

Abstract
An intact kidney filter is vital to retention of essential proteins in the blood and removal of waste from the body. Damage to the filtration barrier results in albumin loss in the urine, a hallmark of cardiovascular disease and kidney failure. Here we found that the ion channel TRPC5 mediates filtration barrier injury. Using Trpc5-KO mice, a small-molecule inhibitor of TRPC5, Ca2+ imaging in isolated kidney glomeruli, and live imagining of podocyte actin dynamics, we determined that loss of TRPC5 or its inhibition abrogates podocyte cytoskeletal remodeling. Inhibition or loss of TRPC5 prevented activation of the small GTP-binding protein Rac1 and stabilized synaptopodin. Importantly, genetic deletion or pharmacologic inhibition of TRPC5 protected mice from albuminuria. These data reveal that the Ca2+-permeable channel TRPC5 is an important determinant of albuminuria and identify TRPC5 inhibition as a therapeutic strategy for the prevention or treatment of proteinuric kidney disease.
Introduction
Ion channels are central to virtually all aspects of cell behavior and physiology (1–6), and yet their role in the regulation of kidney filter function remains obscure. The human kidney filter handles 180 l of plasma each day, sorting molecules for passage into the urine or retention in the blood stream (7, 8). Upon filter barrier damage, plasma albumin spills into the urinary space, a pathologic condition known as albuminuria (7, 8). Presently, there are no targeted therapies for the efficient protection of filter barrier function.
Specialized kidney pericytes, known as podocytes (9, 10), extend microtubule-based major processes and actin-rich foot processes (FPs) around the underlying capillaries, relying on dynamic actin remodeling to safeguard glomerular filter function (11). Decades ago, disrupted Ca2+ signaling was postulated as an early event in the development of podocyte injury (12) leading to albuminuria (13). However, the causative Ca2+-permeable channel has not been identified to date.
The principle “time is precious” is pivotal to the filter barrier, as it highlights the necessity to limit its exposure to injurious agents (7, 11). In a number of chronic, acquired diseases, including the metabolic syndrome with associated obesity, hypertension, and diabetes, all components of the kidney filter are compromised, starting with endothelial cells (7), glomerular basement membrane (GBM; ref. 14) and podocytes (13). In other diseases, such as nephrotic syndrome, podocyte injury appears to predominate (15). In all cases, podocytes initially undergo simplification and retraction of podocyte FPs (16). These structural defects, known as FP effacement (FPE), are reversible for a certain time, and thus our therapeutic efforts should be targeted to this window of reversibility (17).
Transient receptor potential (TRP) channels are highly conserved nonselective cationic channels first identified in Drosophila (18). The 6 members of TRP cation channel subfamily C (TRPC) form either homomeric or heteromeric tetramers (19). TRPC5 is highly expressed in brain and kidney (19), and its loss leads to an attenuated fear response in mice (20). TRPC5 serves as a cellular sensor of redox changes with implications for inflammation (21). At the cellular level, Ca2+ influx through homomeric TRPC5 channels modulates the Rho family GTPase Rac1 to disrupt the integrity of the actin cytoskeleton in mesenchymal cells, such as fibroblasts and podocytes (22). Numerous studies have correlated this motile in vitro phenotype with damage to the filtration barrier in vivo (23–25). Accordingly, constitutive Rac1 signaling leads to albuminuria in mice (26). We therefore wondered whether TRPC5 is the long-sought Ca2+-permeable channel whose activity perturbs filter barrier function.
Results
TRPC5 localizes to the kidney filter.
To characterize the localization of TRPC5 at the glomerular filter, we raised a new antibody against an epitope on the N terminal of TRPC5. Specificity for TRPC5 was confirmed by Western blot analysis of lysates from HEK cells transfected with TRPC5-GFP, but not TRPC6-GFP (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI71165DS1). We also confirmed antibody specificity in vivo by the detection of TRPC5 in brain lysates from 9-day-old WT mice, but not Trpc5-KO mice (Supplemental Figure 1B). Immunolocalization studies in kidney sections from WT mice revealed specific labeling for TRPC5 in podocytes and colocalization with the podocyte marker podocin (Supplemental Figure 1C and ref. 27). This labeling was absent in tissue from Trpc5-KO mice (Supplemental Figure 1C). Importantly, TRPC5 also colocalized with the podocyte marker synaptopodin (28) in rat kidney (Figure 1A).
Figure 1. Genetic Trpc5 deletion is protective in 2 models of filter barrier damage.

(A) TRPC5 colocalized with synaptopodin. (B) TEM showed that WT and Trpc5-KO mice had an intact filtration barrier, demonstrated by normally arranged FPs and intervening slit diaphragms. After LPS injection, WT mice developed characteristic FPE, while Trpc5-KO mice maintained an intact filtration barrier. (C) PBS did not induce albuminuria in WT or Trpc5-KO mice. WT mice developed significant albuminuria after LPS injection, whereas Trpc5-KO mice were protected from LPS-induced albuminuria (n = 8–12 per group). (D) Western blot from isolated mouse glomeruli showed intact synaptopodin (Synpo) abundance in PBS-injected animals. LPS-injected WT mice showed synaptopodin degradation, including the appearance of the canonical 75-kDa degradation fragment (asterisk). In contrast, Trpc5-KO glomeruli were protected from synaptopodin degradation. GAPDH served as loading control. (E) WT and Trpc5-KO mice perfused with HBSS had a normal filtration barrier. Whereas WT mice perfused with PS developed FPE, Trpc5-KO mice were protected from PS-induced FPE. (F) HBSS-perfused WT and Trpc5-KO animals showed comparable FP numbers (1.8 ± 0.08 and 1.8 ± 0.01, respectively). In contrast, PS perfusion caused significant FPE in WT mice, whereas Trpc5-KO mice were protected to near-normal FP numbers (1.2 ± 0.08 and 1.7 ± 0.07, respectively; n = 6 mice and 90–150 images per group). Original magnification, ×400 (A), ×15,000 (B and E). ***P < 0.001, ANOVA.
LPS-induced albuminuria is absent in Trpc5-KO mice.
LPS injection in mice has been successfully used to study the early events leading to filtration barrier defects and albuminuria (23, 29, 30). LPS induces albuminuria within 24 hours after injection in mice, which correlates with podocyte cytoskeletal remodeling and FPE (30). These changes are reversible, similar to findings in humans exposed to LPS, who show transient albuminuria (31). Of note, LPS has also been shown to induce filter barrier damage in SCID mice, suggestive of a B cell– and T cell–independent, podocyte-specific role for LPS (23). Therefore, the advantage of this model is that it reflects the earliest steps in filtration barrier damage related to podocyte injury. We thus elected to use this model in Trpc5-KO mice and WT control littermates. After control PBS injections, both WT and Trpc5-KO mice had intact filter barriers, as examined by transmission electron microscopy (TEM; Figure 1B). Podocyte FPs and the intervening slit diaphragms were intact (Figure 1B). However, LPS-injected WT mice displayed severely damaged glomerular architecture, with substantial FPE (Figure 1B). Remarkably, LPS-injected Trpc5-KO mice were protected from these structural changes (Figure 1B).
TRPC5 is an essential mediator of LPS-induced albuminuria.
