischaemic attack and ischaemic brain insult).
Molecular genetics identified R1242G of the Cav1.1 voltage sensor in domain IV; such mutations in Cav1.1 have so far only been related to hypokalaemic periodic paralysis. Although previously identified mutations neutralize the superficial first or second voltage sensor arginines (Jurkat-Rott et al., 2012), the novel R1242G amino acid substitution neutralizes the deeper third voltage sensor arginine. This location is in agreement with Nav1.4 mutations of a third arginine in domain II (R675Q/G/W) (Sokolov et al., 2008) that cause normokalaemic periodic paralysis (Vicart et al., 2004), whereas more superficially located replacements of voltage sensor arginines cause hypokalaemic periodic paralysis (reviewed in Cannon, 2010; Jurkat-Rott et al., 2012).
As typical for all types of periodic paralysis, the patients responded with benefit to the carbonic anhydrase inhibitors acetazolamide and diclofenamide, as well as to the aldosterone antagonist eplerenone. Both drug groups can repolarize and re-establish the excitability of depolarized muscle fibres and thereby wash out intracellular sodium ion and reduce oedema (Jurkat-Rott et al., 2009; Tricarico and Camerino, 2011; Lehmann-Horn et al., 2012). The triple therapy consisting of low to moderate doses of acetazolamide, eplerenone and potassium seems to be a favourable medication with low risk of adverse effects, at least in this family harbouring the novel normokalaemic periodic paralysis mutation Cav1.1-R1242G. The previous statement that acetazolamide worsens periodic paralysis if caused by an R-to-G mutation (Matthews et al., 2011), was not confirmed.
Our data revealed that R1242G currents produced biophysical defects different from those previously reported for periodic paralysis mutations. First, R1242G revealed a reduced central pore inward current and a left-shift of its steady-state inactivation curve. However, Cav1.1-R1242G also revealed a depolarization-induced outward omega current that could at last partially explain these two features. Similarly, the depolarization-activated outward omega current would oppose the inward sodium current during the rising phase of the action potential and contribute to the reduced size and altered shape of action potentials. This would explain the hypoexcitability of the patients’ musculature which furthers the permanent weakness (Gong et al., 2003). Next to the delayed rectifiers, the outward omega current carried by potassium might help maintain the ictal normokalaemia determining the diagnosis.
In addition to the outward omega current, we identified an inward omega current over a range of negative membrane potentials, after a long depolarization of 1 min duration. According to our simulation for patient muscle, the sodium conductance of the omega pores may be high enough to depolarize the membrane beyond the mechanical threshold of −40 mV and thereby induce muscle contracture. This is experienced by the patients as cramps that cannot be relieved by passive stretching and that are characterized by electrical silence in the EMG. We hypothesize that depolarized muscle fibres take up not only sodium through the omega pore but also osmotic water, to cause fibre swelling that may compress nerves, experienced by the patients as pain and drop foot (transient compartment-like syndrome). The sustained membrane depolarization would inactivate Nav1.4 channels and impair the generation of action potentials, an effect experienced by the patients as weakness as in the classical model of periodic paralysis (Jurkat-Rott et al., 2009). Additionally the whole-cell measurements revealed that R1242G recovered faster than wild-type channels from the inactivated state, revealing gain of function and furthering the tendency towards muscle fibre depolarization.
Supplementary Material
Acknowledgements
We thank the very cooperative family and appreciate the support of Drs. Armin Nagel, Teun de Boer and S. Rosenberg.
Funding
Chunxiang Fan is supported by the International Graduate School of Ulm University, sponsored by the Excellence Program of the German Research Foundation (D.F.G.), and supervised by Frank Lehmann-Horn (F.L.H.) and Karin Jurkat-Rott (K.J.R.). F.L.H. and K.J.R. receive grants from the non-profit Else Kröner-Fresenius-Stiftung, the German Federal Ministry of Research (BMBF) for the IonNeurOnet RD project, and the German Society for Muscle Disorders (DGM). F.L.H. is endowed Senior Research Professor of Neurosciences of the non-profit Hertie-Foundation.
Supplementary material
Supplementary material is available at Brain online.
