

Abstract
Genetic modification (GM) by Agrobacterium-mediated transformation is a robust and widely employed method to confer new traits to crops. In this process, a transfer DNA is delivered into the host genome, but it is still unclear how the host genome is altered by this event at single-base resolution. To decipher genomic discrepancy between GM crops and their host, we conducted whole-genome sequencing of a transgenic rice line OSCR11. This rice line expresses a seed-based edible vaccine containing two major pollen allergens, Cry j 1 and Cry j 2, against Japanese cedar pollinosis. We revealed that genetic differences between OSCR11 and its host a123 were significantly less than those between a123 and its precedent cultivar Koshihikari. The pattern of nucleotide base substitution in OSCR11, relative to a123, was consistent with somaclonal variation. Mutations in OSCR11 probably occurred during the cell culture steps. In addition, strand-specific mRNA-Seq revealed similar transcriptomes of a123 and OSCR11, supporting genomic integrity between them.
Keywords: cedar pollen allergy, GM crop, transgenic rice, transcriptome, whole-genome sequence
1. Introduction
Genetic modification (GM) is widely used to engineer organism traits for the purposes of basic research and applied science. In plants, Agrobacterium tumefaciens-mediated transformation is the most widely used system of genetic engineering. Agrobacterium is an endemic soil bacterium and induces crown gall formation in host plants.1 There are numerous challenges in applying GM technologies to agriculture. To date, various types of GM crops, such as plants resistant to biotic and abiotic stresses, bioenergy crops, and biopharmaceutical crops, have been developed. Since the first commercialization of herbicide- or insect-resistant GM crop in 1996, 170 million hectares in 28 countries had been allotted to GM crops in 2012.2
In a previous study, we generated transgenic rice (Oryza sativa) ectopically expressing the major Japanese cedar (Cryptomeria japonica) pollen allergens Cry j 1 and Cry j 2. The transgene products specifically accumulated to high levels in the endosperm of rice grain.3 In mice fed the transgenic rice seed, we observed suppression of specific IgE antibodies and a reduction in the clinical symptoms of pollinosis, such as sneezing frequency and infiltration of inflammatory cells.3 We choose a single plant with a single transfer DNA (T-DNA) insertion site from this transgenic line and named as OSCR11.
Differences between GM crops and non-GM crops are one of the most cited public concerns regarding food safety and security. Genomic changes caused by Agrobacterium-mediated transformation have been reported.4,5 Agrobacterium-mediated transformation of rice requires a cell culture step, in which mutations called somaclonal variations arise. These mutations can be detected by Amplified Fragment Length Polymorphism (AFLP), Random Amplified Microsatellite Polymorphism (RAMP), and Random Amplified Polymorphic DNA (RAPD): polymerase chain reaction (PCR)-based analyses that depend on restriction enzyme sites, microsatellites, and specific primers, as has been reported in Arabidopsis.5 T-DNA integration occurs at regions of microhomology between the T-DNA and host genome sequences and/or during the double-strand break (DSB) repair process. Chromatin factors and histone proteins of host plants are also involved in this process, revealing potential sources of damage to the plant host genome by Agrobacterium-mediated transformation.6 Next-generation sequencing (NGS) approaches such as whole-genome resequencing and RNA sequencing (RNA-Seq) offer the means to study the genomic differences between GM and non-GM crops. The genome sequence of virus-resistant transgenic papaya has been reported, but the differences between GM papaya and non-GM papaya were not addressed.7 Another study using microarray analyses found that gamma-ray mutagenesis altered the transcriptome of rice more profoundly than a transgenic modification.8
As oral administration of OSCR11 seed is expected to have a therapeutic effect via the induction of oral immune tolerance against cedar pollen allergens, homozygous OSCR11 seed will be subjected to clinical trials as a treatment for cedar pollen allergy patients after authorization by the Pharmaceuticals and Medical Devices Agency of Japan. In this study, we conducted whole-genome sequencing and transcriptome analysis of genetically modified OSCR11 rice using NGS. OSCR11 has the background genotype of the Koshihikari low-glutelin mutant a123, which accumulates high levels of recombinant proteins in the endosperm by rebalancing seed storage proteins.9 The a123 genotype is a triple mutant with mutations in three glutelin genes (a1: GluB-4, a2: GluA-2, and a3: GluA-1), and was obtained by crossing a1, a2, and a3 mutants (Fig. 1).10 The a1 and a2 mutations were obtained by gamma-ray treatment, and a3 was obtained by ethyl methanesulfonate (EMS).10 To accurately detect mutations induced by Agrobacterium-mediated transformation, we determined the whole-genome sequences and transcriptomes of a123 and compared them with those of OSCR11.