We asked whether the structural changes in the LPS model correlated with functional differences in albuminuria, the marker of a leaky filter. As expected, at baseline, the amount of albumin in 24-hour urine collections in metabolic cages was minimal, and indistinguishable between Trpc5-KO and WT mice (Figure 1C). LPS injection resulted in albuminuria detection within 24 hours in WT mice (Figure 1C), consistent with previous studies (23, 29, 30). However, Trpc5-KO mice were protected from LPS-induced albuminuria (Figure 1C), consistent with the preservation of glomerular architecture in the TEM images (Figure 1B). In keeping with previous results (23), synaptopodin degradation was observed in glomerular lysates from WT animals harvested 24 hours after LPS injection. In contrast, synaptopodin abundance was preserved in lysates from LPS-injected Trpc5-KO mice (Figure 1D). Taken together, these data indicate a role for TRPC5 in the early events leading to albuminuria.
TRPC5 mediates protamine sulfate–induced filter barrier disruption.
To further bolster our finding that TRPC5 plays a role in the reversible phase of glomerular damage leading to albuminuria, we chose to test the protamine sulfate (PS) model (13, 32). This model is based on seminal work showing that vascular perfusion of the polycation PS, thought to neutralize negative charges on the podocyte plasma membrane (33), leads to FPE (13) in a Ca2+-dependent manner (12). The injury is reversible, since washout of PS with the polyanion heparin restores filter barrier architecture (13, 34). As expected, in control HBSS-perfused animals, there were no observable structural changes (Figure 1E). WT mice perfused with PS showed well-characterized lesions, including extensive FPE (Figure 1E). Remarkably, Trpc5-KO mice were protected from PS-induced damage (Figure 1E). These results were quantified in a blinded fashion by counting the number of FPs over a measured length of GBM in TEM images (n = 90–105 images per group). By this analysis, Trpc5-KO mice showed significant protection from the effects of PS compared with WT littermates (Figure 1F). These findings support the conclusion that TRPC5 plays an important role in the early, reversible phase of filter barrier disruption and identify TRPC5 as the candidate ion channel necessary for these changes.
Ex vivo imaging reveals the contribution of TRPC5 to filter barrier Ca2+ dynamics.
In our previous mechanistic in vitro studies, we revealed TRPC5 as an important mediator of Ca2+ influx in podocytes (22). We thus reasoned that the protective effects of Trpc5 deletion should translate into measurable differences in intracellular Ca2+ in podocytes from WT versus Trpc5-KO glomeruli. We used a technique that allowed us to measure intracellular Ca2+ in podocytes loaded with Fura-2 in situ on acutely isolated mouse glomeruli (35). Fura-2 was loaded efficiently into the podocytes, which lie on the outer surface of the spherical glomerular corpuscle, and this structure can be immobilized on poly-lysine–coated chambers suitable for imaging (Figure 2A). Using this approach, we performed ratiometric Ca2+ imaging experiments in response to perfusion of 100 μg/ml LPS into a chamber containing glomeruli isolated from WT or Trpc5-KO mice (Figure 2B). At Δt = 1 minute (i.e., peak transient amplitude), LPS evoked a rise in Ca2+ in WT podocytes that was markedly diminished in Trpc5-KO podocytes (Figure 2B). Statistical analysis of all data collected from multiple glomeruli isolated from different animals (≥5 per group) revealed a significantly reduced response to LPS in Trpc5-KO versus WT podocytes (Figure 2C). These results identify TRPC5 as a downstream mediator of the LPS-evoked rise in podocyte intracellular Ca2+.
Figure 2. LPS- and PS-induced in situ podocyte Ca2+ transients are reduced in isolated glomeruli from Trpc5-KO mice.

(A) Approach for imaging Ca2+ in podocytes in situ on intact, acutely isolated mouse glomeruli. Podocytes — located on the exterior surface of the isolated glomerulus, as shown — were efficiently loaded with Fura-2 and displayed measurable changes in Ca2+ in response to various stimuli. (B) LPS mediated Ca2+ influx (arrows) in glomeruli of WT mice, but the response was attenuated in Trpc5-KO glomeruli. (C) Quantification of peak transient amplitude (at Δt = 1 min) revealed a significantly greater response in WT (n = 24) versus Trpc5-KO (n = 10) glomeruli, attributed to TRPC5-mediated Ca2+ influx. (D) PS mediated Ca2+ influx (arrows) in WT glomeruli, but the response was attenuated in Trpc5-KO glomeruli. (E) Quantification of peak transient amplitude revealed a significantly greater response in WT versus Trpc5-KO glomeruli (n = 19 per group), attributed to TRPC5-mediated Ca2+ influx. Original magnification, ×400 (A, B, and D). Boxed regions are shown enlarged in B and D (enlarged ×9 and ×3, respectively). **P < 0.01, ***P < 0.001, Student’s t test.
PS evokes TRPC5-mediated Ca2+ transients in isolated glomeruli.
Using the same approach, we next asked whether PS could also evoke measurable changes in podocyte Ca2+. PS-perfused WT glomeruli showed substantial increases in podocyte intracellular Ca2+ at peak transient amplitude (Figure 2D). In contrast, only modest increases in intracellular Ca2+ were measured in Trpc5-KO glomeruli. Statistical analysis of all data collected from multiple glomeruli isolated from different animals (≥5 per group) revealed a significantly reduced response to PS in Trpc5-KO versus WT podocytes (Figure 2E). Together with the in vivo data (Figure 1), these results established TRPC5-mediated Ca2+ influx as an important step in the initiation of filter barrier disruption.
TRPC5-mediated Ca2+ transients are blocked by ML204.
In a complimentary approach, we asked whether pharmacologic inhibition mimics the effects of genetic deletion of Trpc5. For these experiments, we took advantage of the recently identified TRPC5 inhibitor ML204 (36). ML204 acts as a reversible pore blocker of TRPC5 (and its close homolog, TRPC4), with at least 20-fold selectivity against TRPC6 and no appreciable effect on all other TRP channels, nor on voltage-gated sodium, potassium, or Ca2+ channels (36). ML204 is therefore an excellent tool to isolate the contribution of TRPC5 channels in podocytes, because (a) TRPC4 is not expressed in podocytes (22) and (b) TRPC6, which acts antagonistically to TRPC5 channels in podocytes (22), remains unaffected by the range of ML204 concentrations (3–30 μM) shown to block TRPC5 (36). We initially transfected HEK cells with TRPC5 and used LPS to activate the channel. LPS (100 μg/ml) generated whole-cell currents with a doubly rectifying I-V relationship characteristic of TRPC5 (Supplemental Figure 2, A and B). This current was blocked by 3 μM ML204 in a reversible manner (Supplemental Figure 2, A and B, and ref. 36). In contrast, TRPC6 whole-cell current in HEK cells remained unaffected by ML204 (data not shown and ref. 36). Single-channel recordings in the outside-out configuration revealed that LPS enhanced TRPC5 open channel probability (Po; Supplemental Figure 2C), similar to angiotensin II (22), and this effect was abrogated by ML204 in a reversible manner (Figure 3A). These data suggest that LPS-induced TRPC5 channel activity can be blocked by ML204 (36).
Figure 3. ML204 unmasks the contribution of TRPC5 to podocyte Ca2+ dynamics.

