

Abstract
Circadian rhythms govern nearly every physiological process in our brains and bodies. At the most basic level, the molecular clockwork in each cell interacts with metabolic cycles to influence the redox state, allowing for increased cellular activity at specific times of day. In this issue of the JCI, Musiek et al. show that genetic disruptions in the positive arm of the molecular clock can lead to severe astrogliosis, which likely occurs through disruptions in output genes that keep oxidative stress in check. This study demonstrates the importance of proper circadian protein function in the maintenance of neuronal integrity.
It has become apparent in recent years that disruptions to the circadian system can lead to serious medical problems. For example, shift workers have high rates of metabolic syndrome, obesity, sleep disorders, cardiovascular disease, depression, and cancer (1). In addition, circadian rhythms deteriorate with age, and this deterioration likely contributes to age-related neurological disorders such as Alzheimer and Parkinson diseases (2). Neuronal damage and degradation associated with aging are often the result of abnormal levels of reactive oxygen species (ROS) generation (3). Recent studies indicate that basic cellular metabolism and ROS equilibrium are regulated by a 24-hour rhythm in the peroxiredoxin cycle (4). The cyclic accumulation and expulsion of ROS indirectly feeds into the core circadian transcriptional feedback loop in the nucleus. ROS modulates the ratio of NAD(P)H to NAD(P)+, which binds to and activates the histone deacetylase sirtuin 1 (SIRT1) (5). SIRT1 then binds the CLOCK (or NPAS2)/BMAL1 transcription complex, which alters the expression of circadian rhythm–associated genes. In turn, this circadian transcriptional feedback loop regulates the rhythmic expression of genes involved in redox pathways, allowing for bidirectional control of the cellular metabolic state (Figure 1). It has been hypothesized that desynchrony between the core circadian transcriptional cycle and the peroxiredoxin cycle results in oxidative stress and DNA damage, which potentially leads to neuronal death. The extent to which the proteins that comprise the transcriptional circadian loop impact neuronal survival has not yet been determined.
Figure 1. Crosstalk between the transcriptional and peroxiredoxin cycles.
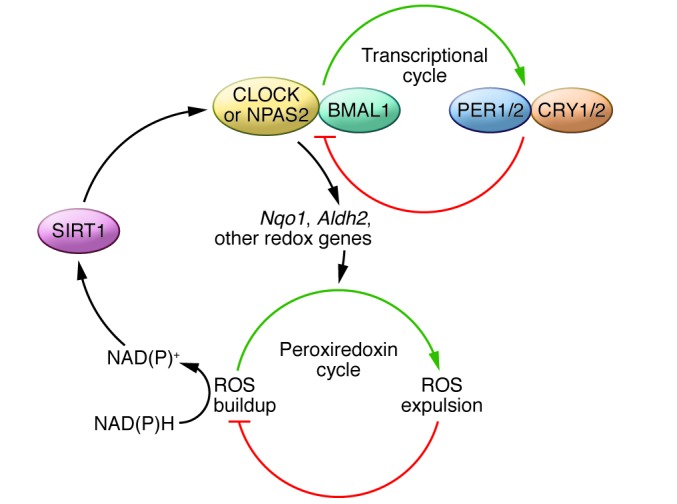
CLOCK or NPAS2 (yellow) and BMAL1 (aqua), which make up the positive limb of the circadian clock, control the expression of negative limb proteins Period (PER) (blue) and cryptochrome (CRY) (orange), which feed back to inhibit the activity of the positive limb. CLOCK(NPAS2)/BMAL1 controls the expression of several output genes (such as Nqo1 and Aldh2) that influence the rhythm in ROS accumulation in the peroxiredoxin cycle. The ratio of NAD(P) to NAD(P)H as an output of this cycle influences SIRT1 (purple) activity (which is NAD+ dependent). When bound to NAD+, SIRT1 regulates the activity of CLOCK (NPAS2):BMAL1 heterodimers.
What happens to the brain when you delete BMAL1?