Figure 1.
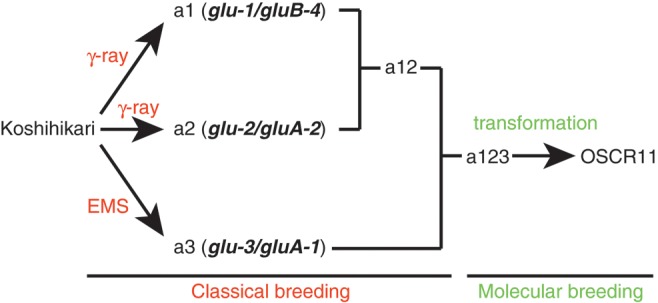
Origin of a123 and OSCR11. a1/gluB-4 and a2/gluA-2 mutations occurred after gamma-ray treatment, and the a3/gluA-1 mutation occurred after EMS treatment of the rice cultivar Koshihikari. a12 with both a1 and a2 mutations was obtained by crossing. a123 with all three mutations was obtained by crossing a12 and a3. OSCR11 was obtained by performing Agrobacterium-mediated transformation on a123.
2. Materials and Methods
2.1. Whole-genome sequencing, sequence alignment, and screening of variant candidates
The generation of transgenic rice expressing Japanese cedar pollen allergens was described previously.3 OSCR11, a rice line containing only a single T-DNA insertion site, was obtained by segregation. The host rice lines such as a123 and OSCR11 were cultivated in a greenhouse under natural light conditions for 2 weeks. Genomic DNA was extracted from leaves using a DNeasy plant mini kit (Qiagen). Paired-end libraries with 450–500 bp insert sizes were constructed and sequenced on a HiSeq2000 (Illumina) instrument using a single lane for each sample and a 100-bp paired-end sequencing protocol. Sequence data are deposited into the DNA Data Bank of Japan Sequence Read Archive (accession no. DRA000918). Adaptor sequences were trimmed using cutadapt 1.011 with parameters: −e 0.1 –O 5 –m 20. Low-quality bases with a Phred Score of <Q20 at either side of each read were also trimmed. Reads shorter than 20 bp in length were discarded. Trimmed paired-end reads were aligned either to the Agrobacterium C58 genome sequence using BWA, or to the reference rice Nipponbare genome sequence (Os-Nipponbare-Reference-IRGSP-1.0) using the CLC Genomics Workbench 5.1 (CLC bio) with the following parameters: similarity, 0.9; length fraction, 0.9; insertion cost, 3; deletion cost, 3; mismatch cost, 3; non-specific matching, ignored. Structural variations (SVs) in OSCR11 were screened using the CLC Genomics Workbench 5.1 with the following parameters: P-value threshold, 1.0E−4; paired-read orientation, yes. Reads properly mapped in pair were extracted, and realigned to the reference with the same parameters. Whole-genome sequencing reads derived from Koshihikari rice (DRR000022–DRR000030) were downloaded from the Sequence Read Archive, and single-end 32-bp reads were aligned to the reference using the same parameters. After removal of duplicated reads, coverage was analysed using SAMtools 0.1.18. Single-nucleotide polymorphism (SNP) candidates were screened using the CLC Genomics Workbench 5.1 with the following parameters: window length, 11; maximum gap and mismatch count, 3; minimum coverage, 1; minimum variant frequency, 75%. Deletion/insertion polymorphism (DIP) candidates were screened by using the CLC Genomics Workbench 5.1 with the following parameters: minimum coverage, 1; minimum frequency, 50%.
2.2. mRNA-Seq analysis
Total RNA was isolated from developing rice seeds at 10–20 days after flowering from each of three independent a123 and OSCR11 plants using a standard phenol/SDS method.12 Following DNase I treatment, mRNAs were enriched using two iterations of a Dynabeads mRNA purification kit (Life Technologies). Libraries were generated using an Ion total RNA-Seq v2 kit (Life Technologies) and sequenced by Ion Torrent PGM (Life Technologies) with an Ion 318 Chip (Life Technologies). Low-quality nucleotides (Phred <Q20) were trimmed, and reads were then mapped onto the RAP-DB annotation13 of the IRGSP-1.0 genome using the CLC Genomics Workbench 5.1, allowing two mismatches for short reads, and requiring a minimum length and similarity of 0.9 for long reads. Non-uniquely aligning reads were randomly mapped. Three biological replicates were analysed for each line, and differentially expressed genes (DEGs) were identified with the DESeq R package using a 5% false discovery rate (FDR) cut-off.14
3. Results and Discussion
3.1. Rice genome re-sequencing
Whole-genome sequencing of a homozygous T5 transgenic OSCR11 plant and its parental line a123 was performed on the Illumina HiSeq2000 platform. We also obtained the genome sequence data of Koshihikari, the background cultivar of a123, from the previous report.15 We obtained 259, 223, and 268 million high-quality reads for Koshihikari, a123, and OSCR11, respectively (Supplementary Table S1). Several reports have shown that Agrobacterium genome sequences are occasionally integrated into the host genome together with T-DNA.16,17 To address this issue, we aligned OSCR11 reads to the Agrobacterium C58 strain reference genome18 using the BWA software.19 We did not detect any OSCR11 whole-genome sequence reads that aligned with C58, indicating that no portions of the Agrobacterium genome had been integrated into the OSCR11 rice genome.