(A) Representative traces of single-channel recordings in the outside-out configuration in HEK cells expressing TRPC5-GFP confirmed that LPS increased TRPC5 single-channel activity by increasing Po. This was blocked by 3 μM ML204 in a reversible manner. o, open channel; c, closed channel. (B) PS activated Ca2+ transients in podocytes (n = 15 cells), whose peak amplitude was efficiently reduced by bath perfusion of 3 μM ML204 (n = 40 cells). (C) PS-mediated Ca2+ influx (arrows) in WT glomeruli was attenuated by 3 μM ML204. (D) Quantification of Ca2+ responses revealed a significantly greater response in PS versus PS+ML204 glomeruli (n = 9–10 per group), attributed to TRPC5-mediated Ca2+ influx. Scale bar: 50 μm (C). *P < 0.02, ***P < 0.001, Student’s t test.
We next explored the effect of ML204 on PS-evoked Ca2+ dynamics in cultured podocytes and isolated mouse glomeruli. ML204 (3 μM) inhibited PS-mediated increases in podocyte intracellular Ca2+ both in vitro (Figure 3B) and in podocytes in situ on isolated glomeruli (Figure 3, C and D). Importantly, average peak transient amplitude from multiple glomeruli isolated from different animals (≥5 per group) was significantly reduced in ML204-treated podocytes in situ (Figure 3D), similar to cultured cells in vitro (Figure 3B). The decrease in intracellular Ca2+ due to ML204 was in line with our experiments in Trpc5-KO mice (Figure 2, D and E). Taken together, these data bolster the conclusion that TRPC5 activity mediates filter barrier disruption.
TRPC5 inhibition rescues podocytes from cytoskeletal remodeling.
A decade of work has converged on podocyte cytoskeletal dysregulation as a central event in albuminuria (37), but the upstream channels that mediate these changes were largely unknown. Our recent mechanistic in vitro work revealed that TRPC5 mediates podocyte cytoskeletal remodeling in response to angiotensin II (22). Here we sought to characterize the effects of TRPC5 activity downstream of PS in order to provide a molecular and cell biological explanation for our in vivo observations (Figure 1, E and F). Pharmacologic inhibition of TRPC5 with ML204 rescued podocytes from PS-mediated cytoskeletal remodeling and protected synaptopodin abundance in a dose-dependent manner (Figure 4, A and B, and Supplemental Figure 3, A and B). This is consistent with our prior work, which defined this concentration range of ML204 to be specific for TRPC5 channels (36). To further confirm the specificity of these findings, we repeated the assay in Trpc5-depleted cells. Similar to ML204 treatment, gene silencing of Trpc5 protected the actin cytoskeleton and synaptopodin abundance against the effects of PS (Figure 4, C and D), confirming the overall conclusion that TRPC5 is necessary for the PS-mediated cytoskeletal remodeling. We validated this further by Western blotting and found that ML204 prevented the PS-mediated degradation of synaptopodin in a dose-dependent manner (Figure 4, E and F). Based on our previous work showing a conserved role for TRPC5 in Rac1 activation in podocytes and fibroblasts (22), we asked whether PS, in addition to synaptopodin degradation, increases Rac1 activity. GST pulldown assays confirmed this hypothesis, showing that ML204 prevented the PS-mediated and TRPC5-dependent activation of Rac1 (Figure 4, G and H). Finally, we repeated the assay in podocytes exposed to LPS and found that ML204 rescued actin stress fibers and restored the integrity of the cytoskeleton, similar to PS (Supplemental Figure 3, C and D). These data show that, in response to noxious stimuli, TRPC5 promotes remodeling of the podocyte actin cytoskeleton. Additionally, they provide mechanistic insight on the effects downstream of TRPC5 activity, which center on the degradation of synaptopodin and concomitant activation of Rac1.
Figure 4. TRPC5 inhibition protects podocytes from cytoskeletal remodeling.

(A) Treatment of podocytes with 300 μg/ml PS resulted in disrupted actin (red) and loss of stress fibers. Under these conditions, podocytes were largely devoid of synaptopodin (green). ML204 treatment blocked synaptopodin loss and cytoskeletal disruption. (B) Quantification of cytoskeletal remodeling (defined as no visible actin fibers) showed that ML204 rescued podocytes in a dose-dependent manner (n = 90 images/condition). (C) Loss of synaptopodin abundance and cytoskeletal remodeling by 300 μg/ml PS was prevented by TRPC5 shRNA–mediated Trpc5 gene silencing. Scr, scrambled shRNA control. (D) Quantification of cytoskeletal remodeling showed that TRPC5 shRNA–mediated Trpc5 depletion rescued podocytes, in contrast to scrambled control. n = 90 images/condition (10 images × 3 repeats × 3 independent trials). (E) Cell lysates from podocytes treated with PS or PS+ML204 showed that synaptopodin abundance, attenuated by 300 μg/ml PS compared with PBS-treated controls, was rescued by 10 or 30 μM ML204 in a dose-dependent manner. (F) Quantification of 4 Western blots from 4 independent experiments showed that 30 μM ML204 rescued synaptopodin abundance for PS-mediated degradation. Treatment with 10 μM ML204 trended in the same direction, but did not achieve statistical significance. (G) Rac1 activity (Rac1-GTP) increased in podocytes treated with 300 μg/ml PS compared with PBS-treated controls; 30 μM ML204 abrogated this activation. (H) Quantification of 3 Western blots from 3 independent experiments showed that 30 μM ML204 significantly blocked PS-mediated Rac1 activation. Original magnification, ×400 (A and C). *P < 0.05, ****P < 0.00001, ANOVA.
Real-time imaging of podocyte actin dynamics reveals the sequence of events leading to cytoskeletal remodeling.
To explore the temporal and spatial dynamics of cytoskeletal remodeling in podocytes, we performed real-time live confocal microscopy using LifeAct (38). This approach revealed that PS induced a program of cytoskeletal remodeling, which began with increased cell membrane ruffling and lamellipodia formation (consistent with increased Rac1 activity) and proceeded to stress fiber retraction (consistent with synaptopodin degradation), leading to ultimate cytoskeletal involution and cellular collapse (Figure 5 and Supplemental Video 1). At baseline, podocytes had minimal membrane ruffling activity. Within 10–15 minutes of the addition of PS, increased ruffling activity was observed (Figure 5), consistent with enhanced (and likely Rac1-driven; Figure 4, G and H) lamellipodia formation. At 20–30 minutes after the addition of PS, we observed loss or retraction of stress fibers. Subsequent to this, podocytes displayed a characteristic involution, and the cytoskeleton was reduced to a highly fluorescent condensed actin structure at the center of what was previously a well-developed, ramified podocyte (Figure 5). In contrast, podocytes imaged under identical conditions in the presence of ML204 were protected from these cytoskeletal changes (Figure 5 and Supplemental Video 1). These real-time observations of podocyte actin dynamics strengthened our conclusion that TRPC5 inhibition confers protection from podocyte injury.
Figure 5. Real-time imaging revealed the dynamics of TRPC5 inhibition on PS-induced actin remodeling.