In this issue of the JCI, Musiek and colleagues (6) investigated the role of core circadian clock function in redox homeostasis regulation in the brain. They hypothesized that genetic disruptions in circadian function might facilitate neuronal injury and neurodegeneration. Mice with a targeted mutation in the circadian clock gene Bmal1 (Bmal1–/–) (7) have outward signs of premature aging including weight loss, organ shrinkage, cataracts, and sarcopenia, as well as increased levels of ROS in most tissues (8). The effects of Bmal1 mutation on the structural integrity of the brain or markers of neurodegeneration have not been evaluated. The authors found that the brains of Bmal1–/– mice appeared structurally normal at 6 months of age; however, staining for glial fibrillary acidic protein (GFAP), which is a general marker of brain injury, revealed intense astrocyte activation. This activation was most severe in cortical regions of the brain and the hippocampus, though less so in the thalamus and brainstem. This astrocytosis was not present at 2 weeks of age, but was evident at 2.5 months and had progressed at 6 months. Furthermore, astrocytosis was associated with increased expression of proinflammatory cytokines and loss of discrete presynaptic axonal terminals. Using mass spectrometry, the authors also found that cortical F4-neuroprostanes (markers of lipid peroxidation) were increased in Bmal1–/– mice, further indicating neuronal damage. Optical intrinsic signal functional connectivity imaging (fcOIS) was used to visualize alterations in regional cortical blood flow in anesthetized mice and to generate maps of resting-state neuronal connectivity. The Bmal1–/– mice had reduced functional connectivity throughout the cortex, indicating that the observed neuronal damage leads to functional changes in overall brain communication. It is interesting that the degree of astrogliosis and oxidative stress were not perfectly correlated, indicating that certain brain regions are more susceptible to neuronal injury than others when faced with the loss of Bmal1 expression.
What causes the damage associated with lack of Bmal1?
After the initial assessment of Bmal1–/– mice, the authors had a few key questions that needed answers. First, Bmal1 is expressed throughout the brain and body and is involved in regulating a number of periphery-associated metabolic processes that might impact brain function (9). Is Bmal1 expression in the brain or in the periphery important for maintaining neuronal integrity? Second, is the observed neuronal damage a result of disrupted sleep-wake rhythms (which were prominent in the Bmal1–/– mice), or a consequence of local Bmal1 reduction that is independent of central rhythm changes? To answer these questions, Musiek et al. generated mice with brain-specific deletion of Bmal1 by crossing a NestinCre line with a Bmal1flox/flox line (6). Importantly, these mice retained sufficient Bmal1 expression in the core pacemaker of the suprachiasmatic nucleus (SCN) to produce relatively normal circadian rhythms associated with locomotor activity and sleep (10); however, circadian gene expression in the cortex of these mice was severely disrupted. Despite having normal sleep-wake rhythms and normal peripheral oscillators, the NestinCre+;Bmal1f/f mice exhibited age-dependent astrogliosis in cortical regions of the brain and the hippocampus. This neuronal damage might be relevant to cognition and behavior, since the NestinCre+;Bmal1f/f mice had an abnormal response to a change in environment. These results indicate that a reduction in local Bmal1 expression, rather than peripheral expression or disruptions in sleep-wake rhythms, is the key contributing factor to the neuronal survival outcomes that are indicative of premature aging. Future studies that examine more complex measures of cognitive function will be necessary to determine the extent of cognitive decline as a result of Bmal1 mutation. It will also be important to develop more brain region–specific Cre-expressing mouse lines or viral approaches to ascertain whether the neuronal damage in the cortex, for example, is truly a direct result of local Bmal1 knockdown or the consequence of a disruption in Bmal1-driven rhythms in another brain circuit.
What mechanisms lead to neuronal damage?
The authors identified a number of genes involved in redox sensing and response, which according to existing microarray databases, have a diurnal rhythm in their expression. Examination of gene expression in the cortex of Bmal1–/– mice and the NestinCre+;Bmal1f/f mice revealed that the expression of NAD(P)H dehydrogenase, quinone 1 (Nqo1), and aldehyde dehydrogenase 2 (Aldh2) was drastically reduced. ALDH2 has been shown to protect neurons from oxidative stress by scavenging reactive aldehydes produced during mitochondrial respiration (11, 12). NQO1 reduces toxic quinones to protect against oxidative damage (13). The authors demonstrated that BMAL1 binds the promoters of both of these genes, indicating that they are direct transcriptional targets. Since both are involved in neuroprotection and the oxidative stress response, it is reasonable to hypothesize that decreased expression of genes associated with Bmal1 knockdown directly promotes neuronal damage; however, this needs to be tested directly in future experiments.
Does disruption of other circadian genes induce neuropathology?
To determine whether the neuropathological effects associated with loss of Bmal1 are common to mice with mutations in other core molecular clock components, the authors examined mice lacking CLOCK and NPAS2. Previous studies indicate that these two proteins have redundant functions in the control of locomotor rhythms in the SCN (14). Clock and Npas2 single-knockout mice had very little astroglial activation; however, the Clock/Npas2 double-knockout mice recapitulated the Bmal1–/– phenotype. Surprisingly, mice with mutations in both negative regulators of the circadian clock, Per1 and Per2, had no evidence of age-dependent astrogliosis. In fact, deletion of Per1 and Per2 enhanced the expression of BMAL1 target genes including Nqo1 and Aldh2, likely due to the removal of breaks in BMAL1 activity. These data further solidify the idea that it is not a disruption in circadian rhythms per se that leads to neuronal injury, since Per1/Per2 double-mutant mice also exhibit disrupted behavioral rhythms, similar to those observed in Bmal1–/– mice (15). This study indicates that local CLOCK(NPAS2):BMAL1 heterodimers are involved in the direct regulation of genes important for the production of ROS, which is tightly controlled by the circadian cycle.