3.2. Detection of structural variations
To detect SVs, we mapped all of our rice sequence reads against the Nipponbare reference genome (Os-Nipponbare-Reference-IRGSP-1.0) using the CLC Genomics Workbench 5.1. We screened SV candidates in OSCR11 by comparing the computed distances of the paired-end reads with their locations within the Nipponbare reference. A total of 149 insertions, 280 deletions, 12 inversions, and 39 instances of interchromosome swapping were called. We validated these SVs by manual inspection of read alignments and found that all of the detected insertions, inversions, and interchromosome swappings were false positives. Although in each case several sequence reads supported the detected polymorphism, these regions were also covered by paired-end reads that supported the Nipponbare reference genome arrangement. Following manual inspection of the sequence alignments, a total of 187 higher-confidence deletions remained. Deletion length varied from 67 bp to 54 kb, and no deletions were detected on chr06 (Supplementary Fig. S1). We compared the mapping results obtained with OSCR11, a123, and Koshihikari for deletions, and found an OSCR11 specific deletion of 810 bp on chr02 of OSCR11 (Fig. 2A). The deletion was confirmed by genomic PCR and DNA sequencing (Fig. 2B). Active transposons have been reported in rice callus.20 The deleted region on chr02 of OSCR11 contains several repeat units that are similar to transposons, but the junction sequences revealed that transposon translocation was not involved in this deletion (Fig. 2A). This result contrasts to previous reports analysing somaclonal variations in Arabidopsis and rice, in which SVs were not detected.21,22 However, since a deletion was only detected at a single locus and deletions in repeat regions are common,23 we could not conclude the deletion was caused by Agrobacterium infection. There are no annotated rice genes within the 10-kb region around the deletion, suggesting that the deletion is unlikely to alter the expression of nearby genes.
Figure 2.

OSCR11-specific 810-bp deletion. (A) Comparison of mapped reads between Koshihikari, a123, and OSCR11. Data were visualized using an Integrative Genomics Viewer.33 A schematic representation of the deleted region is shown below. (B) Confirmation of the OSCR11-specific deletion by PCR using primers indicated in A.
3.3. Detection of transformation- and mutagenesis-induced polymorphisms
To identify specific SNPs and DIPs caused by Agrobacterium-mediated transformation procedures and mutagenesis by gamma ray and EMS, we used the Nipponbare genome as a reference to map our high-quality and clonal reads (Supplementary Tables S1 and S2). For OSCR11 and a123, paired reads with a mapping distance of ∼500 bp were used. Reads that mapped non-uniquely to multiple genomic positions were discarded in this analysis. Uniquely mapped reads covered 81–95% of the reference genome with 11.3–33.2× coverage. Both coverage and depth of coverage for each chromosome were similar (Supplementary Table S2). Rice SNP candidates supported by four or more reads using Illumina HiSeq sequencing and the IRGSP-1.0 genome as a reference are considered to be high-confidence SNPs.21 Therefore, we screened putative SNP using this criterion. Because the depth of coverage across the genome was different between lines, it was difficult to specifically determine whether the SNPs are unique to any particular genotype. To discriminate transformation-induced polymorphisms from false positives, we excluded polymorphisms (a) whose location was not covered by a123 reads, or (b) that were called in Koshihikari or a123 and supported by at least one SNP-containing read in OSCR11. Similarly, to discriminate mutagenesis-induced polymorphisms from false positives, we excluded polymorphisms (a) whose location was not covered in Koshihikari and OSCR11 or (b) that were called in Koshihikari.