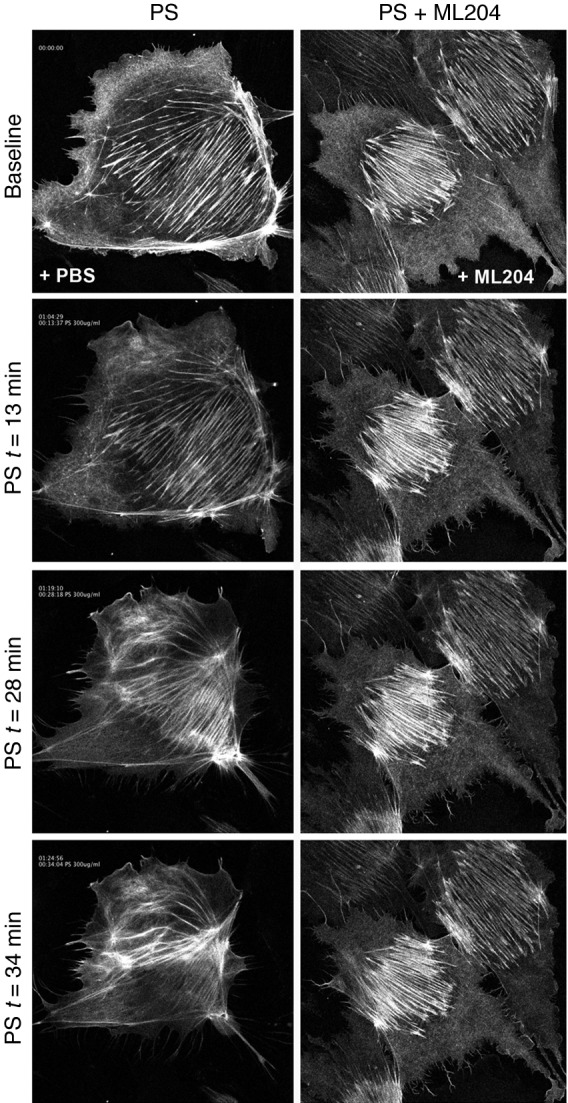
LifeAct technology and confocal microscopy were used to assay the timing and severity of cytoskeletal remodeling in the presence of PS. PS induced increased ruffling activity and lamellipodia formation within 13 minutes of its initial application. By 28 minutes, actin stress fibers were disrupted and no longer extended across the span of the cell’s cytoplasm, as they did before PS application. This cytoskeletal remodeling resulted in the involution of the cell onto itself after 34 minutes, with the appearance of an intensely fluorescent structure at the center of a previously healthy podocyte. This PS-mediated actin remodeling was blocked by 10 μM ML204, resulting in the preservation of stress fibers and the cell’s cytoskeleton throughout the same time frame. Representative images are shown (see Supplemental Video 1). Original magnification, ×400.
Muscarinic receptor–mediated TRPC5 activation at the filter barrier.
TRPCs are receptor-operated channels, susceptible to activation by G protein–coupled receptors, among others (19, 39, 40). Previous work has shown that M5 muscarinic receptor activation in podocytes results in a rise in intracellular Ca2+, which is not inhibited by the trivalent ion lanthanum (La3+) (41). TRPC5 channels in podocytes mediate a La3+-insensitive current (22). We therefore wondered about the effects of carbachol (Cch), a muscarinic receptor agonist, on TRPC5-mediated Ca2+ signaling and actin dynamics. Using Ca2+ imaging in isolated glomeruli, we found that 100 μM Cch evoked a marked rise in podocyte intracellular Ca2+ in WT glomeruli that was significantly smaller in Trpc5-KO glomeruli (Figure 6, A and B). Similarly, ML204 blocked a significant portion of Cch-induced Ca2+ transients in WT glomeruli (Figure 6, C and D), in line with our findings in Trpc5-KO glomeruli. In control experiments, depletion of podocyte internal stores by 10 μM thapsigargin had no significant effect on the peak amplitude of Cch-induced Ca2+ transients (Supplemental Figure 4). These data lend further support to the conclusion that ML204 is a specific inhibitor of TRPC5 at the filter barrier, capable of blocking channel function downstream of receptor-operated pathways.
Figure 6. Inhibition of muscarinic receptor–mediated TRPC5 activity prevents podocyte damage.

(A) The muscarinic receptor agonist Cch induces a significant increase in Ca2+ transients in podocytes located on acutely isolated glomeruli from WT mice but not from Trpc5-KO mice. (B) Quantification of averaged peak amplitude at Δt = 1 min (n = 11 glomeruli per group). (C) Peak amplitude of Cch-induced Ca2+ transients in podocytes on acutely isolated glomeruli was attenuated by 3 μM ML204. (D) Quantification of averaged peak amplitude at Δt = 1 min (n = 12 glomeruli [Cch], 10 glomeruli [Cch+ML204]). (E) Loss of stress fibers induced by 100 μM Cch was blocked by ML204. (F) Significant rescue of stress fiber formation by ML204 (n = 90 images/condition). Scale bar: 50 μm (C). Original magnification, ×400 (E). **P < 0.01, ***P < 0.001, Student’s t test (B and D); ****P < 0.00001, ANOVA (F).
ML204 prevents Cch-induced disruption of the actin cytoskeleton.
The ex vivo Ca2+ imaging Cch experiments suggested that muscarinic receptor stimulation confers susceptibility to Ca2+-induced podocyte injury through TRPC5. To test this hypothesis, we explored the effects of muscarinic receptor–mediated TRPC5 activity on podocyte actin dynamics. Similar to PS and LPS (Figure 4 and Supplemental Figure 3, C and D), 100 μM Cch induced a significant loss of stress fibers and reduction of synaptopodin protein abundance (Figure 6, E and F). Inhibition of TRPC5 with ML204 rescued podocytes from Cch-mediated cytoskeletal remodeling and protected synaptopodin abundance (Figure 6, E and F), in line with our previous experiments (Figure 4 and Supplemental Figure 3, C and D). These data further bolster the conclusion that TRPC5 mediates podocyte injury through its effects on the actin cytoskeleleton and that ML204 is a specific inhibitor of TRPC5-mediated injury, regardless of the upstream stimulus.
TRPC5 inhibition protects the integrity of the glomerular filter barrier.
Finally, having established the protective role of TRPC5 inhibition in vitro and ex vivo with regard to filter barrier stability, we sought to establish the in vivo efficacy of ML204 as an inhibitor of PS-mediated injury. HBSS alone, PS alone, or PS plus 10 μM ML204 were continuously perfused in WT mice (see Methods). As expected, in control HBSS-perfused animals, there were no observable structural changes (Figure 7A). WT mice perfused with PS showed typical lesions, including extensive FPE (Figure 7A and refs. 13, 32). Notably, ML204-perfused mice were protected from PS-induced FPE (Figure 7A). These results were quantified in a blinded fashion by counting the number of FPs over a measured length of GBM in TEM images (n = 90–105 images per group). By this analysis, ML204-perfused mice showed significant protection from the effects of PS (Figure 7B). These results are in line with our observations in Trpc5-KO mice (Figure 1, D and E).
Figure 7. TRPC5 inhibition protects against PS- or LPS-induced filter barrier disruption.

