

Abstract
Thermodynamic parameters were determined for complex formation between the Grb2 SH2 domain and tripeptides of the general form Ac-pTyr-Xaa-Asn in which the Xaa residue bears a linear alkyl chain varying in length from 1–5 carbon atoms. Binding affinity increases upon adding a methylene group to the Ala derivative, but further chain extension gives no extra enhancement in potency. The thermodynamic signatures of the ethyl and n-propyl derivatives are virtually identical as are those for the n-butyl and n-pentyl analogues. Crystallographic analysis of the complexes reveals a high degree of similarity in the structure of the domain and the bound ligands with the notable exception that there is a gauche interaction in the side chains in the bound conformations of ligands having n-propyl, n-butyl, and n-pentyl groups. However, eliminating this unfavorable interaction by introducing a Z-double bond into the side chain of the n-propyl analogue does not result in an increase in affinity. Increases in the amount of nonpolar surface that is buried upon ligand binding correlate with favorable changes in ΔH°, but these are usually offset by corresponding unfavorable changes in −TΔS°; there is little correlation of ΔCp with changes in the amount of buried nonpolar surface.
Keywords: protein-ligand interactions, binding energetics, isothermal titration calorimetry, nonpolar surface area
Predicting how altering the structure of a small molecule affects its affinity for a target protein is a critical goal in molecular recognition in biological systems and during the lead optimization phase of drug discovery.1−6 However, estimating even relative protein binding affinities is a significant challenge because there is a lack of experimental information detailing how the enthalpic, ΔH°, and entropic, ΔS°, components of protein binding free energies, ΔG°, are affected by incrementally varying structures of ligands in well-characterized biological systems.7−17 Even when such data are available, correlating variations in thermodynamic parameters with differences in structural features in the respective complexes is problematic, in part because of enthalpy/entropy compensation18−20 and the frequent lack of a correlation between ΔG° and either ΔH° or −TΔS°.21
Toward enhancing the knowledge base that will lead to a better understanding of energetics and structure in protein–ligand interactions, we have conducted a number of systematic investigations in which binding free energies, enthalpies, and entropies were determined and correlated with structural data for proteins complexed with congeneric small molecules.22−28 Studies of how changes in the structures of peptides derived from Ac-pTyr-Val-Asn-NH2 affect binding thermodynamics in their associations with the SH2 domain of growth receptor binding protein 2 (Grb2)29 have led to some unexpected discoveries. For example, we found that introducing a cyclic constraint to stabilize the bound conformation of small molecules in solution does not necessarily result in relatively more favorable binding entropies, even though potencies may increase.24−27 This finding is contrary to commonly held beliefs regarding the putative effects of ligand preorganization.30 In another study, we determined binding energetics for complexes of the Grb2 SH2 domain with tripeptides Ac-pTyr-Xaa-Asn-NH2 in which the Xaa residues were α,α-cycloaliphatic amino acids having different ring sizes and found that adding methylene groups enhanced binding affinity because increasingly favorable binding enthalpies dominated increasingly less favorable binding entropies.28 Although this discovery is not unprecedented in protein–ligand associations,9,31−33 it is not in accord with the entropy driven hydrophobic effects that might be expected to accompany adding nonpolar surface area to small molecules.34−36
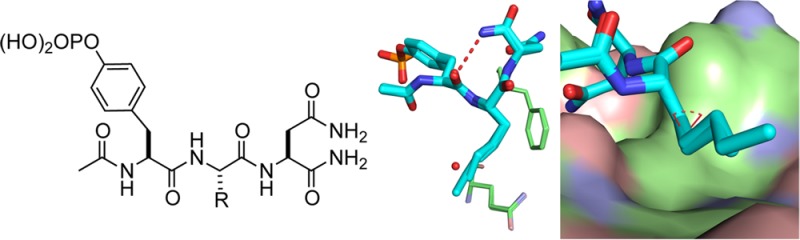
In order to further explore this enthalpy-driven hydrophobic effect, we prepared compounds 1–5, which are analogues of Ac-pTyr-Xaa-Asn-NH2 in which the Xaa residues bear n-alkyl groups. We then determined their binding parameters (Ka, ΔG°, ΔH°, ΔS°, and ΔCp) for the Grb2 SH2 domain using isothermal titration calorimetry (ITC) and the structures of their respective complexes with the domain by X-ray crystallography.