In agreement with the results of a previous study,15 63 480 SNPs relative to the Nipponbare reference were detected by four or more reads in Koshihikari, and 67 721 and 93 110 SNPs relative to Nipponbare were detected in a123 and OSCR11, respectively (Table 1). We identified 167 transformation-induced SNPs and 939 mutagenesis-induced SNPs, covering 95.3% and 93.1% of the genome, respectively (Tables 1 and 2). The ratio of transitions-to-transversions in transformation-induced SNPs was 1.0, which is similar to the ratio detected during somaclonal variation (Fig. 3A).21,22 In contrast, the transition : transversion ratio in mutagenesis-induced SNPs was 2.1, which is similar to the 2.4∼2.7 ratio reported for spontaneous mutations in Arabidopsis (Fig. 3A).24 Most mutations caused by EMS are transitions, which reflect the chemical properties of this mutagen.25 Morita et al.26 reported that base substitutions accounted for 12.5% of mutations caused by gamma ray-inducing DSB in rice, and all of them were transversions. The relatively high transition-to-transversion rate in this study may reflect these tendencies.
Table 1.
Summary of polymorphisms
| Effectiveness | Transformation-induced |
Mutagenesis-induced |
|||
|---|---|---|---|---|---|
| SNP | DIP | SNP | DIP | ||
| No polymorphism sites | Yes | 355 561 133 | 355 657 126 | 347 347 999 | 347 436 870 |
| Polymorphism sites | 93 110 | 6953 | 67 721 | 6903 | |
| Non-unique | Yes | 91 035 | 6290 | 58 316 | 3042 |
| Not covered in other lines | No | 1908 | 635 | 8466 | 714 |
| Unique | Yes | 167 | 28 | 939 | 3147 |
| Covered sites | 355 652 335 | 355 677 204 | 347 407 254 | 347 446 813 | |
| Covered sites (%) | 95.3 | 95.3 | 93.1 | 93.1 | |
Effectiveness indicates whether the sites were used for polymorphism calling and comparison.
Covered sites were the sum of effective sites.
Table 2.
Annotation of SNPs and DIPs
| Mutagenesis | Transformation | |
|---|---|---|
| SNPs | ||
| Intergenic | 776 | 137 |
| Genic | 163 | 30 |
| Exon | 116 | 24 |
| Synonymous | 76 | 13 |
| Non-synonymous | 40 | 11 |
| Intron | 16 | 3 |
| UTR | 31 | 3 |
| Total | 939 | 167 |
| DIPs | ||
| Deletion | 1473 | 17 |
| Insertion | 1675 | 11 |
| Intergenic | 2426 | 23 |
| Genic | 722 | 5 |
| Exon | 118 | 0 |
| Synonymous | 0 | 0 |
| Non-synonymous | 118 | 0 |
| Intron | 400 | 2 |
| UTR | 204 | 3 |
| Total | 3147 | 28 |
Figure 3.

Distribution of specific classes of mutation. (A) Distribution of specific classes of mutagenesis-specific (black) and transformation-specific (white) base substitution mutation. Y-axis indicates the ratio of the number of each type of base change to that of total base changes. (B) Distribution of sizes of transformation-specific deletions (black), transformation-specific insertions (dark gray), mutagenesis-specific deletions (light grey), and mutagenesis-specific insertions (white). Y-axis indicates the ratio of the number of each length of deletion or insertion to that of total deletion or insertion. (C) Transgenic-specific deletion and insertion mutations. Locations and flanking sequences are shown. Mono- or di-nucleotide context are highlighted by black letters. Lower case and bold letters indicate deletion and insertion, respectively.
We found 1386, 6903, and 6953 DIPs relative to the Nipponbare reference in Koshihikari, a123, and OSCR11, respectively (Table 1). DIPs preferentially occurred in repetitive regions including homopolymers and microsatellites, as previously reported.22 We detected 28 transformation-induced DIPs and 3147 mutagenesis-induced DIPs, covering 95.3% and 93.1% of the genome, respectively (Tables 1 and 2). Transformation-induced DIPs were 1 or 2 bp in size except for one 5-bp deletion (Fig. 3B). Among them, all of the 1- or 2-bp DIPs occurred in a mono- or di-nucleotide context, but the 5-bp deletion (chr09: 13005724) did not occur in a polymeric context (Fig. 3C). One or two bp DIPs may be generated by slippage during DNA replication. In Arabidopsis, somaclonal DIPs are 1 or 2 bp insertions or deletions in a polymeric context.22 The 5-bp deletion might have been the result of the improper repair of a DSB caused by Agrobacterium infection, but without T-DNA integration. However, since 5-bp deletions in a non-polymeric context have been detected among mutagenesis-induced DIPs (Fig. 3B), it might happen spontaneously.