(A) PS induced FPE. Coperfusion of 10 μM ML204 in PS-perfused mice preserved FPs, similar to HBSS controls. (B) ML204 treatment conferred protection from FPE (HBSS, 1.84 ± 0.08 FPs/μm GBM; PS, 1.16 ± 0.08 FPs/μm GBM; PS+ML204, 1.78 ± 0.2 FPs/μm GBM) (n = 6–7 animals and 90–150 images per group). (C) LPS injection led to FPE. Treatment of LPS-injected mice with 20 mg/kg/d ML204 i.p. mitigated FPE. (D) FP numbers in ML204-treated animals (PBS, 2.21 ± 0.07 FPs/μm GBM; LPS, 1.67 ± 0.10 FPs/μm GBM; LPS+ML204, 2.04 ± 0.04 FPs/μm GBM) (n = 3–4 animals and 60 images per group). (E) PBS or PBS+ML204 injection did not induce albuminuria in WT mice. After LPS injection, WT mice developed significant albuminuria, which was significantly reduced after ML204 treatment (n = 5–12 animals/group). Original magnification, ×15,000 (A and C). *P < 0.05, ***P < 0.001, ****P < 0.0001, ANOVA.
Next, we tested the effect of ML204 in the LPS model. ML204 was injected (20 mg/kg/d i.p.) twice at 12-hour intervals after injection of LPS. Control PBS-injected mice had no observable structural changes and no albuminuria (Figure 7, C–E). LPS injection resulted in FPE (Figure 7D), although this was milder than the PS-induced FPE. Importantly, the ML204-treated mice were protected from LPS-induced FPE and albuminuria (Figure 7, D and E).
Discussion
Despite numerous studies in genetically deleted TRPC mice, to date, little insight has been gained into the targeting of TRPC channels for therapeutic benefit. A possible explanation may be that TRPC channels are drivers of change when there is insult or stress in physiology or disease (42). Their role may therefore be unmasked once their activity is challenged in appropriate disease models. In addition, there is a lack of specific pharmacologic tools to help distinguish the effects of specific channels in a physiologic setting. Here, with a combination of Trpc5-KO mice, advanced ex vivo physiology, live imaging, 2 animals models of disease, and a new TRPC5-selective pharmacologic inhibitor, we overcame these challenges to reveal TRPC5 inhibition as a strategy to protect the glomerular filter barrier (Figure 8).
Figure 8. Model for (dys)regulation of kidney filter function by TRPC5.

Top-down view of podocyte interdigitating FPs composed of parallel actin bundles. TRPC5 activation in response to injury leads to Rac1 activation (Rac1-GTP) and synaptopodin degradation. These events promote podocyte cytoskeletal remodeling. Loss of filter integrity ensues, leading to albuminuria, a potentially reversible condition. TRPC5 inhibition therefore emerges as a therapeutic strategy for the stabilization of the human filter barrier.
Ion channels are widely recognized as key regulators of virtually every vital physiologic and pathologic process, yet their role in the regulation of the filter barrier has remained obscure. Our ability to compare the results of ex vivo Ca2+ imaging in intact glomeruli from WT and Trpc5-KO mice has afforded us a valuable toolbox to explore Ca2+ dynamics in this uniquely specialized tissue. From an ion channel biology perspective, the most exciting discovery in the present study is the identification of TRPC5 as the long-sought Ca2+ channel necessary for the reversible, early phase of filter barrier damage.
TRPC5 can modulate Rac1 activity and actin dynamics in cells of mesenchymal origin, such as fibroblasts and podocytes (22), but the in vivo significance of this finding has been obscure. Notably, the 2 molecules are linked by a positive feedback loop, whereby Rac1 activity is required for the vesicular insertion of TRPC5 into the plasma membrane (22, 39, 40). As an important in vivo correlate to these in vitro findings, the present study showed that loss of TRPC5 or its pharmacologic inhibition protected against albuminuria. Our findings also extended previous work showing that constitutive Rac1 signaling in Arhgdia-KO mice leads to filter barrier damage and kidney disease (26, 43). Thus, the detailed mechanistic understanding of the interplay between TRPC5 and Rac1 activities at the cellular level is now translated to the mouse, paving the way for the validation of TRPC5-targeted therapies.
Cell-specific mechanisms and synergistic versus antagonistic effects distinct from tissue to tissue are the key to placing our findings in the context of prior work. For example, TRPC6 is a mediator of LPS-induced lung vascular permeability and inflammation (44), whereas our data here revealed TRPC5 as a mediator of LPS-induced glomerular filter damage. LPS has also been shown to have effects on other channels, including Kir in monocytes (45) and TRPV2 in macrophages (46). Thus, LPS can signal to different ion channels in a context-dependent manner in diverse cells and tissues. Indeed, the antagonistic relationship between TRPC5/Rac1 and TRPC6/RhoA signaling in podocytes and fibroblasts (22) does not appear to be at play in endothelial cells, where TRPC5 and TRPC6 act synergistically to activate RhoA (47, 48). Podocyte-specific TRPC5/Rac1 signaling thus emerges as the key, specific, and specifically inducible pathway necessary for acute, and possibly reversible, filter barrier disruption.
Our data show that TRPC5 promotes the degradation of synaptopodin, leading to cytoskeletal remodeling both in vitro and in vivo, in 2 models of disease. This is an important finding in support of a paradigm shift in our understanding of TRPC5 in acute/reversible versus chronic/irreversible injury of the glomerular filter. LPS- or PS-mediated activation of the Ca2+-dependent phosphatase calcineurin displaces 14-3-3 from synaptopodin, thus allowing for synaptopodin cleavage by cathepsin L (23, 49). This is efficiently inhibited by the calcineurin inhibitor cyclosporine A (23). Synaptopodin also promotes RhoA activity (50), and thus protects the podocyte cytoskeleton in vitro and safeguards against filter barrier damage in mice (23, 50). The present study extends prior work to reveal that TRPC5-induced synaptopodin degradation mediated acute injury (e.g., after exposure to LPS, PS, and Cch).
The identification of TRPC5 as a mediator of podocyte injury may belie its putative role under physiologic conditions. It is tempting to speculate that the podocyte may require the ability to transiently increase glomerular permeability as a protective mechanism for the successful clearance of the filter barrier from accumulated pathogens, toxins, or debris. Therefore, the limited activation of TRPC5/Rac1 by the transient, locally increased abundance of LPS (due to clinically occult bacteremia, for example) may result in a desirable increase in filter barrier permeability, which in turn might allow for the efficient clearance of LPS and/or associated toxins. Future studies will address these important questions.
Our results showed that genetic deletion or pharmacologic inhibition of TRPC5 protected the filter barrier from acute, reversible injury (e.g., LPS and PS). This is particularly interesting in juxtaposition with what is known about TRPC6. Humans with gain-of-function TRPC6 mutations develop kidney disease, most often well into adulthood (51), and mice expressing the same gain-of-function TRPC6 mutants have no proteinuria early on, but develop kidney disease at 6 months of age, a late onset in the mouse life cycle (52). It is thus intriguing to speculate that TRPC6 (over)activity promotes chronic, irreversible kidney injury, whereas acute disruption of kidney filter function is driven by inducible TRPC5 activity. Our present observations support the notion that TRPC5 plays a role distinct from that of TRPC6 in filter barrier Ca2+ dynamics; thus, targeting of TRPC5 channels within the window of reversibility emerges as a viable therapeutic strategy for albuminuria.