The results of the ITC studies (Table 1) reveal that 2 binds with a free energy change that is 0.8 kcal·mol–1 greater than 1. The enhanced potency of 2 relative to 1 arises because a more favorable binding enthalpy (ΔΔH° = −1.9 kcal·mol–1) for 2 dominates a relatively less favorable binding entropy (−TΔΔS° = +1.1 kcal·mol–1). However, subsequent addition of methylene groups to give 3–5 provides no further increase in affinity for the Grb2 SH2 domain. The thermodynamic signatures of 2 and 3 are virtually identical as are those for 4 and 5. Although the observed ΔH° for binding of 4 and 5 to the domain are each about 0.5 kcal·mol–1 more favorable than those of 2 and 3, this enthalpic advantage is completely offset by binding entropy changes for 4 and 5 that are 0.5 kcal·mol–1 less favorable than for 2 and 3. The observed binding affinities for 2–5 are thus identical within experimental error. The heat capacity changes, ΔCp, for the associations of 1–5 with the Grb2 SH2 domain are negative as expected for the burial of nonpolar surfaces upon complex formation,34,37−39 but there is no meaningful relationship between the value of ΔCp and the number of methylene groups in the alkyl chain.
Table 1. Thermodynamic Data and Summary of Polar and Nonpolar Contacts for Complexes of 1–6 and the Grb2 SH2 Domaina,b.
| total polar contactsc |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| ligand | Ka (× 105 M–1) | ΔG° (kcal·mol–1) | ΔH° (kcal·mol–1) | –TΔS° (kcal·mol–1) | ΔCp (cal·mol–1K–1) | direct | water-mediated | pTyr+1 vdW contactsd | ΔCSAnp (Å2) |
| 1 | 2.2 ± 0.1 | –7.3 ± 0.1 | –4.9 ± 0.3 | –2.4 ± 0.1 | –123 ± 9 | 14 | 5 | 4 | 180 |
| 2 | 8.6 ± 0.2 | –8.1 ± 0.1 | –6.8 ± 0.5 | –1.3 ± 0.1 | –170 ± 15 | 14 | 5 | 9f | 189 |
| 3 | 7.6 ± 1.0 | –8.0 ± 0.1 | –6.7 ± 0.5 | –1.3 ± 0.3 | –173 ± 13 | 14 | 4e | 12 | 202 |
| 4 | 8.4 ± 0.6 | –8.1 ± 0.1 | –7.3 ± 0.3 | –0.8 ± 0.2 | –138 ± 12 | 14 | 3 | 14 | 220 |
| 5 | 7.8 ± 0.6 | –8.0 ± 0.1 | –7.2 ± 0.3 | –0.8 ± 0.2 | –148 ± 7 | 13 | 4e | 14 | 217 |
| 6 | 6.1 ± 0.2 | –7.9 ± 0.1 | –7.0 ± 0.2 | –0.9 ± 0.1 | –176 ± 7 | 13 | 2e | 9f | 204 |
ITC experiments were conducted at 25 °C in HEPES (50 mM) with NaCl (150 mM) at pH 7.45 ± 0.05 as previously reported.24
Three or more experiments were performed for each ligand, and the averages are reported following normalization of the n values for each experiment by adjusting ligand concentration (See Supporting Information). Errors in the thermodynamic values were determined by the method of Krishnamurthy.10
Total direct and single water-mediated hydrogen bonding contacts between protein and ligand for which non-hydrogen donor–acceptor distances are within the range 2.5–3.5 Å.
A vdW contact exists when the interatomic distance between a methylene or a methyl group in the pTyr+1 residue and a carbon, nitrogen, or oxygen atom belonging to Gln106, His107, and Phe108 of the domain is in the range of 3.4–4.2 Å.
A water-mediated contact is not counted because the non-hydrogen donor–acceptor distance is 3.6 or 3.7 Å, whereas this distance is 3.5 Å for corresponding contacts in other complexes.
An interaction with a contact distance of 4.27 Å is not counted as a vdW contact.