3.4. Comparison of mutation rates
We identified a total of 195 transformation-induced mutations and 4086 mutagenesis-induced mutations, and mutation rates (number of mutations/covered sites) were 5.5 × 10−7 and 1.2 × 10−5, respectively. Both transformation-induced and mutagenesis-induced mutations were distributed across the genome in a largely uniform pattern, suggesting that they were randomly produced at a genome-wide level (Supplementary Fig. S2). The calculated mutagenesis rates are 91- and 2000-fold higher than the spontaneous mutation rate of 6.0 × 10−9 mutations/effective site calculated for Arabidopsis.24 Jiang et al.22 reported a theoretical mutation rate in Arabidopsis by multiplying the detected mutation rate by 4, resulting in a mutation rate of 1.1 × 10−6. The detected somaclonal mutation rate in Arabidopsis after 3 weeks of cell culture is ∼2.6 × 10−7. Miyao et al.21 reported a somaclonal mutation rate of 1.7 × 10−6 in rice after 5 months of cell culture. In the transformation process used for establishing the transgenic OSCR11 line, cell culture was performed for ∼2 months (callus induction: ∼1 month, Agrobacterium infection: 3 days, and selection: ∼1 month). Interestingly, somaclonal mutation rate per cell culture week (Arabidopsis: 0.86 × 10−7 and rice: 0.85 × 10−7)21,22 and the transformation mutation rate per cell culture week (0.68 × 10−7: in this study) were similar. Considering the base substitution rate of transformation-induced SNPs was highly comparable with the rate induced by somaclonal variation, transformation-induced SNPs cannot be readily distinguished from somaclonal variants. These results support previous reports, showing that cell culture is a primary mutation source during Agrobacterium-mediated transformation.5
Among the transformation-induced mutations, 5.6% (11 SNPs) were non-synonymous, and altered the amino acid sequences of 11 genes (Tables 1, 2, and Supplementary Table S3). Among the mutagenesis-induced mutations, 3.9% (40 SNPs, 44 deletions, and 74 insertions) were non-synonymous, and altered the amino acid sequences of 140 genes (Table 1 and Supplementary Table S4). Numbers of non-synonymous mutations caused by transformation and mutagenesis were much less than those between japonica rice cultivars Koshihikari (794 genes) or Omachi (1017 genes) and Nipponbare.15,27 Although we did not observe deleterious phenotypes in our rice lines, the non-synonymous mutations induced by transformation might cause some phenotypes under specific conditions. The mutations detected showed a largely uniform distribution and did not preferentially accumulate near the T-DNA insertion site or near loci involved in the glutelin-reduced trait (Supplementary Fig. S2). Thus, undesired mutations can be easily eliminated by crossing.
3.5. Transcriptome analysis of the a123 and OSCR11 endosperms
To investigate the influence of Agrobacterium-mediated transformation on the rice transcriptome, we performed strand-specific mRNA-Seq in the maturing endosperm of a123 and OSCR11 lines using Ion Torrent PGM. When OSCR11 seeds are provided to allergy patients as an edible vaccine, the embryo is removed by polishing; therefore, RNA was extracted from de-hulled endosperms whose embryos had been cut out. Since activities of promoters used for transgene expression are highest at 10–20 days after flowering, we chose this stage to analyse. We used single-end reads that mapped to known annotated exons (Supplementary Table S5). We identified only 28 DEGs between OSCR11 and a123 using a FDR of <0.05 (Table 3 and Supplementary Fig. S3), which is comparable with the small number of DEGs detected previously between transgenic rice and wild-type rice using microarray analyses. Among the DEGs, 11 were related to quality control proteins in the endoplasmic reticulum (ER), such as chaperones28,29 (Table 3). The altered expression of these genes is likely caused by the accumulation of recombinant proteins (cedar pollen allergens) that induce ER stress. OsbZIP50 is a cytoplasmic splice variant regulated by OsIRE1and is a molecular marker of strong ER stress.28 However, we did not detect this spliced transcript, indicating that the OSCR11 endosperm is not under strong ER stress. In OSCR11, the T-DNA was inserted 27 bp upstream of the Os08g0107400 5′-UTR, suggesting that the upregulation of Os08g0107400 was probably caused by the T-DNA insertion. Increased expression of GluA-2/Os10g0400200 and Os10g0207500, components of T-DNA,3 were directly derived from the transformed T-DNA. Half of DEGs, described above, could be explained by T-DNA insertion and introduced genes, or expected.
Table 3.
