

Abstract
Lung transplantation remains the only effective therapy for patients with end-stage pulmonary diseases. Unfortunately, acute rejection of the lung remains a frequent complication and is an important cause of morbidity and mortality. The induction of transplant tolerance is thought to be dependent, in part, on the balance between allograft effector mechanisms mediated by effector T lymphocytes (Teff), and regulatory mechanisms mediated by FOXP3+ regulatory T cells (Treg). In this study, we explored an approach to tip the balance in favor of regulatory mechanisms by modulating chemokine activity. We demonstrate in an adoptive transfer model of lung rejection that CXCR3-deficient CD8+ Teff have impaired migration into the lungs compared with wild-type Teff, which results in a dramatic reduction in fatal pulmonary inflammation. The lungs of surviving mice contained tolerized CXCR3-deficient Teff, as well as a large increase in Treg. We confirmed that Treg were needed for tolerance and that their ability to induce tolerance was dependent on their numbers in the lung relative to the numbers of Teff. These data suggest that transplantation tolerance can be achieved by reducing the recruitment of some, but not necessarily all, CD8+ Teff into the target organ and suggest a novel approach to achieve transplant tolerance.
Lung transplantation remains the only effective therapy for the large number of patients with end-stage lung disease (1, 2). However, despite advances in immunosuppressive therapies and surgical techniques, overall median survival after lung transplantation is only 5 y, with actuarial survival at 77, 61, and 50% at 1, 3, and 5 y, respectively (3). Clinical studies have implicated lung injury from acute rejection (AR) as a major causative factor of morbidity and mortality after lung transplantation (4-6).
The holy grail of the transplantation field is to induce donor-specific tolerance. In animal models, long-term graft survival and transplantation tolerance without immunosuppression can be induced by a number of methods (7). Most of these strategies have relied on the concept that deletion or inhibition of donor-specific effector T lymphocytes (Teff) is necessary to prevent rejection and achieve tolerance (8, 9). Recently, it has become clear that regulatory T cells (Treg) play a crucial role in suppressing alloimmune responses directed against transplanted tissues (10). Naturally occurring and induced Treg have been identified as CD4+CD25+ T lymphocytes that specifically express the forkhead family transcription factor FOXP3 (11-13) and are critical regulators of autoimmunity (14) and peripheral tolerance (15, 16). A large body of data has emerged suggesting tolerance depends on a balance between effector mechanisms mediated by Teff and suppressive mechanisms mediated by Treg (17). In the presence of low effector cell numbers, regulatory mechanisms are thought to suppress effector mechanisms, keeping them in check, resulting in transplant tolerance. Unfortunately, many of the immunosuppressive techniques used to prevent rejection also inhibit Treg, thus preventing the induction of tolerance (18, 19).
Central to the development of AR is the recruitment of Teff into the transplanted lung (20-22). Leukocyte recruitment into tissue is orchestrated by chemoattractants, such as chemokines, a superfamily of secreted chemotactic cytokines, as well as lipid mediators, which regulate cell migration through G protein-coupled chemoattractant receptors expressed on immune cells (23-26). AR of an organ is a complex and intense inflammatory response with many chemokines and their receptors implicated in the process. Research in humans and animals has demonstrated that the chemokine receptor CXCR3 and its ligands, CXCL9 and CXCL10, play important roles in this process (27-29). In animal models of heart and small bowel transplantation, the inhibition or deletion of CXCL9, CXCL10, and CXCR3 significantly prolonged graft survival (30-33), which conceptualized their importance in organ rejection. Recent findings using tracheal transplantation in mice as a model of lung transplantation have shown similar importance for CXCR3 and its ligands (34). However, other studies have questioned the importance of CXCR3 in heart transplantation (35, 36). CXCR3 is expressed on multiple cell types in addition to Teff, such as NK and NKT cells, dendritic cells, B cells, and Treg (29, 37-39). The prior studies described above did not examine inhibition of CXCR3 on individual cell types, which may explain some of the variability in the literature. Using our recently developed adoptive transfer mouse model of lung rejection (21), in the current study we address this possible confounding issue by having CD8+ T cells be the only cell type deficient in CXCR3.
In our previous study, we were able to partially inhibit Teff recruitment into the lung during AR and prolong allograft survival by specifically deleting the leukotriene B4 receptor BLT1 only on Teff (21, 34). In the current study, we evaluated the ability of CXCR3 to mediate Teff recruitment in our model of acute lung rejection, which has advantages over established murine models by utilizing the whole lung and having survival as an end point (21). In addition, our adoptive transfer transgenic mouse model allowed us to specifically isolate a role for CXCR3 in the trafficking of Ag-specific Teff. We found that deleting CXCR3 on Teff also partially reduced Teff homing into the lung, and this was sufficient to induce tolerance and prevent rejection despite recruitment of some Ag-specific Teff into the lung. This occurred without inhibiting T cell activation and without general immunosuppression. In addition, we observed a large increase in endogenous Treg specifically in the lungs of these mice, and these cells were essential in inducing tolerance as Treg-deficient mice were not able to tolerize Teff and did not survive. Taken together, these studies suggest the novel concept that manipulation of chemoattractant-induced T cell recruitment into the lung can generate a microenvironment advantageous to endogenous Treg. In fact, even partial inhibition of Teff homing into the lung can tip the balance in favor of Treg and allograft tolerance induction, thus providing a novel therapeutic approach to solid organ transplantation.
Materials and Methods
Mice
Wild-type (WT) C57BL/6 mice were purchased from the National Cancer Institute, National Institutes of Health (Bethesda, MD). CXCR3-deficient mice (CXCR3−/−) in the C57BL/6 background (32) were provided by C. Gerard (Children′s Hospital, Boston, MA) and bred in our facility. OT-I TCR mice in the C57BL/6 background were obtained from The Jackson Laboratory (Bar Harbor, ME) and crossed with CXCR3−/− mice to generate CXCR3−/−/OT-I mice. CC10-OVA mice in the C57BL/6 background were generated and maintained in our laboratory (21). Thy1.1+Thy1.2+ double-positive CC10-OVA mice were generated by mating CC10-OVA with B6.PL-Thy1a/CyJ (The Jackson Laboratory). CD80−/−/CD86−/−/CC10-OVA mice were generated by mating CC10-OVA with B6.129S4-Cd80tm1ShrCd86tm2Shr/J (The Jackson Laboratory). DEREG (Depletion of Regulatory T cell) mice in C57BL/6 background (40) were crossed with CC10-OVA mice to generate DEREG/CC10-OVA in our facility. All protocols were approved by the Massachusetts General Hospital Subcommittee on Research and Animal Care.