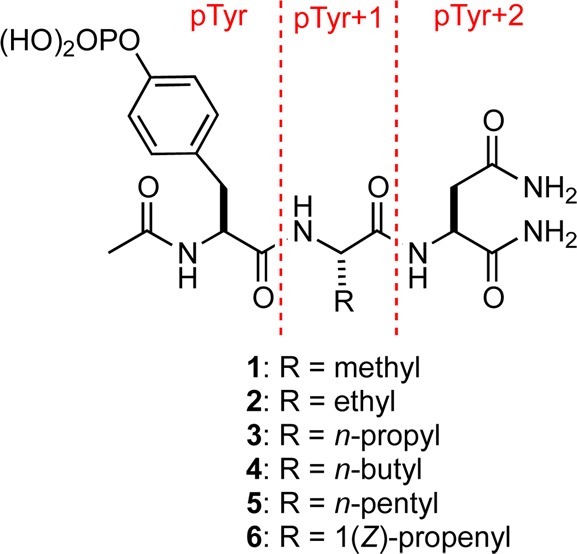
The structures of 1–5 bound to the Grb2 SH2 domain were determined by X-ray crystallography to resolutions of 1.6–1.8 Å. Pairwise alignment of the backbone atoms belonging to the domain in the complex of 1 with the complexes of 2–5 yielded average root-mean-square deviations (rmsds) of <0.2 Å (Figure 1a). Similarly, the rmsds for all non-hydrogen, equivalent atoms belonging to 1–5 and to the domain residues, that make van der Waals (vdW) contacts with the pY + 1 residue of the bound ligands are <0.2 Å (Figure 1b). Because these values are less than the average atomic coordinate error associated with the molecular models,40 these differences are not considered significant. Thus, with the obvious exception of the side-chain atoms in the pTyr+1 replacement, the complexes are virtually identical. Each ligand adopts a β-turn conformation in which the carbonyl oxygen atom of the pTyr residue forms an intramolecular hydrogen bond with the NH2 group of the C-terminal amide (Figure 1b, dashed, red line). It is noteworthy that there is a gauche interaction about the Cβ–Cγ bond in the n-alkyl side chains in the pTyr+1 residue of the bound forms of 3–5 (Figure 1d, dashed, red arc). This energetically unfavorable conformation is presumably adopted in order to avoid the close contacts with the side-chains of Gln106 and Phe108 that would be required if the side chains were in extended conformations.
Figure 1.
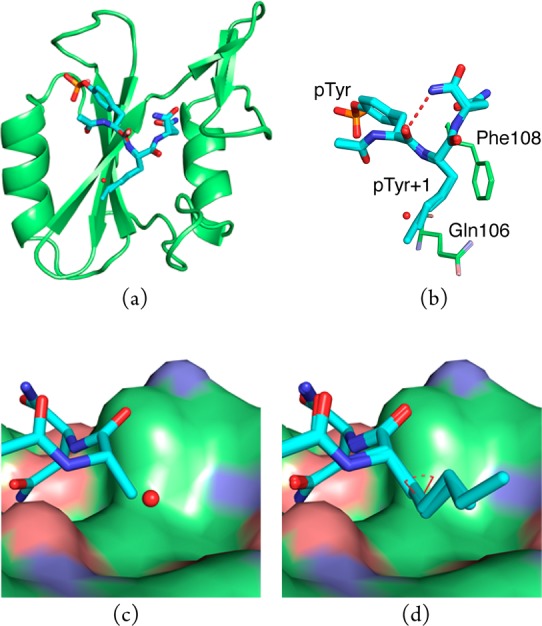
(a) Overlay of complexes of the Grb2 SH2 domain with 1–5. Oxygen, nitrogen, and phosphorus atoms are colored red, blue, and orange, respectively. Carbon atoms belonging to the ligands (sticks) are colored cyan, while those belonging to the domain (cartoon) are colored green. (b) Overlay of 1–5 (thick sticks) showing side chains of domain residues Gln106 and Phe108 (thin sticks) involved in vdW contacts. Water molecule (red sphere) is present only in the complex of 1. (c) Compounds 1 and (d) 3–5 bound to the Grb2 SH2 domain (vdW surface).
Having thermodynamic and structural data for a series of related complexes offers an opportunity to correlate structure and energetics. Toward this end, we analyzed the nonbonded, polar contacts within 2.5–3.5 Å between non-hydrogen donor and acceptor atoms in the complexes of 1–5. The number of direct polar contacts is the same in all of these complexes with the exception of 5, for which there is one less such contact (Table 1). This difference arises because the distance between one of the phosphate oxygen atoms of the ligand and a side-chain nitrogen atom of Arg86 is 3.6 Å in the complex of 5 compared with 3.3–3.5 Å in the other complexes (Figures S5–S9, Supporting Information). These variations in contact distances are within coordinate error, so there is arguably no meaningful difference in the total number of direct polar contacts in the complexes of 1–5.