The lack of potent selective pharmacological inhibitors for TRPC channels has limited our understanding of these channels in native tissues and prevented the development of therapeutics in this area. Therefore, from a clinical perspective, the in vivo validation of ML204 as a TRPC5 channel inhibitor holds promise for the development of future therapeutic approaches for albuminuria targeting TRPC channels. While the weight of evidence suggests an important role for TRPC5 in the early, reversible phase of filter barrier disruption, TRPC5-independent mechanisms should also be investigated. Additionally, it will be important to study the role of TRPC5 channels in chronic models of disease, particularly diabetic kidney disease, which begins with microalbuminuria before progression to kidney failure.
Kidney glomeruli arose through evolution to secure vertebrate survival in freshwater and on land (53). An intact kidney filter is vital for survival in terrestrial mammals (53). In humans, a leaky filter barrier is a risk factor for cardiovascular disease, kidney failure, and death. Due to the current worldwide, rapidly growing epidemic of metabolic disease, there is an unmet, urgent need for effective antiproteinuric treatment options (54, 55). Our present findings offer TRPC5 inhibition as a potential therapeutic strategy for albuminuria.
Methods
Reagents.
Unless otherwise indicated, all chemicals and reagents were purchased from Sigma-Aldrich.
Antibodies.
Guinea pig polyclonal antibodies specific for TRPC5 (NP_033454.1) were generated in the Helmholtz Group for Cell Biology, DKFZ, using the polypeptide RAETELSAEEKAFL–C-KLH (aa position 24–37) for immunization. The polypeptide was synthesized (PSL) and conjugated to keyhole limpet hemocyanin (KLH) via an additional cysteine (C) to trigger and enhance immunoreaction.
Immunofluorescence microcopy of mouse kidney frozen sections.
Adult rats as well as Trpc5-KO (provided by D.E. Clapham, Children’s Hospital, Boston, Massachusetts, USA) and age-matched littermate WT mice were fixed by vascular perfusion with 4% paraformaldehyde in PBS and processed for immunofluorescence microscopy or TEM as described previously (28). Double-labeling fluorescence microscopy of frozen sections for TRPC5 and the podocyte markers synaptopodin (28) and podocin (56) was done as reported previously (28). Images were captured on an upright fluorescence light microscope (Olympus BX51) or a Zeiss LSM 510 confocal microscope as described previously (22).
LPS-induced albuminuria.
Induction of albuminuria in male WT and Trpc5-KO mice (20–25 g BW) by LPS injection was done as previously described (23), with some modifications. At 48 hours prior to injection, baseline urine was collected for 24 hours in metabolic cages. LPS (15 μg/g i.p., 1 mg/ml; Sigma-Aldrich) was injected twice, at the 0- and 24-hour time points. PBS was injected i.p. twice, at 12 and 36 hours, to avoid dehydration. ML204 (20 mg/kg/d i.p.) was injected at 12 and 24 hours. Urine was collected for a 24-hour period beginning 24 hours after initial LPS injection using metabolic cages. To quantify the levels of albuminuria, 10 μl urine was analyzed by SDS-PAGE. Bovine serum albumin standards (0.25, 0.5, 1.0, 2.5, and 5.0 μg) were run on the same gel and used to identify and quantify urinary albumin bands. Coomassie signals were quantified using ImageJ. The resulting values — the product of area size and mean gray value of each albumin standard band — were used for construction of a standard curve and its associated mathematical function. Subsequently, the values of the sample bands were translated into albumin concentrations, which were extrapolated to the 24-hour total urine volume. Results were assessed by ANOVA and Bonferroni’s multiple-comparison test.
For preparation of glomerular lysates for Western blotting, mice were treated as above and killed 36 hours after initial LPS injection. Mouse kidneys were perfused through the renal artery with Dynabeads (Invitrogen) for magnetic isolation of highly purified glomeruli (57). Protein extraction from isolated glomeruli, SDS-PAGE, and Western blotting was done as described previously (28), and proteins were detected with appropriate primary and secondary antibodies.
PS model.
Adult WT (n = 15) and Trpc5-KO (n = 7) littermate mice were anesthetized with pentobarbital and placed on a heat pad set at 37°C, and their kidneys were perfused in situ through the renal artery at a pressure of approximately 240 mm Hg and an infusion rate of 9 ml/min as previously described (58), with some modifications. First, kidneys were flushed with HBSS or with HBSS plus 10 μM ML204 at 37°C for 2 minutes, followed by perfusion with 2 mg/ml PS in HBSS or with PS plus ML204 at 37°C for 15 minutes. All vascular perfusion solutions were kept at 37°C throughout the duration of the experiment.
TEM.
At the end of both PS and LPS experiments, kidneys were fixed by perfusion with 4% paraformaldehyde in PBS, removed, and immersed in the same fixative for a maximum of 2 days until further processing for TEM. The degree of FPE was assessed in a series of images taken at ×15,000 magnification by a renal pathologist in a blinded fashion. 5 images per glomerulus × 3 glomeruli per mouse × 6–7 mice per group yielded a total of 90–105 images per animal group for quantification. These micrographs were analyzed in a blinded fashion by 2 independent observers. ImageJ software was used to measure the length of the GBM on each micrograph. The number of FP per μM GBM was subsequently calculated. Results were assessed by ANOVA and Bonferroni’s multiple-comparison test.
Cell culture.
WT mouse podocytes (passages 8–14) were provided by P. Mundel (MGH, Boston, Massachusetts, USA) and cultured as previously described (22).
Lentiviral delivery protocol.
Lentivirus-mediated gene silencing of Trpc5 was done according to recently described protocols (22), with some modifications. For all experiments, parallel infections with scrambled shRNA were used as control. TRPC5 shRNA and scrambled shRNA were constructs from the RNAi Consortium library of the Harvard–Massachusetts Institute of Technology Broad Institute (22). Knockdown efficiency was assessed as previously described (22) and confirmed before each experiment by quantitative PCR (Supplemental Figure 1D), according to standard protocols. TRPC5 (and all other TRPC knockdown constructs) were previously verified at the mRNA and protein levels (22). We had previously also tested for efficient functional knockdown by Ca2+ imaging and electrophysiology as well as by rescue experiments in this setting, in which TRPC5-mediated Ca2+ influx was rescued by TRPC5 overexpression in TRPC5-depleted podocytes (22). Notably, TRPC5 depletion has no effect of TRPC6 levels (22). LifeAct was cloned into the VVPW lentiviral vector to optimize expression levels. All experiments were performed at least 96 hours after infection of podocytes that were thermoshifted 5–7 days before the assay.
PS, LPS, and Cch treatment of cultured podocytes.