OT-I cell preparation and adoptive transfer
Isolation and preparation of OT-I and CXCR3−/−/OT-I CD8+ Teff was performed as described previously (41). Briefly, spleens were harvested from OT-I TCR transgenic mice, single-cell suspensions were prepared, and CD8+ cells were purified using MACS CD8a MicroBeads kit (Miltenyi Biotech). Effector OT-I cells were prepared by placing the purified CD8+ OT-I cells in culture for 5 d with irradiated APCs prepared from spleens of C57BL/6 mice with 700 ng/ml SIINFEKL peptide, 2 μg/ml anti-CD28, 10 ng/ml recombinant IL-2, and 10 ng/ml recombinant IL-12. Effector CD8+ OT-I cells were then resuspended in PBS and injected i.p.
OT-I cell cytotoxicity
Cytotoxicity was measured using a commercially available kit according to the manufacturer′s protocol (CyToxiLux Plus, OncoImmunin, Gaithers-burg, MD).
CC10-OVA mouse tissue sampling and processing
Animals were sacrificed with a lethal injection of ketamine (100 mg/kg). The lungs were lavaged with six 0.5-ml aliquots of PBS containing 0.6 mM EDTA. The spleen, thoracic lymph nodes, and inguinal lymph nodes were removed. The lungs were flushed free of blood by slowly injecting 10 ml PBS into the right ventricle prior to excision and digested for 45 min in RPMI 1640 with 0.28 Wunsch U/ml Liberase Blendzyme (Roche, Indianapolis, IN) and DNase 30 U/ml (Sigma-Aldrich, St. Louis, MO) at 37°C. The digested lungs were then extruded through a mesh strainer.
Histopathologic examination
Tissue was placed into 10% buffered formalin. Multiple paraffin-embedded 5-μm sections were prepared and stained with H&E. The slides were evaluated by light microscopy.
Flow cytometry and cell sorting
Cells recovered from cell culture, the bronchoalveolar lavage (BAL) fluid, or single-cell suspensions of lung, lymph node, or spleen were blocked, stained, and analyzed as previously described (42). The class I tetramer specific for OT-I cells was obtained from Beckman Coulter (Fullerton, CA). The staining kit for Foxp3+ Treg was obtained from eBioscience (San Diego, CA). Fluorescently labeled anti-CD3, anti-CD8, anti-Thy1.1, and anti-Thy1.2 Abs were obtained from BD Pharmingen. Fluorescently labeled anti-CXCR3, anti-CCR4, anti-CCR6, and anti-CCR7 were obtained from BioLegend (San Diego, CA).
Quantitative real-time PCR
RNA was purified using a purification column (RNeasy; Qiagen, Valencia, CA). After a DNase step, 1 μg of RNA was converted to cDNA (Applied Biosystems, Warrington, UK). Specific primers used for sequence detection of message for the CXCL10 (IP-10) gene were 5′-GCCGTCATTTTCTGCCTCA-3′ and 5′-CGTCCTTGCGAGAGGGATC-3′, for CXCL9 (MIG) gene were 5′-AATGCACGATGCTCCTGCA-3′ and 5′-AGGTCTTTGAGGGATTTGTAGTGG-3′, for CXCL11 (ITAC) gene were 5′-AATTTACCCGAGTAACGGCTG-3′ and 5′-ATTATGAGGCGAGCTTGCTTG-3′, and for the GAPDH gene were 5′-GGCAAATTCAACGGCACAGT-3′ and 5′-AGATGGTGATGGGCTTCCC-3′. Samples underwent amplification in the presence of SYBR Green (Applied Biosystems). The reaction was analyzed in real-time during amplification by the PCR machine (MX-4000; Stratagene, La Jolla, CA).
Proliferation assay
Lung and spleen from surviving CC10-OVA mice adoptively transferred with CXCR3−/−/OT-I cells for 2 wk were made into single-cell suspensions, and the CXCR3−/−/OT-I cells were isolated by cell sorting as responder cells in two separate experiments. We used class I tetramer to sort for CXCR3−/−/OT-I cells from CC10-OVA mice in the first experiment and anti-Thy1.2 and anti-Thy1.1 Abs in the second experiment to sort for Thy1.2+ CXCR3−/−/OT-I cells from Thy1.1+Thy1.2+ double-positive CC10-OVA mice. In vitro OT-I cells were taken from freshly prepared effector OT-I cells as described above. Spleen cells from C57BL/6 mice were used as stimulator cells. Stimulator cells were incubated with or without SIINFEKL peptide at 700 ng/ml. Responder and stimulator cells were incubated together in a 96-well plate in triplicate for each group for 2 d at 37°C. One group also received recombinant IL-2 at 10 ng/ml. [3H] (0.5 μl) was added to each well and then incubated overnight before the plate was harvested and read in a scintillation plate counter machine (TopCount NXT; Packard Bioscience).
Selective Treg depletion
The DEREG mouse is a transgenic mouse carrying a DTR-eGFP transgene under the control of an additional Foxp3 promoter (40). DEREG/CC10-OVA mice were i.p. injected with 1 μg diphtheria toxin (EMD Biosciences, San Diego, CA) for 2 consecutive days starting 2 d before Teff adoptive transfer and a third diphtheria toxin injection 3 d later.