Differentially expressed genes between a123 and OSCR11
| RAP-ID | Average of RPKM |
Fold change | Annotation | |
|---|---|---|---|---|
| a123 | OSCR11 | |||
| Os03g0115800 | 0.8 | 155.0 | 7.5 | Conserved hypothetical protein |
| Os08g0107400 | 1.0 | 72.4 | 6.1 | Similar to GDP-mannose transporterb |
| Os10g0400200 | 132.3 | 2293.5 | 4.1 | GluA-2b |
| Os09g0538000 | 0.0 | 8.1 | N/A | Ribonuclease T2 family protein (OsRNS5) |
| Os01g0537250 | 0.5 | 115.1 | 8.0 | Protein of unknown function DUF3778 domain-containing protein |
| Os09g0512700 | 9.1 | 40.1 | 2.1 | Armadillo-like helical domain-containing protein (Fes1-like)a |
| Os01g0917100 | 7.5 | 0.3 | −4.7 | Conserved hypothetical protein |
| Os04g0641101 | 150.6 | 48.1 | −1.7 | Non-protein coding transcript |
| Os06g0593100 | 82.3 | 219.7 | 1.3 | Similar to UDP-galactose/UDP-glucose transportera |
| Os01g0293000 | 0.0 | 4.3 | N/A | Similar to S-adenosylmethionine synthetase 1 (EC 2.5.1.6) |
| Os06g0622700 | 63.8 | 160.7 | 1.3 | OsbZIP50a |
| Os01g0880800 | 81.9 | 32.2 | −1.4 | Acyl-[acyl-carrier-protein] desaturase |
| Os09g0451500 | 109.7 | 274.3 | 1.3 | OsPDI2;3a |
| Os03g0182800 | 30.4 | 12.0 | −1.4 | OsERF3 |
| Os03g0293000 | 19.4 | 48.9 | 1.3 | Similar to DnaJ domain-containing proteina |
| Os12g0504900 | 15.0 | 1.2 | −3.7 | Glucoamylasea |
| Os10g0207500 | 0.0 | 5.1 | N/A | Similar to TPD1a |
| Os09g0491100 | 0.0 | 2.0 | N/A | Similar to beta-primeverosidase (EC 3.2.1.149)a |
| Os07g0679400 | 4.2 | 0.8 | −2.3 | Conserved hypothetical protein |
| Os01g0693600 | 0.1 | 3.4 | 4.8 | Conserved hypothetical protein |
| Os02g0710900 | 12.1 | 26.8 | 1.1 | Heat shock protein Hsp70 family proteina |
| Os04g0498900 | 4.2 | 0.5 | −3.0 | Conserved hypothetical protein |
| Os05g0156500 | 65.9 | 145.2 | 1.1 | Similar to Apobec-1-binding protein 2 (ERdj3B-like)a |
| Os09g0451000 | 3.1 | 17.0 | 2.3 | OsACO1 |
| Os08g0142900 | 64.6 | 20.6 | −1.8 | Conserved hypothetical protein |
| Os01g0149200 | 5.7 | 13.3 | 1.1 | Similar to Metallothionein-like protein type 2 |
| Os02g0565200 | 50.3 | 106.2 | 1.0 | Microsomal signal peptidase 25-kDa subunit family proteina |
| Os06g0716700 | 61.9 | 129.8 | 1.0 | Similar to heat shock protein 90a |
Fold changes are represented as log2 phase (OSCR11 to a123).
Expression levels are normalized to Reads Per Kilobase of exon per Million mapped reads (RPKM).
N/A in Fold change indicating RPKM in a line was >0, but the other was 0.
aGenes associated with ER stress.
bGenes directly related to integrated T-DNA.
We detected no transformation-induced SNPs or DIPs within 2 kb upstream of any DEG, suggesting that chromosome damage caused by the transformation event had little effect on the endosperm transcriptome. Signalling pathways operating downstream of a few polymorphic genes, or the ER stress that accompanies transgene transformation, may be responsible for the differential gene expression in OSCR11. However, we cannot exclude the possibility that epigenetic memory of stress or other factors are responsible for the DEGs that were not directly or indirectly caused by the T-DNA insertion.
If there are significant damages on host genome by Agrobacterium-mediated transformation, there should be many base transversions and numerous DSB repair traces such as deletions in non-polymeric contexts, like gamma-ray treatment causes them. We demonstrated that mutations caused by the transformation event used in developing a transgenic OSCR11 rice line were mainly induced during the cell culture step and were almost indistinguishable from those associated with somaclonal variation, suggesting that there was an undetectable level of genomic damage, if any, caused by Agrobacterium infection other than T-DNA insertion. Furthermore, the calculated mutation rate between OSCR11 and the host rice line a123 was significantly lower than that between a123 and the original cultivar Koshihikari. Thus, the observed low frequency of mutation is consistent with only a subtle transcriptome difference between OSCR11 and a123. The transformation procedures employed in this study are standard practices in many laboratories, but the culture media were optimized for Koshihikari and its derivative cultivars. We did not perform any backcrosses or screening based on the agronomic traits of OSCR11, yet OSCR11 was almost identical to a123 at the genomic level. Although it will be necessary to analyse more lines to establish the general differences between GM crops and non-GM crops, we concluded that this transgenic line is almost identical to a123 at the genomic level.