There are some minor differences in the number of water-mediated contacts in the complexes of 1–5. These dissimilarities result from slight variations in contact distances that are within experimental error and from differences in the number of ordered, interfacial water molecules. Namely, a water-mediated contact between the pTyr+3 amide nitrogen atoms of 3 and 5 and the side-chain of Lys109 in their respective complexes is 3.7 Å, whereas in other complexes the corresponding distance is 3.5 Å (Figures S5–S9, Supporting Information). Relative to the other complexes, there are two fewer ordered water molecules in the complex of 4. There is also a water molecule in the complex of 1 that is located 3.9 Å from the Cβ carbon atom of the pTyr+1 residue, but this water molecule is not apparent in complexes of 2–5 (Figure 1b,c). Although this water molecule does not mediate any protein–ligand contact, its presence in the binding pocket will incur an entropic penalty.41 However, such a penalty is not reflected in the observed relative binding entropy for 1, which is more favorable than for 2–5, suggesting that the specific environment of bound water molecules is important. Because only ordered water molecules are visible by crystallography and some water molecules are not observed in all complexes, correlating the number of water-mediated contacts and interfacial water molecules in the complexes with binding thermodynamics can be problematic.
We inventoried the vdW contacts in the complexes of 1–5 with the Grb2 SH2 domain using a distance criterion of 3.4–4.2 Å for nonbonded atoms (Table 1).42 The number of such contacts between the domain and the pTyr and pTyr+2 residues of 1–5 are identical. However, there are significant differences in the number of vdW contacts between the domain and the alkyl side chains of the pTyr+1 residues of 1–5. Specifically, the total number of vdW contacts increases from four in the complex with 1 to 14 in each of the complexes with 4 and 5. Consistent with the more favorable ΔH° that is observed for 2 relative to 1, the greatest increase in vdW contacts is observed upon adding the first methylene group to give 2, the terminal methyl group of which interacts with a shallow pocket in the domain.
In order to determine whether there was any correlation between thermodynamic binding parameters and buried nonpolar Connolly surface area (CSAnp),43 values for ΔCSAnp were determined using the structures of the complexes 1–5 as previously described.28 The value of ΔCSAnp for each complex correlates positively with ΔH° (R2 = 0.74) and negatively with −TΔS° (R2 = 0.87). It is notable that increases in the amount of nonpolar surface that is buried upon ligand binding thus correlate with favorable changes in ΔH° but less favorable changes in –TΔS°. This finding is inconsistent with conventional wisdom regarding the thermodynamic effects of burying nonpolar surface. Moreover, there is little correlation of ΔCSAnp with ΔCp (R2 = 0.25), suggesting that ΔCp may not be a reliable indicator for the amount of nonpolar surface excluded from solvent upon complex formation.37
The preferred conformations of the n-alkyl side chains of 3–5 in solution are presumably extended, so the gauche interaction found in their bound forms might be anticipated to be accompanied by an enthalpic penalty of up to about 0.9 kcal·mol–1. Modeling suggested that the effects of A1,3-strain would preferentially orient the 1-(Z)-propenyl side chain in 6 in a conformation that corresponds with the n-propyl group in the bound structure of 3. The olefinic side chain would thus be expected to attenuate the adverse enthalpic consequences associated with conformational differences in the pTyr+1 residue in the free and bound ligands. Moreover, preorganization of the side chain might be expected to have a favorable entropic consequence.44
Accordingly, we prepared 6, and although we discovered that it does bind to the domain with a slightly more favorable enthalpy than 3, an unexpected entropic penalty offsets this advantage, and 3 and 6 are equipotent. Toward correlating structure and energetics for interactions of 3 and 6 with the Grb2 SH2 domain, the structure of the complex of 6 with the Grb2 SH2 domain was obtained at 1.6 Å resolution. The domain backbone atoms in the complexes of 3 and 6 align with an average rmsd of <0.2 Å, and although the three carbon atoms in the respective side chains of 3 and 6 align with an rmsd of 0.48 Å, all other atoms in 3 and 6 align with an average rmsd of <0.2 Å.