Results
Deficiency of CXCR3 on CD8+ Teff diminish mortality and lung inflammation
We previously developed a novel transgenic model of acute lung rejection where C57BL/6 mice express a membrane-bound form of chicken egg albumin (OVA) in the airway lining cells of the lung (CC10-OVA mice) (21). Transfer of 5 × 105 in vitro-activated CD8+ Teff with a TCR specific for OVA (isolated from the OT-I C57BL/6 TCR-transgenic mouse) into CC10-OVA mice induced airway injury, inflammation, and death within 5–9 d of transfer. In this study, we evaluated the ability of CXCR3 to mediate Teff recruitment in the CC10-OVA lung rejection model. CD8+ T cells from the spleens of OT-I and CXCR3-deficient OT-I mice were isolated and activated in vitro with IL-2 and IL-12 to generate Teff, as previously described (21, 41). Flow cytometry demonstrated an equivalent effector phenotype for WT and CXCR3-deficient OT-I Teff: low CD62L, high CD25, high IFN-γ, and positive perforin expression (Fig. 1A), along with high cytotoxic ability (Fig. 1B). CC10-OVA mice that received in vitro-activated CXCR3−/− OT-I Teff had a dramatic reduction in mortality compared with CC10-OVA mice that received WT OT-I Teff (Fig. 1C). Two weeks after Teff adoptive transfer, 91% of CC10-OVA mice that received CXCR3−/− Teff were alive, whereas only 9% of CC10-OVA mice that received WT Teff survived. Histological analysis of the lungs 3 d after adoptive transfer of WT OT-I Teff demonstrated perivascular and peribronchial inflammation (Fig. 1Di, 1Dii). In contrast, mice that received CXCR3−/− OT-I Teff showed minimal inflammation around the lung vasculature and almost no involvement of the airways (Fig. 1Diii, 1Div). To determine the ligands that mediate the recruitment of Teff through CXCR3 signaling in our model, we isolated the lungs of CC10-OVA mice and C57BL/6 controls that received adoptively transferred OT-I cells 3 d prior and performed quantitative real-time PCR for CXCR3 ligand expression. As can be seen in Fig. 1E, CXCL10 and CXCL9 were highly induced (12- and 8-fold, respectively) in the CC10-OVA lungs compared with C57BL/6 control lungs. The expression of these chemokines has been shown to correlate with T cell recruitment into transplanted organs during AR (28, 30, 33, 43). CXCL11 RNA expression was absent in both strains.
FIGURE 1.
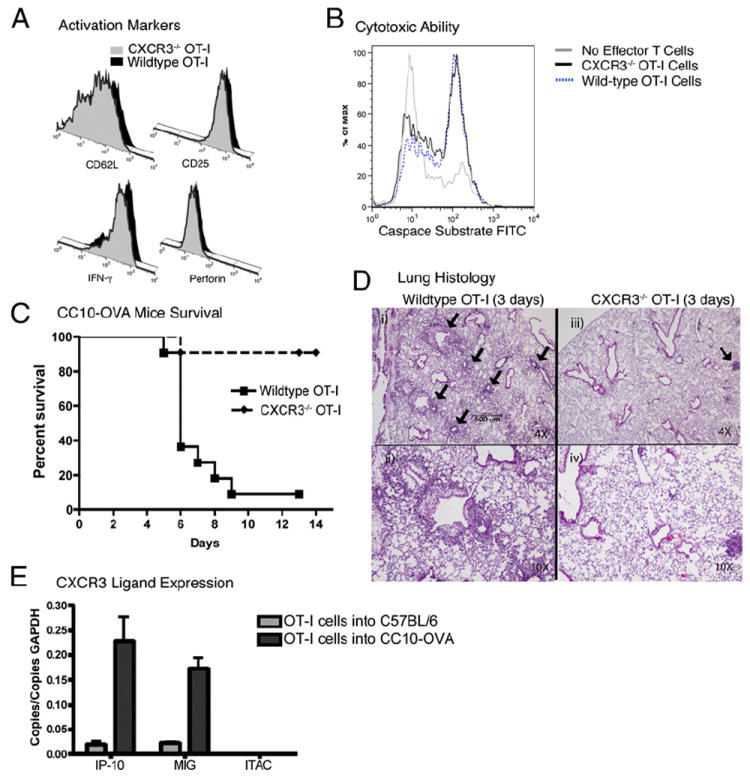
Reduced mortality and pulmonary inflammation after adoptive transfer of CXCR3−/− OT-I into CC10-OVA mice. A, In vitro-activated CD8+ cells from CXCR3−/− or WT OT-I mice stained for activation markers (CD62L, CD25, IFN-γ, and perforin). B, Cytotoxicity assay of in vitro-activated CXCR3−/− or WT OT-I cells using the CyToxiLux kit with EL-4 cells as targets. C, Mortality of CC10-OVA transgenic mice injected with WT OT-I Teff or CXCR3−/− OT-I Teff (n = 11 per group). Curves were significantly different by log-rank test. D, Representative histologies of lungs from CC10-OVA transgenic mice 3 d after adoptive transfer of WT OT-I Teff stained with H&E (i, ii) or CXCR3−/− OT-I Teff (iii, iv). Original magnification: low power at ×4 (i, iii) and high power at ×40 (ii, iv). Black arrows indicate apparent perivascular and peribronchial inflammation. E, CXCR3 ligand expression in the lungs of C57BL/6 and CC10-OVA mice 3 d after the adoptive transfer of in vitro-activated OT-I cells determined by quantitative real-time PCR.
CXCR3-deficent Teff have impaired homing into the lung
To compare the trafficking of CXCR3−/− Teff to WT Teff in our mouse model, we performed competitive homing assays (21). Briefly, CXCR3−/− OT-I cells expressing Thy1.2 and WT OT-I cells expressing Thy1.1 were both adoptively transferred into the same CC10-OVA mouse congenic for both Thy1.1 and Thy1.2. This allowed us to track the individual populations of transferred WT OT-I (Thy1.1+) and CXCR3−/− OT-1 (Thy1.2+) cells in the same recipient mouse as well as distinguish endogenous T cells (Thy1.1+/Thy1.2+). Analysis of BAL fluid and lung tissue 4 d after cotransfer revealed a 40% decrease in the accumulation of CXCR3−/− OT-I cells in the lung and BAL compared with WT OT-I cells (Fig. 2A, 2B). In contrast, the peripheral organs, such as the spleen and inguinal lymph nodes, contained 2-fold and 1.7-fold more CXCR3−/− OT-I cells than WT OT-I, respectively. These data indicate that CXCR3 plays a role in Teff trafficking into the lung and that inhibition of CXCR3-mediated Teff recruitment in this model can reduce mortality.
FIGURE 2.