Rapid progress has been made in gene targeting and recombination technologies to specifically edit target loci within the rice genome,30–32 but the potential side effects of Agrobacterium-mediated transformation have not been fully explored. To obtain transgenic crops whose genome is identical to that of the host in every respect except for specific target loci, multiple backcrosses will be necessary. Our study provides valuable information for researchers who use transgenic approaches to improve crop yields and other traits. Classical molecular techniques such as Southern blotting are still useful and necessary to compare genetic identity, but NGS platforms have the added potential of whole-genome analysis of transgenic plants. Combining whole-genome sequencing with transcriptome analysis is a robust method to assess the genome integrity of a transgenic plant in relation to its host crop.
Supplementary data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work is supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan, ‘Agri-Health Translational Research Project’ grant to F.T., and ‘Development of Genome Information Database System for Innovation of Crop and Livestock Production’ to T.I.
Supplementary Material
Acknowledgements
We thank E. Domon and Y. Katayose for helpful discussions of this work.
Footnotes
Edited by Prof. Kazuhiro Sato
References
- 1.Van Montagu M. It is a long way to GM agriculture. Ann. Rev. Plant Biol. 2011;62:1–23. doi: 10.1146/annurev-arplant-042110-103906. [DOI] [PubMed] [Google Scholar]
- 2.Clive J. Ithaca, NY: ISAAA; 2011. Global Status of Commercialized Biotech/GM Crops: 2011. ISAAA Brief. [Google Scholar]
- 3.Wakasa Y., Takagi H., Hirose S., et al. Oral immunotherapy with transgenic rice seed containing destructed Japanese cedar pollen allergens, Cry j 1 and Cry j 2, against Japanese cedar pollinosis. Plant Biotechnol. J. 2013;11:66–76. doi: 10.1111/pbi.12007. [DOI] [PubMed] [Google Scholar]
- 4.Labra M., Savini C., Bracale M., et al. Genomic changes in transgenic rice (Oryza sativa L.) plants produced by infecting calli with Agrobacterium tumefaciens. Plant Cell Rep. 2001;20:325–30. [Google Scholar]
- 5.Labra M., Vannini C., Grassi F., et al. Genomic stability in Arabidopsis thaliana transgenic plants obtained by floral dip. Theor. Appl. Genet. 2004;109:1512–8. doi: 10.1007/s00122-004-1773-y. [DOI] [PubMed] [Google Scholar]
- 6.Gelvin S.B. Plant proteins involved in Agrobacterium-mediated genetic transformation. Ann. Rev. Phytopathol. 2010;48:45–68. doi: 10.1146/annurev-phyto-080508-081852. [DOI] [PubMed] [Google Scholar]
- 7.Ming R., Hou S., Feng Y., et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus) Nature. 2008;452:991–6. doi: 10.1038/nature06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batista R., Saibo N., Lourenco T., Oliveira M.M. Microarray analyses reveal that plant mutagenesis may induce more transcriptomic changes than transgene insertion. Proc. Natl Acad. Sci. USA. 2008;105:3640–45. doi: 10.1073/pnas.0707881105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tada Y., Utsumi S., Takaiwa F. Foreign gene products can be enhanced by introduction into low storage protein mutants. Plant Biotechnol. J. 2003;1:411–22. doi: 10.1046/j.1467-7652.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- 10.Iida S., Kusaba M., Nishio T. Mutants lacking glutelin subunits in rice: mapping and combination of mutated glutelin genes. Theor. Appl. Genet. 1997;94:177–83. [Google Scholar]
- 11.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2. [Google Scholar]
- 12.Takaiwa F., Kikuchi S., Oono K. A rice glutelin gene family—a major type of glutelin messenger-RNAs can be divided into 2 classes. Mol. Genet. Genomics. 1987;208:15–22. [Google Scholar]
- 13.Sakai H., Lee S.S., Tanaka T., et al. Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol. 2013;54:e6. doi: 10.1093/pcp/pcs183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto T., Nagasaki H., Yonemaru J., et al. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genomics. 2010;11:267. doi: 10.1186/1471-2164-11-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulker B., Li Y., Rosso M.G., Logemann E., Somssich I.E., Weisshaar B. T-DNA-mediated transfer of Agrobacterium tumefaciens chromosomal DNA into plants. Nature Biotechnol. 2008;26:1015–7. doi: 10.1038/nbt.1491. [DOI] [PubMed] [Google Scholar]