References
- Amarteifio E, Nagel AM, Weber MA, Jurkat-Rott K, Lehmann-Horn F. Hyperkalemic periodic paralysis and permanent weakness: 3-T MR imaging depicts intracellular 23Na overload–initial results. Radiology. 2012;264:154–63. doi: 10.1148/radiol.12110980. [DOI] [PubMed] [Google Scholar]
- Cannon SC. Voltage-sensor mutations in channelopathies of skeletal muscle. J Physiol. 2010;588:1887–95. doi: 10.1113/jphysiol.2010.186874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–28. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Walls TJ, Hanna MG, Bates D, Fawcett PR. Normokalemic periodic paralysis revisited: does it exist? Ann Neurol. 2002;52:251–2. doi: 10.1002/ana.10257. [DOI] [PubMed] [Google Scholar]
- Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3–23. doi: 10.1016/S0065-2660(08)01001-8. [DOI] [PubMed] [Google Scholar]
- Francis DG, Rybalchenko V, Struyk A, Cannon SC. Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurology. 2011;76:1635–41. doi: 10.1212/WNL.0b013e318219fb57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Legault D, Miki T, Seino S, Renaud JM. KATP channels depress force by reducing action potential amplitude in mouse EDL and soleus muscle. Am J Physiol Cell Physiol. 2003;285:C1464–74. doi: 10.1152/ajpcell.00278.2003. [DOI] [PubMed] [Google Scholar]
- Grabner M, Dirksen RT, Beam KG. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc Natl Acad Sci USA. 1998;95:1903–8. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara S, Takahashi K. The anomalous rectification and cation selectivity of the membrane of a starfish egg cell. J Membr Biol. 1974;18:61–80. doi: 10.1007/BF01870103. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott K, Groome J, Lehmann-Horn F. Pathophysiological role of omega pore current in channelopathies. Front Pharmacol. 2012;3:112. doi: 10.3389/fphar.2012.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat-Rott K, Lehmann-Horn F, Elbaz A, Heine R, Gregg RG, Hogan K, et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet. 1994;3:1415–9. doi: 10.1093/hmg/3.8.1415. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott K, Uetz U, Pika-Hartlaub U, Powell J, Fontaine B, Melzer W, et al. Calcium currents and transients of native and heterologously expressed mutant skeletal muscle DHP receptor alpha1 subunits (R528H) FEBS Lett. 1998;423:198–204. doi: 10.1016/s0014-5793(98)00090-8. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott K, Weber MA, Fauler M, Guo XH, Holzherr BD, Paczulla A, et al. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci USA. 2009;106:4036–41. doi: 10.1073/pnas.0811277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiecinski H, Lehmann-Horn F, Rüdel R. The resting membrane parameters of human intercostal muscle at low, normal, and high extracellular potassium. Muscle Nerve. 1984;7:60–5. doi: 10.1002/mus.880070110. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Weber MA, Nagel AM, Meinck HM, Breitenbach S, Scharrer J, et al. Rationale for treating oedema in Duchenne muscular dystrophy with eplerenone. Acta Myol. 2012;31:31–9. [PMC free article] [PubMed] [Google Scholar]
- Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. 2009;72:1544–7. doi: 10.1212/01.wnl.0000342387.65477.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews E, Portaro S, Ke Q, Sud R, Haworth A, Davis MB, et al. Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. Neurology. 2011;77:1960–4. doi: 10.1212/WNL.0b013e31823a0cb6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricker K, Rüdel R, Lehmann-Horn F, Küther G. Muscle stiffness and electrical activity in paramyotonia congenita. Muscle Nerve. 1986;9:299–305. doi: 10.1002/mus.880090403. [DOI] [PubMed] [Google Scholar]
- Ruff RL. Insulin acts in hypokalemic periodic paralysis by reducing inward rectifier K+ current. Neurology. 1999;53:1556–63. doi: 10.1212/wnl.53.7.1556. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007;446:76–8. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, Catterall WA. Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci USA. 2008;105:19980–5. doi: 10.1073/pnas.0810562105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, Catterall WA. Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J Gen Physiol. 2010;136:225–36. doi: 10.1085/jgp.201010414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song YW, Kim SJ, Heo TH, Kim MH, Kim JB. Normokalemic periodic paralysis is not a distinct disease. Muscle Nerve. 2012;46:914–6. doi: 10.1002/mus.23441. [DOI] [PubMed] [Google Scholar]
- Struyk AF, Cannon SC. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol. 2007;130:11–20. doi: 10.1085/jgp.200709755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struyk AF, Markin VS, Francis D, Cannon SC. Gating pore currents in DIIS4 mutations of NaV1.4 associated with periodic paralysis: saturation of ion flux and implications for disease pathogenesis. J Gen Physiol. 2008;132:447–64. doi: 10.1085/jgp.200809967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengan CH, Antunes AC, Gabbai AA, Manzano GM. The exercise test as a monitor of disease status in hypokalaemic periodic paralysis. J Neurol Neurosurg Psychiatry. 2004;75:497–9. doi: 10.1136/jnnp.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricarico D, Camerino DC. Recent advances in the pathogenesis and drug action in periodic paralyses and related channelopathies. Front Pharmacol. 2011;2:8. doi: 10.3389/fphar.2011.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicart S, Sternberg D, Fournier E, Ochsner F, Laforet P, Kuntzer T, et al. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology. 2004;63:2120–7. doi: 10.1212/01.wnl.0000145768.09934.ec. [DOI] [PubMed] [Google Scholar]
- Weber MA, Nielles-Vallespin S, Essig M, Jurkat-Rott K, Kauczor HU, Lehmann-Horn F. Muscle Na+ channelopathies: MRI detects intracellular 23Na accumulation during episodic weakness. Neurology. 2006;67:1151–8. doi: 10.1212/01.wnl.0000233841.75824.0f. [DOI] [PubMed] [Google Scholar]
- Wu F, Mi W, Burns DK, Fu Y, Gray HF, Struyk AF, et al. A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis. J Clin Invest. 2011;121:4082–94. doi: 10.1172/JCI57398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Mi W, Hernandez-Ochoa EO, Burns DK, Fu Y, Gray HF, et al. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J Clin Invest. 2012;122:4580–91. doi: 10.1172/JCI66091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.