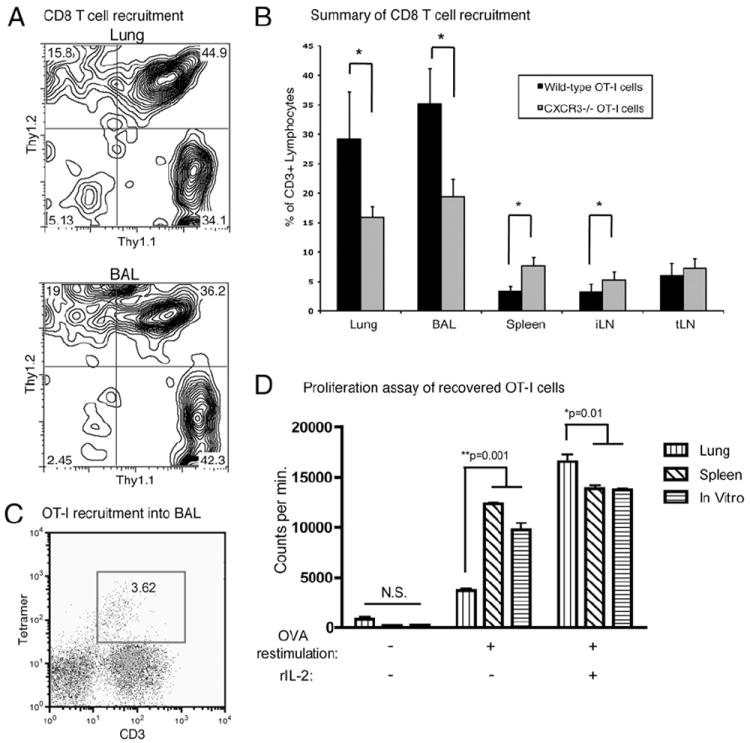
Decreased recruitment and responsiveness of CXCR3−/− OT-I cells in the airways. A, Representative flow cytometry of CD3+ lymphocytes revealing WT OT-I (Thy1.1+) and CXCR3−/− OT-I (Thy1.2+) Teff recruitment into the lung and BAL 4 d after cotransfer into CC10-OVA mice (Thy1.1+Thy1.2+). B, Summary of flow cytometry data of Thy1.1+ WT OT-I and Thy1.2+ CXCR3−/− OT-I recruited into the lung, BAL, spleen, inguinal lymph node (iLN), and thoracic lymph node (tLN) 4 d after cotransfer into CC10-OVA mice (n = 4 mice). *p < 0.04. C, Representative flow cytometry of BAL for OT-I cells 2 wk after adoptive transfer into CC10-OVA mice. The transferred cells are identified by tetramer staining specific for the OT-I TCR and anti-CD3 staining. D, [3H] proliferation assay of recovered CXCR3−/− OT-I Teff isolated from the lung and spleen of CC10-OVA survivors 2 wk after adoptive transfer by cell flow sorter and activated WT OT-I cells from 5-d culture. The cells were, or were not, restimulated with OVA peptide and exogenous rIL-2.
Teff are anergic in the lungs of survivors
The competitive homing assay revealed that some CXCR3−/− OT-I cells still reached the airways and accumulated in surviving mice (Fig. 2B). We therefore transferred only CXCR3−/− OT-I Teff into CC10-OVA mice and analyzed the BAL of surviving mice 2 wk after adoptive transfer for the presence of these transferred Teff. As a secondary method of identifying these adoptively transferred CXCR3−/− OT-I Teff, we used fluorochrome-conjugated class I tetramers specific for the OT-I TCR and anti-CD3. This analysis revealed that the Teff were still present in the airways (Fig. 2C) but were apparently not causing overt signs of pulmonary damage. We then isolated the CXCR3−/− OT-I cells from the lungs and spleens of surviving CC10-OVA mice 2 wk after adoptive transfer using FACS and determined their ability to proliferate to OVA restimulation in vitro. CXCR3−/− OT-I cells isolated from the lung showed a 3.3-fold decrease in OVA-induced proliferation compared with cells isolated from the spleen of the same recipient mouse, as well as from in vitro-activated WT OT-I cells (Fig. 2D). However, the addition of exogenous IL-2 to CXCR3−/− Teff isolated from the lung dramatically overcame the proliferative defect (Fig. 2D). These data demonstrate that CXCR3−/− OT-I cells in the lungs of surviving mice were unresponsive to their target Ag and that the effect was specific to the lungs.
Teff induce Treg in the lungs
Analysis of surviving CC10-OVA mice 2 wk after adoptive transfer of CXCR3−/− OT-I cells revealed a 2-fold increase in FOXP3+ Treg in the lungs compared with WT C57BL/6 mice that also received the CXCR3−/− OT-I cells or compared with untreated CC10-OVA mice (Fig. 3A). This increase in Treg was specific to the lung, as there was no difference in the number of FOXP3+ Treg in the spleen among the different groups (Fig. 3A). We also examined an earlier time point after the adoptive transfer of CXCR3−/− and WT OT-I Teff. We reasoned that if CXCR3−/− Teff that entered the lung induced Treg accumulation in the lung, then CXCR3+/+ Teff that entered the lung might also induce Treg accumulation. We found a 2-fold increase in CD4+FOXP3+ cells in the lung 3–4 d after WT OT-I Teff transfer compared with untreated mice (Fig. 3B). In contrast, the lungs of CC10-OVA mice that received CXCR3−/− OT-I Teff showed no increase in Treg at this early period. Consistent with the specificity for the lung, the spleen showed no early increase in Treg after either WT or CXCR3−/− OT-I transfer compared with the control (Fig. 3B).
FIGURE 3.