On the basis of the distance criterion used for direct polar contacts, there is one fewer such contact in the complex of 6 than 3 (Table 1) because the distance between a side-chain nitrogen atom of Arg86 and one of the phosphate oxygen atoms of 6 is 3.6 and 3.4 Å in the complex of 3 (cf. Figures S7 and S10, Supporting Information). Inasmuch as this difference is within coordinate error, the number of direct polar contacts in the complexes of 3 and 6 are arguably the same. However, there are significant dissimilarities in the water networks in these complexes because two ordered water molecules present in the complex of 3 are absent in the complex of 6 (Figure 2a,b). In the complex of 3, these water molecules mediate interactions between the side chain of Lys109 of the domain and the pTyr+3 amide moiety of 3. Within experimental error, the ΔH° and −TΔS° for binding of these two ligands are closely similar, thereby reflecting the difficulties associated with correlating differences in water networks with observed binding energetics.45 There are fewer vdW contacts between the pTyr+1 residue in 6 than in 3 because the propenyl side chain of 6 is displaced slightly away from the side chain of the Gln106 residue of the domain (Figure 2c).
Figure 2.
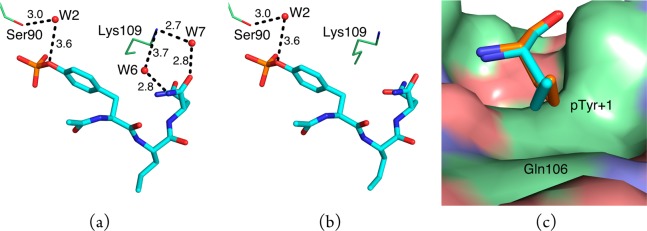
Selected water networks and vdW contacts in the complexes of 3 and 6. Oxygen, nitrogen, and phosphorus atoms are colored red, blue, and orange, respectively, and carbon atoms are colored cyan, green, or orange. Some water-mediated contact distances exceed the 3.5 Å cutoff used to define polar contacts, but the differences in corresponding distances are within coordinate error. (a) Compound 3 (cyan sticks) makes water-mediated contacts (black dashed lines) to the side-chains of residues Ser90 and Lys109 of the domain (green lines) via conserved water molecules W2, W6, and W7 (red spheres). (b) Compound 6 (cyan sticks) makes a water-mediated contact (black dashed lines) to the side-chain of Ser90 (green lines). (c) The propenyl side-chain of the pTyr+1 residue of 6 (orange sticks) is turned slightly away from the side-chain of Gln106 of the domain (green surface) relative to the propyl side-chain of the pTyr+1 residue of 3 (cyan sticks).
In summary, the binding affinities of the congeneric series of phosphotyrosine-derived ligands 1–6, which differ incrementally in the nature of their side chains at the pTyr+1 site, for the Grb2 SH2 domain were determined, and the structure of each complex was solved by X-ray crystallography. Although some differences in thermodynamic parameters and polar and vdW contact distances are nearly identical within experimental error, some correlations between structure and energetics are possible. Although 2 binds with a free energy change that is 0.8 kcal·mol–1 greater than 1, further increases in chain length are not accompanied by any significant changes in binding affinities because any favorable change in ΔH° is offset by a correspondingly less favorable change in −TΔS°. This finding may be contrasted with our previous study in which we observed that adding methylene groups to cyclic amino acid pTyr+1 replacements resulted in increasingly more favorable binding free energies and enthalpies.28 Even though 3 makes more vdW contacts with the domain than 2, the thermodynamic signatures of 2 and its homologue 3 are indistinguishable. Toward assessing whether a gauche interaction in the side chain of the bound form of 3 might mitigate the benefit of these additional vdW contacts, a Z-double bond was introduced into the side chain leading to 6, which was equipotent with 3. Analysis of the structure of the complex of 6 with the domain suggests that the enthalpic advantage expected from removing the gauche interaction may have been attenuated because the side chain of 6 makes fewer vdW contacts with the domain than does that of 3. Moreover, there are two fewer interfacial water molecules mediating polar contacts in the complex of 6. The binding energetics of 4 and 5 are identical, and the fact that their binding enthalpies are more favorable than for 3 is consistent with the observation that the side chains of 4 and 5 each make more vdW contacts with the domain than 3. The less favorable entropy of binding for these homologues relative to 3 may arise from the entropic cost associated with binding small molecules having the greater number of rotatable bonds that accompany the addition of methylene groups.46 Like some of our previous work,28 these studies show that changes in ΔCp may not correlate well with differences in buried nonpolar surface area for a series of related protein–ligand complexes. Studies directed toward resolving some of the questions raised by these investigations are in progress, and the results will be reported in due course.
Acknowledgments
We are thankful to Dr. Stuart Black (Schering-Plough Research Institute) for the Grb2 SH2 expression vector. We also thank Ms. Andrea Beckham for experimental and technical assistance. Instrumentation and technical assistance for this work were provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at the University of Texas at Austin.