Is the neuronal damage due to loss of BMAL1 in neurons or glia?
Using a cell culture model, the authors found that knockdown of Bmal1 with lentiviral-delivered shRNA in primary neuron–enriched cultures led to spontaneous neurite degeneration and cell death, which was exacerbated with low-dose H2O2. In contrast, siRNA-mediated knockdown of Bmal1 in primary astrocyte cultures had no impact on cell viability or inflammatory markers, as seen in the Bmal1–/– mice. These data suggest that Bmal1 expression in neurons, and not astrocytes, is important for neuronal viability. Finally, to address the question of whether gene dosage of Bmal1 might be important in oxidative neurodegeneration, they treated heterozygous Bmal1+/– mice, which had no baseline pathology, with the mitochondrial complex III inhibitor 3-nitroproprionic acid (3-NP), a substance that produces oxidative injury and striatal lesions. They found that the Bmal1+/– mice had larger lesions than the wild-type mice, suggesting that a 50% reduction in Bmal1 expression could make individuals more vulnerable to neuronal damage in response to insult.
Conclusions
The results of this study demonstrate that genetic mutations in the positive arm of the circadian system are deleterious to the integrity of neurons. This loss of neuronal integrity may be directly responsible for premature aging and neurodegeneration. These data also suggest a potential mechanism whereby specific circadian genes impact neuronal survival. Since we know that levels of BMAL1, CLOCK, and NPAS2 are tightly linked to light exposure and sleep deprivation, chronic circadian disruptions through activities such as shift work, travel, nighttime light exposure, or other environmental factors could have serious consequences for individuals predisposed to neurological disorders. Furthermore, these data indicate that those currently suffering from neurological disorders would certainly benefit from a stable sleep-wake and light-dark schedule. It will be interesting for future studies to determine whether any of these neuropathologies can be prevented through direct, local rescue of BMAL1 function or through manipulation of important clock target genes. Such studies might provide an avenue toward future therapeutic developments.
Acknowledgments
I would like to thank Ryan Logan and Trey Williams for helpful comments on this article.
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(12):4994–4996. doi:10.1172/JCI73059.
See the related article beginning on page 5389.
References
- 1.Herichova I. Changes of physiological functions induced by shift work. Endocr Regul. 2013;47(3):159–170. doi: 10.4149/endo_2013_03_159. [DOI] [PubMed] [Google Scholar]
- 2.Kondratova AA, Kondratov RV. The circadian clock and pathology of the ageing brain. Nat Rev Neurosci. 2012;13(5):325–335. doi: 10.1038/nrn3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lizama-Manibusan B, McLaughlin B. Redox modification of proteins as essential mediators of CNS autophagy and mitophagy. FEBS Lett. 2013;587(15):2291–2298. doi: 10.1016/j.febslet.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bass J, Takahashi JS. Circadian rhythms: redox redux. Nature. 2011;469(7331):476–478. doi: 10.1038/469476a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellet MM, Orozco-Solis R, Sahar S, Eckel-Mahan K, Sassone-Corsi P. The time of metabolism: NAD+, SIRT1, and the circadian clock. . Cold Spring Harb Symp Quant Biol. 2011;76:31–38. doi: 10.1101/sqb.2011.76.010520. [DOI] [PubMed] [Google Scholar]
- 6.Musiek ES, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest. 2013;123(12):5389–5400. doi: 10.1172/JCI70317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunger MK, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103(7):1009–1017. doi: 10.1016/S0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006;20(14):1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013;(217):3–27. doi: 10.1007/978-3-642-25950-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mieda M, Sakurai T. Bmal1 in the nervous system is essential for normal adaptation of circadian locomotor activity and food intake to periodic feeding. J Neurosci. 2011;31(43):15391–15396. doi: 10.1523/JNEUROSCI.2801-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohsawa I, Nishimaki K, Murakami Y, Suzuki Y, Ishikawa M, Ohta S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J Neurosci. 2008;28(24):6239–6249. doi: 10.1523/JNEUROSCI.4956-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzmaurice AG, et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci U S A. 2013;110(2):636–641. doi: 10.1073/pnas.1220399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010;501(1):116–123. doi: 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci. 2007;10(5):543–545. doi: 10.1038/nn1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR. Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron. 2001;30(2):525–536. doi: 10.1016/S0896-6273(01)00302-6. [DOI] [PubMed] [Google Scholar]