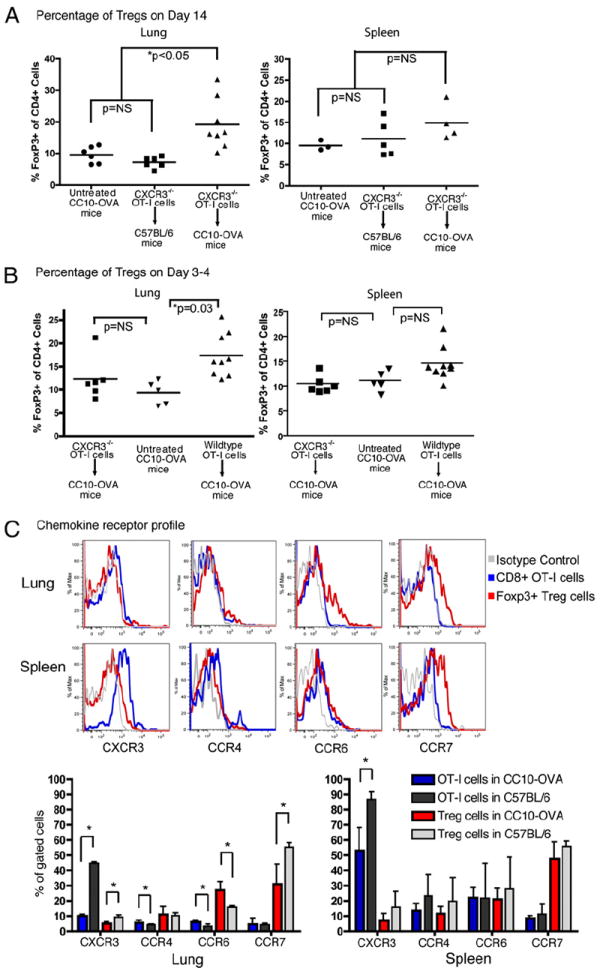
Foxp3+ Treg increased in the lungs of CC10-OVA survivors. A, Summary graphs of the percentage of Foxp3+ cells within gated CD4+ lymphocytes in the lungs and spleens of untreated CC10-OVA mice and WT C57BL/6 mice and CC10-OVA survivors 2 wk after injection with CXCR3−/− OT-I Teff. B, Summary graphs of the percentage of Foxp3+ cells in the lungs and spleens of CC10-OVA mice 3–4 d after adoptive transfer of WT or CXCR3−/− OT-I Teff and untreated CC10-OVA mice. C, Representative flow cytometry histograms of chemokine receptor cell surface expression on CD8+ OT-I Teff and CD4+ Foxp3+ Treg recovered from the lungs and spleens of CC10-OVA mice 3 d after adoptive transfer of WT OT-I (Thy1.1+) Teff. Summary graphs presented below histograms exhibit the mean percentage and SD of CD8+ Thy1.1+ OT-I cells and CD4+ Foxp3+ Treg from CC10-OVA or C57BL/6 mice expressing CXCR3, CCR4, CCR6, or CCR7 chemokine receptors (n = 6 CC10-OVA, 3 C57BL/6). *p < 0.05.
To determine if there is similar chemokine signaling for the recruitment of Teff and Treg in the lung, we ascertained the chemokine receptor profile on WT OT-I Teff and Treg isolated from the lung and spleen of CC10-OVA or C57BL/6 mice 3 d after OT-I adoptive transfer by flow cytometry (Fig. 3C). OT-I Teff recovered from the spleen expressed high levels of CXCR3 and low levels of CCR7, similar to in vitro-generated Teff prior to injection, as we have previously reported (44). In contrast, Treg recovered from the spleen expressed low levels of CXCR3 and high levels of CCR7. CCR4 and CCR6 were expressed to similar levels on Teff and Treg recovered from the spleen. In the lung, Teff recovered from C57BL/6 mice also expressed high levels of CXCR3. However, CXCR3 was downregulated (4.5-fold) on Teff recovered from the lungs of CC10-OVA mice compared with C57BL/6 mice. Treg recovered from the lungs of C57BL/6 and CC10-OVA mice showed low expression of CXCR3 with levels in the CC10-OVA mice 1.9-fold less than in C57BL/6 mice. Both Teff and Treg in the lung of CC10-OVA mice showed a slight increase in CCR6 expression (1.9- and 1.7-fold, respectively) compared with those from C57BL/6 mice. These data suggest that the recruitment of Teff and Treg are likely controlled by different chemokine pathways in this model.
Lung rejections depend on the number of Teff recruited to the airways
We next determined if more CXCR3−/− Teff adoptively transferred into CC10-OVA mice would result in an increase in their accumulation in the lung and overcome regulatory mechanisms and induce rejection. We found that a 3-fold increase in the number of CXCR3−/− Teff did indeed induce 100% mortality compared with 30% mortality observed with the standard dose of 5 × 105 cells (Fig. 4A). As a further means to determine if the number of Teff reaching the lung was a crucial determinant for organ rejection, we reversed our approach and asked if reducing the number of WT OT-I Teff adoptively transferred into the CC10-OVA mice would prevent death. As seen in Fig. 4B, the standard number of 5 × 105 cells predictively resulted in 100% mortality 6 d after transfer. Lowering the number of transferred Teff by more than half to 2 × 105 cells delayed death to 9 d after transfer. Further reduction to 1 × 105 cells, one-fifth the standard number of transferred cells, resulted in only 14% mortality. We next determined if the transfer of lower numbers of WT Teff also induced Treg in surviving mice. The lungs from surviving CC10-OVA mice showed nearly a 3-fold increase in FOXP3+ Treg compared with their WT C57BL/6 controls (Fig. 4C), indicating that at one-fifth the standard dose, WT OT-I cells reached the lungs and induced the accumulation of Treg. The draining thoracic lymph nodes from surviving CC10-OVA mice also showed an increase in the FOXP3+ cells by 1.5-fold. In contrast, as was seen with the transfer of CXCR3−/− Teff, the spleen and inguinal lymph nodes of CC10-OVA mice that received 1 × 105 WT OT-I cells did not show any difference in FOXP3+ cells compared with their C57BL/6 controls (Fig. 4C).
FIGURE 4.