- 17.Kim S.R., An G. Bacterial transposons are co-transferred with T-DNA to rice chromosomes during Agrobacterium-mediated transformation. Mol. Cells. 2012;33:583–9. doi: 10.1007/s10059-012-0010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood D.W., Setubal J.C., Kaul R., et al. The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science. 2001;294:2317–23. doi: 10.1126/science.1066804. [DOI] [PubMed] [Google Scholar]
- 19.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sabot F., Picault N., El-Baidouri M., et al. Transpositional landscape of the rice genome revealed by paired-end mapping of high-throughput re-sequencing data. Plant J Cell Mol. Biol. 2011;66:241–6. doi: 10.1111/j.1365-313X.2011.04492.x. [DOI] [PubMed] [Google Scholar]
- 21.Miyao A., Nakagome M., Ohnuma T., et al. Molecular spectrum of somaclonal variation in regenerated rice revealed by whole-genome sequencing. Plant Cell Physiol. 2012;53:256–64. doi: 10.1093/pcp/pcr172. [DOI] [PubMed] [Google Scholar]
- 22.Jiang C., Mithani A., Gan X., et al. Regenerant Arabidopsis lineages display a distinct genome-wide spectrum of mutations conferring variant phenotypes. Curr. Biol. 2011;21:1385–90. doi: 10.1016/j.cub.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurwitz B.L., Kudrna D., Yu Y., et al. Rice structural variation: a comparative analysis of structural variation between rice and three of its closest relatives in the genus Oryza. Plant J Cell Mol Biol. 2010;63:990–1003. doi: 10.1111/j.1365-313X.2010.04293.x. [DOI] [PubMed] [Google Scholar]
- 24.Ossowski S., Schneeberger K., Lucas-Lledo J.I., et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–4. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greene E.A., Codomo C.A., Taylor N.E., et al. Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics. 2003;164:731–40. doi: 10.1093/genetics/164.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morita R., Kusaba M., Iida S., Yamaguchi H., Nishio T., Nishimura M. Molecular characterization of mutations induced by gamma irradiation in rice. Genes Genet. Syst. 2009;84:361–70. doi: 10.1266/ggs.84.361. [DOI] [PubMed] [Google Scholar]
- 27.Arai-Kichise Y., Shiwa Y., Nagasaki H., et al. Discovery of genome-wide DNA polymorphisms in a landrace cultivar of Japonica rice by whole-genome sequencing. Plant Cell Physiol. 2011;52:274–82. doi: 10.1093/pcp/pcr003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi S., Wakasa Y., Takahashi H., Kawakatsu T., Takaiwa F. Signal transduction by IRE1-mediated splicing of bZIP50 and other stress sensors in the endoplasmic reticulum stress response of rice. Plant J. 2012;69:946–56. doi: 10.1111/j.1365-313X.2011.04844.x. [DOI] [PubMed] [Google Scholar]
- 29.Oono Y., Wakasa Y., Hirose S., Yang L., Sakuta C., Takaiwa F. Analysis of ER stress in developing rice endosperm accumulating beta-amyloid peptide. Plant Biotechnol. J. 2010;8:691–718. doi: 10.1111/j.1467-7652.2010.00502.x. [DOI] [PubMed] [Google Scholar]
- 30.Li T., Liu B., Spalding M.H., Weeks D.P., Yang B. High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 2012;30:390–92. doi: 10.1038/nbt.2199. [DOI] [PubMed] [Google Scholar]
- 31.Terada R., Urawa H., Inagaki Y., Tsugane K., Iida S. Efficient gene targeting by homologous recombination in rice. Nat. Biotechnol. 2002;20:1030–34. doi: 10.1038/nbt737. [DOI] [PubMed] [Google Scholar]
- 32.Ozawa K., Wakasa Y., Ogo Y., Matsuo K., Kawahigashi H., Takaiwa F. Development of an efficient Agrobacterium-mediated gene targeting system for rice and analysis of rice knockouts lacking granule-bound starch synthase (Waxy) and beta1,2-xylosyltransferase. Plant Cell Physiol. 2012;53:755–61. doi: 10.1093/pcp/pcs016. [DOI] [PubMed] [Google Scholar]
- 33.Robinson J.T., Thorvaldsdottir H., Winckler W., et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.