Supporting Information Available
Experimental procedures, spectral data, and copies of 1H and 13C NMR spectra for 1–6, complete diffraction data and refinement statistics, density difference maps and omit maps of the ligands in the complexes, protein–ligand polar contact diagrams, vdW surface representations, and tables listing the number of direct and water-mediated protein–ligand contacts, vdW contacts, buried total and nonpolar surface area, and average atomic B-factors. This material is available free of charge via the Internet at http://pubs.acs.org.
We thank the National Institutes of Health (GM 84965), National Science Foundation (CHE 0750329), Robert A. Welch Foundation (F-0652), Norman Hackerman Advanced Research Program, and Texas Institute for Drug and Diagnostic Development through the Welch Foundation Grant #H-F-0032 for support of this research. J.E.D. thanks the ACS Division of Medicinal Chemistry, Pfizer, Inc., and the Dorothy B. Banks Foundation for predoctoral fellowships.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gohlke H.; Klebe G. Approaches to the Description and Prediction of the Binding Affinity of Small-Molecule Ligands to Macromolecular Receptors. Angew. Chem., Int. Ed. 2002, 41, 2644–2676. [DOI] [PubMed] [Google Scholar]
- Mobley D. L.; Dill K. A. Binding of Small Molecule Ligands to Proteins: “What You See” is Not Always “What You Get. Structure 2009, 17, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire E. A. A Thermodynamic Approach to the Affinity Optimization of Drug Candidates. Chem. Biol. Drug Des. 2009, 74, 468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-X.; Gilson M. K. Theory of Free Energy and Entropy in Noncovalent Binding. Chem. Rev. 2009, 109, 4092–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladbury J. E.; Klebe G.; Freire E. Adding Calorimetric Data to Decision Making in Lead Discovery: A Hot Tip. Nat. Rev. Drug Discovery 2010, 9, 23–27. [DOI] [PubMed] [Google Scholar]
- Marshall G. R. Limiting Assumptions in Structure-Based Design: Binding Entropy. J. Comput.-Aided Mol. Des. 2012, 26, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh S. H.; Seavers P. R.; Wilkinson A. J.; Ladbury J. E.; Tame J. R. H. Crystallographic and Calorimetric Analysis of Peptide Binding to OppA Protein. J. Mol. Biol. 1999, 291, 393–415. [DOI] [PubMed] [Google Scholar]
- Talhout R.; Villa A.; Mark A. E.; Engberts J. B. F. N. Understanding Binding Affinity: A Combined Isothermal Titration Calorimetry/Molecular Dynamics Study of the Binding of a Series of Hydrophobically Modified Benzamidinium Chloride Inhibitors to Trypsin. J. Am. Chem. Soc. 2003, 125, 10570–10579. [DOI] [PubMed] [Google Scholar]
- Malham R.; Johnstone S.; Bingham R. J.; Barratt E.; Phillips S. E. V.; Laughton C. A.; Homans S. W. Strong Solute–Solute Dispersive Interactions in a Protein–Ligand Complex. J. Am. Chem. Soc. 2005, 127, 17061–17067. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy V. M.; Bohall B. R.; Semetey V.; Whitesides G. M. The Paradoxical Thermodynamic Basis for the Interaction of Ethylene Glycol, Glycine, and Sarcosine Chains with Bovine Carbonic Anhydrase II: An Unexpected Manifestation of Enthalpy/Entropy Compensation Steuber. J. Am. Chem. Soc. 2006, 128, 5802–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum B.; Mohamed M.; Zayed M.; Gerlach C.; Heine A.; Hangauer D.; Klebe G. More than a Simple Lipophilic Contact: A Detailed Thermodynamic Analysis of Nonbasic Residues in the S1 Pocket of Thrombin. J. Mol. Biol. 2009, 390, 56–69. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y.; Chufan E. E.; Lafont V.; Hidaka K.; Kiso Y.; Amzel L. M.; Freire E. How Much Binding Affinity Can Be Gained by Filling a Cavity?. Chem. Biol. Drug Des. 2010, 75, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum B.; Muley L.; Smolinski M.; Heine A.; Hangauer D.; Klebe G. Non-Additivity of Functional Group Contributions in Protein-Ligand Binding: A Comprehensive Study by Crystallography and Isothermal Titration Calorimetry. J. Mol. Biol. 2010, 397, 1042–1054. [DOI] [PubMed] [Google Scholar]