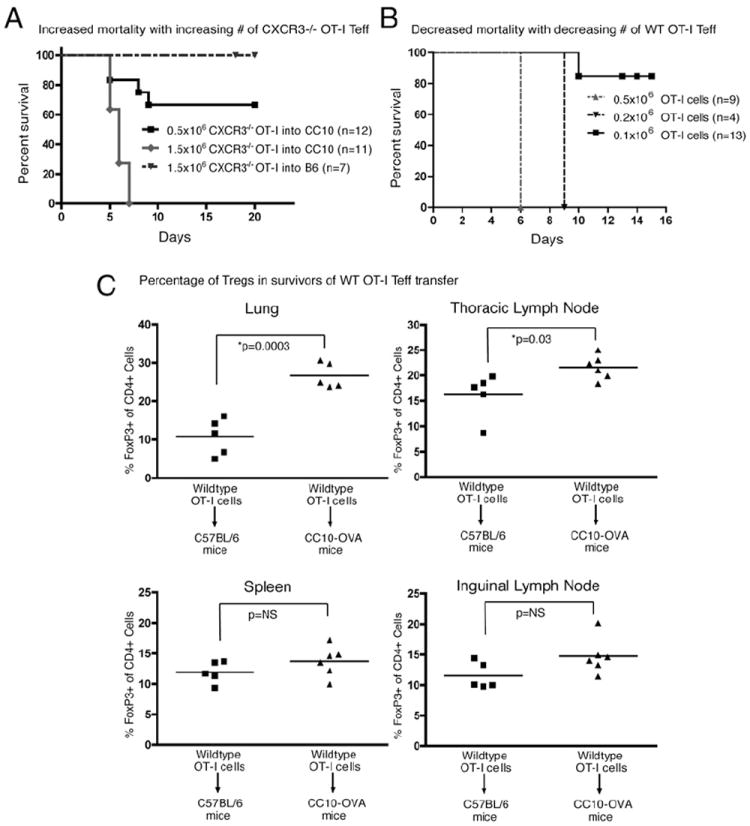
CC10-OVA survival dependent on number of Teff recruited to airways. A, Mortality of CC10-OVA mice injected with CXCR3−/− OT-I Teff at doses of 0.5 × 106 or 1.5 × 106 cells. Curves were significantly different by log-rank test (p = 0.0002). Mortality curve of WT C57BL/6 mice injected with 1.5 × 106 CXCR3−/− OT-I Teff was not significantly different from that of CC10-OVA mice injected with 0.5 × 106 CXCR3−/− OT-I cells by log-rank test. B, Mortality of CC10-OVA mice adoptively transferred with WT OT-I Teff at three different doses: 0.5 × 106, 0.2 × 106, and 0.1 × 106. The lowest-dose curve was significantly different from that of the other two curves by log-rank test (p < 0.0001). C, Summary graphs of the percentage of Foxp3+ cells within gated CD4+ lymphocytes from the lung, spleen, thoracic and inguinal lymph nodes of C57BL/6 and CC10-OVA mice 2 wk after injection with 0.1 × 106 WT OT-I Teff.
Treg are essential to prevent acute lung rejection and induce tolerance
We demonstrated that a low dose of effector CD8+ OT-I cells in the CC10-OVA mice induced the accumulation of Treg specifically in the lung. To demonstrate that these regulatory cells were truly protective against Teff-induced rejection, we took three complementary approaches: 1) determined if the increase in Treg in CC10-OVA mice after low-dose Teff transfer, as shown in Fig. 4B and 4C, would prevent rejection from a subsequent normally lethal Teff dose; 2) used Treg-deficient CD80−/−/CD86−/−/CC10-OVA mice; and 3) used selective Treg-depletable DEREG/CC10-OVA mice to specifically deplete Treg.
In our first approach, CC10-OVA mice were adoptively transferred with 1 × 105 WT OT-I cells, as described above, but after 2 wk a second dose of WT OT-I cells that would normally be fatal was given to the same recipient CC10-OVA mice (Fig. 5A). We hypothesized that the large increase in FOXP3+ Treg that accumulated in the lung after the low-dose OT-I transfer would modulate the effector functions of subsequent OT-I cells given to the CC10-OVA mice and prevent fatal pulmonary inflammation. Whereas only 20% of the control CC10-OVA mice given 2.5 × 105 OT-I cells survived after 7 d, 100% of the CC10-OVA mice first given 1 × 105 OT-I cells survived for more than 30 d after receiving a second dose of 2.5 × 105 OT-I cells (Fig. 5A). We next determined if a higher number of OT-I cells in the second dose could also be prevented from inducing mortality. As can be seen in Fig. 5A, 100% of the control mice given 5 × 105 OT-I cells died, as previously shown (Figs. 1C, 4B), but the number of deaths was dramatically reduced to 14% when the mice were first given a low dose of 1 × 105 OT-I cells before the standard dose of 5 × 105 OT-I cells. Greater than 25% of the CD8+ cells in the lungs of the survivors were found to be the adoptively transferred OT-I Teff (Supplemental Fig. 1). These results demonstrate that a nonlethal dose of WT Teff was protective against a subsequently higher, normally lethal dose of WT Teff.
FIGURE 5.
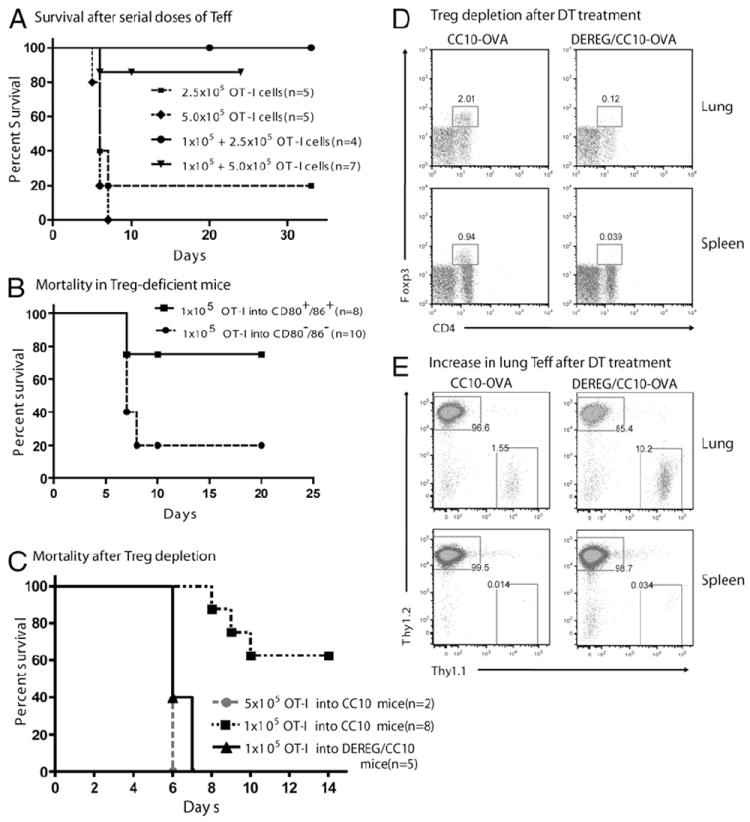
Resident Treg required to prevent lung rejection. A, Mortality of CC10-OVA mice injected i.p. with serial doses of WT OT-I Teff; first low dose of 1 × 105 cells was given 2 wk before the second dose of 2.5 × 105 or 5.0 × 105 cells on day 0. Control CC10-OVA mice were given a single dose of 2.5 × 105 or 5.0 × 105 OT-I Teff on day 0. B, Mortality of CC10-OVA and CD80−/−CD86−/−/CC10-OVA littermates injected with 1 × 105 OT-I Teff. C, Mortality of DEREG/CC10-OVA mice treated with DT to deplete Treg and injected with 1 × 105 OT-I Teff on day 0. Littermate CC10-OVA control mice were injected with either 1 × 105 or 5 × 105 OT-I Teff on day 0. D and E, CC10-OVA and DEREG/CC10-OVA littermates were injected i.p. with low-dose WT in vitro-activated OT-I (Thy1.1+) cells on day 0. On day 10, survivors of both strains were treated with DT for 2 consecutive days. On day 13, spleens and lungs were harvested and pooled from two CC10-OVA and two DEREG/CC10-OVA mice. D, Flow cytometry for CD4+FOXP3+ cells gated from lymphocytes after DT treatment. E, Flow cytometry for Thy1.1+ OT-I cells gated from CD8+ cells.