Differentiated cultured podocytes grown at >90% confluence were incubated with 1–30 μM ML204 for 20 minutes, and then exposed to 300 μg/ml PS, 100 μg/ml LPS, or 100 μM Cch as appropriate. For PS experiments, as soon as changes in cell morphology could be seen by light microscopy (70–90 min), cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 15 minutes before permeabilization with 0.1% Triton X-100 for 10 minutes. For LPS and Cch experiments, cells were fixed as above at 24 hours after treatment. For immunostaining, podocytes were incubated with synaptopodin “NT” antibody (55) and detected with Alexa Fluor 488–conjugated secondary antibody. Actin structures were labeled with Alexa Fluor 594–conjugated phalloidin as described previously (22). 3 independent trials were analyzed, with 3 dishes per condition in each trial and 10 images per dish with comparable cell density. A total of 1,600–2,000 cells was analyzed for the PS/ML204 experiment and 1,400–1,600 cells for the PS/KD experiments. A total of 1,000 cells was analyzed for the LPS experiment and 1,400 cells for the Cch experiment. The number of cells was counted by DAPI staining and analysis with an automated script in ImageJ, which was subsequently corrected manually. Affected cells were defined as either collapsed with a very bright, condensed actin staining (for PS experiments), or cells without clearly visible stress fibers (for LPS and Cch experiments), as previously described (22). Images were acquired with a Zeiss LSM510 upright confocal microscope. Images from an optical slice of 3–4 μm were acquired using Zeiss Pascal software. Statistical significance was evaluated by ANOVA and Dunnett’s multiple-comparison test.
Live imaging.
For LifeAct imaging, podocytes expressing LifeAct (38) were placed in Phenol Red–free media in an appropriate imaging chamber. Time-lapse images were collected every 30 seconds using a ×40 water immersion lens on a Zeiss LSM510 confocal microscope. Images from an optical slice of 3–4 μm were acquired using Zeiss Pascal software. Image analysis was done using ImageJ.
Rac1 activation assay.
Rac1 activation assays were done as previously described (22), with some modifications. Podocytes were treated with 300 μg/ml PS for 1 hour, followed by harvest and Rac1 pulldown experiments. In the ML204 experiments, cells were pretreated with 30 μM ML204 for 20 minutes before PS was applied. Activated Rac1 was analyzed with a commercial Rac1 activation assay kit (Millipore) using a GST-tagged fusion protein corresponding to the p21-binding domain (PBD; residues 67–150) of human PAK-1, according to the manufacturer’s instructions. After pulldown, the eluted active Rac1 was detected by immunoblotting using a mouse monoclonal Rac1 antibody (BD). Total Rac1 and GADPH were measured in the cell lysates used for the pulldown studies and served as loading controls.
Isolation of glomeruli for imaging.
Isolation of glomeruli for Ca2+ imaging was done by serial sieving, as previously described (28), with some modifications. Emphasis was placed on the integrity of the glomeruli rather than the purity of the preparation, so the protocol was modified to exclude the use of collagenase, using gravity and serial washes with ice-cold PBS to isolate glomeruli. After visual inspection, isolated glomeruli were placed in RPMI medium in 37°C for 20 minutes before loading with Fura-2 for imaging experiments.
Ca2+ imaging.
The ratiometric, cell-permeable Ca2+ indicator Fura-2 (Molecular Probes) was used in all experiments. For podocytes in vitro, 2 μM Fura-2 was loaded for 20 minutes at 37°C in podocytes grown to 90% confluence on collagen-coated coverslips. Glomeruli isolated ex vivo were loaded with Fura-2 for 30 minutes at 37°C in poly-lysine–coated dishes. After Fura-2 incubation, cells/glomeruli were washed gently 3 times with PBS. Images were acquired in 2-second intervals using MetaMorph software on an Olympus IX71 inverted microscope. Cells were stimulated by switching of solutions with a 4-channel gravity-driven perfusion system. Images were analyzed using MetaMorph and Clampfit software.
Electrophysiology.
Patch-clamp electrophysiology (Axopatch 200B amplifier; Axon Instruments) was performed in the whole-cell configuration or on outside-out patches. Patch pipettes with resistances of 3–4 MΩ were pulled from borosilicate glass with a P-97 puller (Sutter Instruments) and filled with a solution containing 135 mM CH3SO3Cs, 10 mM CsCl, 3 mM MgATP, 0.2 mM NaGTP, 0.2 mM EGTA, 0.13 mM CaCl2, and 10 mM HEPES (pH 7.3) with CsOH. The bath solution contained 135 mM CH3SO3Na, 5 mM CsCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose (pH 7.4) with NaOH. Angiotensin II (500 nM), LPS (100 μg/ml), and ML204 (10 μM) were applied to the bath solution. Whole-cell currents were recorded from –100 mV to +100 mV voltage ramps over 400 ms and a holding potential of –60 mV. For single-channel recordings in the outside-out configuration, we used a voltage step protocol from –100 mV to +100 mV delivered at 20-mV intervals and a holding potential of 0 mV. Average pipette resistance filled with pipette solution was 3–5 MΩ. Data were sampled at 10 kHz and filtered at 5 kHz. Single-channel data were further off-line filtered at 500 Hz before analysis. In single-channel traces, currents were idealized using a manually defined amplitude criterion to assign ion channel opening and closing transitions. Ensemble averages were expressed as Po (average current divided by unitary current amplitude and number of channels per patch) and plotted as histograms. All data were acquired at room temperature and analyzed using pClamp 10 (Axon Instruments).
Statistics.
All data are presented as mean ± SEM. Standard ANOVA using Prism Software was employed for multiple-means comparisons. 2-tailed Student’s t test was used in all other cases. A P value less than 0.05 was considered significant.
Study approval.
All animal studies were approved by the MGH Subcommittee on Research Animal Care (SRAC).
Supplementary Material
Acknowledgments
ML204 was discovered as part of the MLPCN initiative and was supported by NIH/MLPCN grant U54 MH084659 (to C.W. Linsley). This work was supported by a fellowship from Boehringer Ingelheim Fonds (to T. Schaldecker), NIH grant DK093783 (to A. Weins), the Swedish Research Council and Swedish Governmental Agency for Innovation Systems (to L. Buvall), DFG Forschungsstipendium HA6334/1-1 (to S. Hakroush), an ASN Gottschalk Career Development Award (to A. Greka), a Harvard/MGH Shore Fellowship/Claflin Distinguished Scholar Award (to A. Greka), a BADERC/NIDDK P&F grant (to A. Greka, from NIH grant P30DK057521), and NIH grants DK083511 and DK093746 (to A. Greka).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(12):5298–5309. doi:10.1172/JCI71165.