- Snyder P. W.; Mecinović J.; Moustakas D. T.; Thomas S. W. III; Harder M.; Mack E. T.; Lockett M. R.; Héroux A.; Sherman W.; Whitesides G. M. Mechanism of the Hydrophobic Effect in the Bimolecular Recognition of Arylsulfonamides by Carbonic Anhydrase. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 17889–17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecinović J.; Snyder P. W.; Mirica K. A.; Bai S.; Mack E. T.; Kwant R. L.; Moustakas D. T.; Héroux A.; Whitesides G. M. Fluoroalkyl and Alkyl Chains Have Similar Hydrophobicities in Binding to the “Hydrophobic Wall” of Carbonic Anhydrase. J. Am. Chem. Soc. 2011, 133, 14017–14026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt T.; Holzmann N.; Muley L.; Khayat M.; Wegscheid-Gerlach C.; Baum B.; Heine A.; Hangauer D.; Klebe G. Congeneric but Still Distinct: How Closely Related Trypsin Ligands Exhibit Different Thermodynamic and Structural Properties. J. Mol. Biol. 2011, 405, 1170–1187. [DOI] [PubMed] [Google Scholar]
- For a review, see:Martin S. F.; Clements J. H. Correlating Structure and Energetics in Protein–Ligand Interactions: Paradigms and Paradoxes. Annu. Rev. Biochem. 2013, 82, 267–29. [DOI] [PubMed] [Google Scholar]
- Dunitz J. D. Win Some, Lose Some: Enthalpy–Entropy Compensation in Weak Intermolecular Interactions. Chem. Biol. 1995, 2, 709–712. [DOI] [PubMed] [Google Scholar]
- Cooper A.; Johnson C. M.; Lakey J. H.; Nöllmann M. Heat Does Not Come in Different Colours: Enthropy–Enthalpy Compensation, Free Energy Windows, Quantum Confinement, Pressure Perturbation Calorimetry, Solvation and the Multiple Causes of Heat Capacity Effects in Bimolecular Interactions. Biophys. Chem. 2001, 93, 215–230. [DOI] [PubMed] [Google Scholar]
- Olsson T. S. G.; Ladbury J. E.; Pitt W. R.; Williams M. A. Extent of Enthalpy–Entropy Compensation in Protein–Ligand Interactions. Protein Sci. 2011, 20, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C. H.; Holloway M. K. Thermodynamics of Ligand Binding and Efficiency. ACS Med. Chem. Lett. 2011, 2, 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson J. P.; Lubman O.; Rose T.; Waksman G.; Martin S. F. Calorimetric and Structural Studies of 1,2,3-Trisubstituted Cyclopropanes as Conformationally Constrained Peptide Inhibitors of Src SH2 Domain Binding. J. Am. Chem. Soc. 2002, 124, 205–215. [DOI] [PubMed] [Google Scholar]
- Ward J. M.; Gorenstein N. M.; Tian J.; Martin S. F.; Post C. B. Constraining Binding Hot Spots: NMR and Molecular Dynamics Simulations Provide a Structural Explanation for Enthalpy–Entropy Compensation in SH2-Ligand Binding. J. Am. Chem. Soc. 2010, 132, 11058–11070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfield A. P.; Teresk M. G.; Plake H. R.; DeLorbe J .E.; Millspaugh L. E.; Martin S. F. Ligand Preorganization May Be Accompanied by Entropic Penalties in Protein–Ligand Interactions. Angew. Chem., Int. Ed. 2006, 45, 6830–6835. [DOI] [PubMed] [Google Scholar]
- DeLorbe J. E.; Clements J. H.; Teresk M. G.; Benfield A. P.; Plake H. R.; Millspaugh L. E.; Martin S. F. Thermodynamic and Structural Effects of Conformational Constraints in Protein–Ligand Interactions. Entropic Paradoxy Associated with Ligand Preorganization. J. Am. Chem. Soc. 2009, 131, 16758–16770. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Zhu C. Z.; Martin S. F.; Ren P. Probing the Effect of Conformational Constraint on Phosphorylated Ligand Binding to an SH2 Domain Using Polarizable Force Field Simulations. J. Phys. Chem. B 2012, 116, 1716–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorbe J. E.; Clements J. H.; Whiddon B. B.; Martin S. F. Thermodynamic and Structural Effects of Macrocyclic Constraints in Protein–Ligand Interactions. ACS Med. Chem. Lett. 2010, 1, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myslinski J. M.; DeLorbe J. E.; Clements J. H.; Martin S. F. Protein–Ligand Interactions: Thermodynamic Effects Associated with Increasing Nonpolar Surface Area. J. Am. Chem. Soc. 2011, 133, 18518–18521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review of SH2 domains, see:Bradshaw J. M.; Waksman G. Molecular Recognition by SH2 Domains. Adv. Protein Chem. 2002, 61, 161–210. [DOI] [PubMed] [Google Scholar]