In our second approach, we used CC10-OVA mice lacking the costimulatory molecules CD80 and CD86 as recipient mice. CD80−/−/CD86−/− mice have a profound deficiency in FOXP3+ Treg (45) and they were mated with our CC10-OVA mice to generate CD80−/−/CD86−/−/CC10-OVA mice. In the spleens of these mice, <1% of CD4+ T cells are FOXP3+ Treg, compared with 5–8% of CD4+ T cells in CC10-OVA mice (Supplemental Fig. 2). Adoptive transfer of 1 × 105 OT-I Teff into Treg-deficient CD80−/−/CD86−/−/CC10-OVA mice resulted in 80% mortality within 7 d, whereas transfer of the same number of Teff into CC10-OVA mice resulted in 20% mortality (Fig. 5B).
We hypothesized that it was the lack of Treg in CD80−/−/CD86−/−/CC10-OVA mice that prevented the suppression of OT-I Teff pathogenicity in the lungs. To confirm this hypothesis, our third approach was to breed the selective Treg-depletable DEREG mouse (40) to our CC10-OVA mouse. Treatment of DEREG mice with two consecutive doses of diphtheria toxin (DT) selectively depleted up to 93% of the FOXP3+ Treg in the spleen, lymph nodes, and lung 1 d after the last treatment (Supplemental Fig. 3). DEREG/CC10-OVA mice were treated with the same two consecutive doses of DT, along with one more dose 3 d after the adoptive transfer of 1 × 105 OT-I Teff. As hypothesized, all of the DT-treated DEREG/CC10-OVA mice died within 7 d (Fig. 5C), whereas, the Treg-containing CC10-OVA mice showed only 37% mortality after receiving 1 × 105 OT-I Teff. Treatment of DEREG/CC10-OVA mice with DT alone showed no mortality (data not shown). In other experiments, DEREG/CC10-OVA and CC10-OVA mice were injected with low-dose OT-I cells, and survivors were then treated with DT for 2 consecutive days. Two days later, lungs and spleens were harvested and analyzed for Treg and Teff numbers (Fig. 5D, 5E). As expected, DT treatment eliminated Treg from the lungs and spleens of DEREG/CC10-OVA mice but not from CC10-OVA mice (Fig. 5D). Of note, along with this Treg depletion, DT treatment increased the number of OT-I cells found in the lungs of DEREG/CC10-OVA mice by 7-fold compared with those found in CC10-OVA mice (Fig. 5E). These data are consistent with our hypothesis that Treg can actively suppress OT-I Teff recruited into the lung to achieve tolerance. Together, these results demonstrate that FOXP3+ Treg in CC10-OVA mice are able to suppress the cytopathic activity of Teff that reach the lung up to a certain threshold.
Discussion
Using a transgenic mouse model of lung rejection, we have delineated a role for CXCR3 specifically on CD8+ Teff in the rejection process. In so doing, we have found that decreasing the number of Teff recruited into the lung allowed endogenous regulatory mechanisms to be activated by these Teff and induce organ-specific tolerance. Deletion of the CXCR3 chemokine receptor pathway on Teff did not completely eliminate their trafficking into the lungs; however, this partial inhibition was sufficient to allow for the effective generation of active tolerance. Thus, even partial interruption of Teff recruitment into the target tissue can generate a graft-specific microenvironment conducive to Treg generation and function.
Animal models of transplantation have increased our understanding of the mechanisms underlying rejection and chronic allograft dysfunction. Well-established murine models of cardiac, renal, and skin transplantation have been used to determine the role of several chemokines in transplantation (46). However, because lung transplantation in small animals is technically difficult, an ideal murine model of lung transplantation does not exist. We developed the CC10-OVA transgenic mouse (21) to use as a model for acute lung transplant rejection. Adoptive transfer of activated CD8+ OT-I cells specific for the OVA peptide leads to respiratory distress and death within 7 d. In this transgenic mouse model, OVA is membrane bound on the airway epithelium, resulting in CD8+ T cell-mediated injury to the airway lining, which closely mimics the pathophysiology of AR. The model exhibits significant perivascular and peribronchial inflammation typically seen in AR and serves as a proof-of-concept for specifically examining Ag-specific effector CD8+ T cells targeted against a specific organ. Similar transgenic mouse models, such as the RIP-OVA mouse, have been successfully used to study T cell tolerance, tissue damage, and T cell trafficking (47, 48).
Previous studies in humans and animal models of transplantation have identified CXCR3 and two of its ligands, CXCL9 and CXCL10, as important mediators of rejection after solid organ transplantation (28, 30, 32, 34, 43, 49). Recently, the role of CXCR3 in organ rejection has become less clear with reports indicating that this chemokine receptor pathway is not essential for the rejection process (35, 36). These prior studies used either CXCR3-deficient recipients or CXCR3 antagonists to interrogate CXCR3 function. This type of experimental design would render CXCR3 non-functional on all cell types, including FOXP3+ Treg, which have recently been shown capable of expressing CXCR3 (38, 50, 51). However, the design of our study enabled us to isolate specifically an important role for CXCR3 on Ag-specific CD8+ Teff in inducing pulmonary AR. We demonstrate that CXCR3 contributes importantly to the ability of Teff to home to the lung and that this CXCR3-dependent Teff trafficking also contributes to pulmonary inflammation and injury and resultant mortality in a model of AR.