References
- 1.Ren D. Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron. 2011;72(6):899–911. doi: 10.1016/j.neuron.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lishko PV, Kirichok Y, Ren D, Navarro B, Chung JJ, Clapham DE. The control of male fertility by spermatozoan ion channels. Annu Rev Physiol. 2012;74:453–475. doi: 10.1146/annurev-physiol-020911-153258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clapham DE. Calcium signaling. Cell. 2007;131(6):1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 5.Fribourg M, et al. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell. 2011;147(5):1011–1023. doi: 10.1016/j.cell.2011.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Logothetis DE, Petrou VI, Adney SK, Mahajan R. Channelopathies linked to plasma membrane phosphoinositides. Pflugers Arch. 2010;460(2):321–341. doi: 10.1007/s00424-010-0828-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88(2):451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 8.Farquhar MG. The glomerular basement membrane: not gone, just forgotten. J Clin Invest. 2006;116(8):2090–2093. doi: 10.1172/JCI29488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin J, et al. Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell. 2012;151(2):384–399. doi: 10.1016/j.cell.2012.08.037. [DOI] [PubMed] [Google Scholar]
- 10.Mundel P, Kriz W. Structure and function of podocytes: an update. Anat Embryol (Berl). 1995;192(5):385–397. doi: 10.1007/BF00240371. [DOI] [PubMed] [Google Scholar]
- 11.Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerjaschki D. Polycation-induced dislocation of slit diaphragms and formation of cell junctions in rat kindey glomeruli. The effects of low temperature, divalent cations, colchicine, and cytochalasin B. Lab Invest. 1978;39(5):430–440. [PubMed] [Google Scholar]
- 13.Seiler MW, Venkatachalam MA, Cotran RS. Glomerular epithelium: structural alterations induced by polycations. Science. 1975;189(4200):390–393. doi: 10.1126/science.1145209. [DOI] [PubMed] [Google Scholar]
- 14.Miner JH. The glomerular basement membrane. Exp Cell Res. 2012;318(9):973–978. doi: 10.1016/j.yexcr.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354(13):1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 16.Haraldsson B, Jeansson M. Glomerular filtration barrier. Curr Opin Nephrol Hypertens. 2009;18(4):331–335. doi: 10.1097/MNH.0b013e32832c9dba. [DOI] [PubMed] [Google Scholar]
- 17.Greka A, Mundel P. Balancing calcium signals through TRPC5 and TRPC6 in podocytes. J Am Soc Nephrol. 2011;22(11):1969–1980. doi: 10.1681/ASN.2011040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montell C. TRP channels in Drosophila photoreceptor cells. J Physiol. 2005;567(Pt 1):45–51. doi: 10.1113/jphysiol.2005.092551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 20.Riccio A, et al. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell. 2009;137(4):761–772. doi: 10.1016/j.cell.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu SZ, et al. TRPC channel activation by extracellular thioredoxin. Nature. 2008;451(7174):69–72. doi: 10.1038/nature06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian D, et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal. 2010;3(145):ra77. doi: 10.1126/scisignal.2001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faul C, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14(9):931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma H, et al. Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J Am Soc Nephrol. 2010;21(7):1145–1156. doi: 10.1681/ASN.2009090991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanagida-Asanuma E, et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol. 2007;171(2):415–427. doi: 10.2353/ajpath.2007.070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Togawa A, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIα. Oncogene. 1999;18(39):5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 27.Schwarz K, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108(11):1621–1629. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mundel P, Heid HW, Mundel TM, Kruger M, Reiser J, Kriz W. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139(1):193–204. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenzen J, et al. The role of osteopontin in the development of albuminuria. J Am Soc Nephrol. 2008;19(5):884–890. doi: 10.1681/ASN.2007040486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiser J, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113(10):1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gosling P, Czyz J, Nightingale P, Manji M. Microalbuminuria in the intensive care unit: Clinical correlates and association with outcomes in 431 patients. Crit Care Med. 2006;34(8):2158–2166. doi: 10.1097/01.CCM.0000228914.73550.BD. [DOI] [PubMed] [Google Scholar]
- 32.George B, et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J Clin Invest. 2012;122(2):674–692. doi: 10.1172/JCI60070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurihara H, Anderson JM, Kerjaschki D, Farquhar MG. The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am J Pathol. 1992;141(4):805–816. [PMC free article] [PubMed] [Google Scholar]
- 34.Seiler MW, Rennke HG, Venkatachalam MA, Cotran RS. Pathogenesis of polycation-induced alterations (“fusion”) of glomerular epithelium. Lab Invest. 1977;36(1):48–61. [PubMed] [Google Scholar]
- 35.Ilatovskaya DV, Levchenko V, Ryan RP, Cowley AW, Jr, Staruschenko A. NSAIDs acutely inhibit TRPC channels in freshly isolated rat glomeruli. Biochem Biophys Res Commun. 2011;408(2):242–247. doi: 10.1016/j.bbrc.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller M, et al. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J Biol Chem. 2011;286(38):33436–33446. doi: 10.1074/jbc.M111.274167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greka A, Mundel P. Calcium regulates podocyte actin dynamics. Semin Nephrol. 2012;32(4):319–326. doi: 10.1016/j.semnephrol.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riedl J, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5(7):605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6(8):709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 40.Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6(8):837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- 41.Nitschke R, et al. Acetylcholine increases the free intracellular calcium concentration in podocytes in intact rat glomeruli via muscarinic M(5) receptors. J Am Soc Nephrol. 2001;12(4):678–687. doi: 10.1681/ASN.V124678. [DOI] [PubMed] [Google Scholar]
- 42.Jiang LH, Gamper N, Beech DJ. Properties and therapeutic potential of transient receptor potential channels with putative roles in adversity: focus on TRPC5, TRPM2, and TRPA1. Curr Drug Targets. 2011;12(5):724–736. doi: 10.2174/138945011795378568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shibata S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14(12):1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 44.Tauseef M, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209(11):1953–1968. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jo HY, et al. Kir3.1 channel is functionally involved in TLR4-mediated signaling. Biochem Biophys Res Commun. 2011;407(4):687–691. doi: 10.1016/j.bbrc.2011.03.076. [DOI] [PubMed] [Google Scholar]
- 46.Yamashiro K, et al. Role of transient receptor potential vanilloid 2 in LPS-induced cytokine production in macrophages. Biochem Biophys Res Commun. 2010;398(2):284–289. doi: 10.1016/j.bbrc.2010.06.082. [DOI] [PubMed] [Google Scholar]
- 47.Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282(11):7833–7843. doi: 10.1074/jbc.M608288200. [DOI] [PubMed] [Google Scholar]
- 48.Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell. 2008;19(8):3203–3211. doi: 10.1091/mbc.E07-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vassiliadis J, Bracken C, Matthews D, O’Brien S, Schiavi S, Wawersik S. Calcium mediates glomerular filtration through calcineurin and mTORC2/Akt signaling. J Am Soc Nephrol. 2011;22(8):1453–1461. doi: 10.1681/ASN.2010080878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8(5):485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 51.Winn MP, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308(5729):1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 52.Krall P, et al. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS One. 2010;5(9):e12859. doi: 10.1371/journal.pone.0012859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gordon MS, Fischer S, Tarifeno E. Aspects of the physiology of terrestrial life in amphibious fishes. II. The Chilean clingfish, Sicyases sanguineus. J Exp Biol. 1970;53(3):559–572. doi: 10.1242/jeb.53.3.559. [DOI] [PubMed] [Google Scholar]
- 54.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world — a growing challenge. N Engl J Med. 2007;356(3):213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 55.Brensing KA, Raab P, Frotscher U. Diabetic nephropathy. N Engl J Med. 2002;347(12):947–948. doi: 10.1056/NEJM200209193471218. [DOI] [PubMed] [Google Scholar]
- 56.Schwartz EJ, et al. Human immunodeficiency virus-1 induces loss of contact inhibition in podocytes. J Am Soc Nephrol. 2001;12(8):1677–1684. doi: 10.1681/ASN.V1281677. [DOI] [PubMed] [Google Scholar]
- 57.Takemoto M, et al. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 2002;161(3):799–805. doi: 10.1016/S0002-9440(10)64239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asanuma K, et al. Synaptopodin regulates the actin-bundling activity of α-actinin in an isoform-specific manner. J Clin Invest. 2005;115(5):1188–1198. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.