- For a leading reference, see:Khan A. R.; Parrish J. C.; Fraser M. E.; Smith W. W.; Bartlett P. A.; James M. N. G. Lowering the Entropic Barrier for Binding Conformationally Flexible Inhibitors to Enzymes. Biochemistry 1998, 37, 16839–16845. [DOI] [PubMed] [Google Scholar]
- Olsson T. S. G.; Williams M. A.; Pitt W. R.; Ladbury J. E. The Thermodynamics of Protein–Ligand Interaction and Solvation: Insights for Ligand Design. J. Mol. Biol. 2008, 384, 1002–1017. [DOI] [PubMed] [Google Scholar]
- Carey C.; Cheng Y.-K.; Rossky P. Hydration Structure of the α-Chymotrypsin Substrate Binding Pocket: The Impact of Constrained Geometry. J. Chem. Phys. 2000, 258, 415–425. [Google Scholar]
- Setny P.; Baron R.; McCammon J. A. How Can Hydrophobic Association Be Enthalpy Driven?. J. Chem. Theory Comput. 2010, 6, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J. The Basis of the Hydrophobic Effect. Biophys. Chem. 2003, 100, 193–203. [DOI] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437, 640–647. [DOI] [PubMed] [Google Scholar]
- Dill K. A.; Truskett T. M.; Vlachy V.; Hribar-Lee B. Modeling Water, The Hydrophobic Effect, and Ion Solvation. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 173–199. [DOI] [PubMed] [Google Scholar]
- Sturtevant J. M. Heat Capacity and Entropy Changes in Processes Involving Proteins. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 2236–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privalov P. L.; Gill S. J. Stability of Protein Structure and Hydrophobic Interaction Adv. Protein Chem. 1988, 39, 191–234. [DOI] [PubMed] [Google Scholar]
- Loladze V. V.; Ermolenko D. N.; Makhatadze G. I. Heat Capacity Changes upon Burial of Polar and Nonpolar Groups in Proteins. Protein Sci. 2001, 10, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzati V. Traitement Statistique des Erreurs dans la Determinaton des Structures Cristallines. Acta Crystallogr. 1952, 5, 802–810. [Google Scholar]
- Dunitz J. D. The Entropic Cost of Bound Water in Crystals and Biomolecules. Science 1994, 264, 670. [DOI] [PubMed] [Google Scholar]
- Li A.-J.; Nussinov R. A Set of van der Waals and Coulombic Radii of Protein Atoms for Molecular and Solvent-Accessible Surface Calculation, Packing Evaluation, and Docking. Proteins 1998, 32, 111–127. [PubMed] [Google Scholar]
- Tuñón I.; Silla E.; Pascual-Ahuir J. L. Molecular Surface Area and Hydrophobic Effect. Protein Eng. 1992, 5, 715–716. [DOI] [PubMed] [Google Scholar]
- For a lead reference of the use of double bonds as conformational constraint in small molecules, see:Harrison B. A.; Gierasch T. M.; Neilan C.; Pasternak G. W.; Verdine G. L. High-Affinity Mu Opioid Receptor Ligands Discovered by the Screeening of an Exhaustively Sterodiversified Library of 1,5-Enediols. J. Am. Chem. Soc. 2002, 124, 13352–13353. [DOI] [PubMed] [Google Scholar]
- Biela A.; Nasief N. N.; Betz M.; Heine A.; Hangauer D.; Klebe G. Dissecting the Hydrophobic Effect on the Molecular Level: The Role of Water, Enthalpy, and Entropy in Ligand Binding to Thermolysin. Angew. Chem., Int. Ed. 2013, 52, 1822–1828and references therein. [DOI] [PubMed] [Google Scholar]
- For example, see:Eldridge M. D.; Murray C. W.; Auton T. R.; Paolini G. V.; Mee R. P. Empirical Scoring Functions: I. The Development of a Fast Empirical Scoring Function to Estimate the Binding Affinity of Ligands in Receptor Complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.