We found that Teff recovered from the lungs of CC10-OVA mice have markedly decreased surface expression of CXCR3 compared with Teff recovered from the spleens of CC10-OVA mice or from the lungs of C57BL/6 mice. We believe that these data indicate that CXCR3 is specifically downregulated on Teff after encounter with its ligands in the lung and support a functional role for CXCR3 in the model. Previous studies have found that CXCR3 ligands induce CXCR3 internalization in vitro as well as in vivo upon entering the lung where high ligand levels were found (52, 53). In contrast to Teff, Treg recovered from lungs and spleens of CC10-OVA and C57BL/6 mice did not express high levels of CXCR3 in any tissue compartment. Instead, Treg expressed high levels of the lymph node-homing chemokine receptor CCR7 as well as moderate levels of CCR6. The large decrease in CCR7 expression on Treg isolated from the lungs of CC10-OVA mice compared with that of those from C57BL/6 mice may indicate the activation of endogenous naive Treg into effector Treg in the inflamed lung (54), as such decrease was not seen in the spleen.
Our study has also uncovered a dynamic interplay between CD8+ Teff and FOXP3+ Treg. The net outcome of the pro- and anti-inflammatory activities mediated by these cells appears to be an important determinant of allograft tolerance versus allograft rejection. We found that early in the rejection process, Teff recruitment into the lung induced an increase in the number of FOXP3+ Treg specifically in the lung (Fig. 3B). However, the suppressive activity of these regulatory cells was not sufficient to inhibit the inflammatory activities of Teff recruited into the lung, and mice ultimately died of acute lung rejection 3–4 d later (Fig. 1C). However, when the number of Teff reaching the lung was decreased by either CXCR3 deficiency or the transfer of fewer WT Teff (Fig. 4), mice were able to survive even with residual Teff present in the lungs as these Teff were now rendered tolerant (Fig. 2D). These data are consistent with a study that found that cardiac allograft survival was prolonged by blocking CXCR3 and CCR5, which was associated with an increase in FOXP3+ Treg in the grafts (55). It is becoming apparent that Treg are like other T cell subsets in that they develop during an immune response (56). Recent studies have shown that Treg can be present simultaneously with Teff in inflamed tissues and serve to balance the toxic effects of microbicidal cytokines (57, 58). The results from our study show that the same mechanism can also be applied to suppress the organ-rejecting activity of CD8+ Teff.
Treg are now recognized as a fundamental component in the development and maintenance of transplantation tolerance as they protect against pathogenic Teff (56, 59). Consistent with this concept, the data from our model of CD8-mediated rejection suggest that an increase in lung Treg or a reduction in Teff can limit lung injury and enhance survival. Notably, the introduction of Teff into the lung was associated with enhanced accumulation of Treg specifically in the lung. This increase in Treg may be secondary to the burst of IL-2 produced by activated Teff in the inflamed tissue (60), as IL-2 has been found to be crucial in the proliferation and maintenance of Treg (61, 62). Treg have also been shown to expand quickly after immune priming before their suppressive properties become apparent (63), and Teff have been shown to alter gene transcriptions in Treg (64, 65). Our study adds to these findings by demonstrating that Teff can boost the function of Treg in vivo.
Rejection and death occur in our model when a certain threshold of Teff reaching the lung is reached and the tolerogenic Treg/Teff ratio is no longer maintained. IL-2 is not only crucial for Treg but is also an important growth factor for the Teff population. As more Teff accumulate in the lung, they may out-compete Treg for a limited pool of IL-2 to a point where Treg can no longer be sustained, and rejection is inevitable. In support of this model, a recent study of mice infected with Toxoplasma gondii demonstrated that a hyperimmune Th1 mucosal immune response could lead to collapse of mucosal Treg, resulting in markedly increased mucosal immunopathology (66). Thus, an overexuberant organ-specific Teff response appears to be detrimental to the survival and function of Treg in that organ.
In conclusion, our data demonstrate that inhibition of CXCR3 on Teff may be therapeutically beneficial by inhibiting enough pathogenic Teff recruitment into the allograft to allow for the inflammation-induced recruitment and expansion of FOXP3+ Treg selectively in the graft to be effective at suppressing the rejection process. Our data also suggest that the complete inhibition of allospecific T cell responses may not be required to induce transplantation tolerance and that allospecific Teff may in fact induce Treg expansion in the graft. Thus, modulating the chemoattractant pathways that participate in the homing of Teff without also inhibiting the resultant accumulation of Treg may be a novel approach to prevent graft rejection and induce transplantation tolerance.
Supplementary Material
Acknowledgments
This work was supported by the Roche Organ Transplantation Research Foundation, by National Institutes of Health Grant R01CA069212 (to A.D.L.), and by the International Society for Heart and Lung Transplantation (to E.S.).
Abbreviations used in this article
- AR
acute rejection
- BAL
bronchoalveolar lavage
- DT
diphtheria toxin
- Teff
effector T lymphocytes
- Treg
regulatory T cells
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Trulock EP. Lung and heart-lung transplantation: overview of results. Semin Respir Crit Care Med. 2001;22:479–488. doi: 10.1055/s-2001-18420. [DOI] [PubMed] [Google Scholar]
- 2.DeMeo DL, Ginns LC. Clinical status of lung transplantation. Transplantation. 2001;72:1713–1724. doi: 10.1097/00007890-200112150-00001. [DOI] [PubMed] [Google Scholar]
- 3.Hertz MI, Aurora P, Christie JD, Dobbels F, Edwards LB, Kirk R, Kucheryavaya AY, Rahmel AO, Rowe AW, Taylor DO. Registry of the International Society for Heart and Lung Transplantation: a quarter century of thoracic transplantation. J Heart Lung Transplant. 2008;27:937–942. doi: 10.1016/j.healun.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 4.Heng D, Sharples LD, McNeil K, Stewart S, Wreghitt T, Wallwork J. Bronchiolitis obliterans syndrome: incidence, natural history, prognosis, and risk factors. J Heart Lung Transplant. 1998;17:1255–1263. [PubMed] [Google Scholar]
- 5.Stewart KC, Patterson GA. Current trends in lung transplantation. Am J Transplant. 2001;1:204–210. doi: 10.1034/j.1600-6143.2001.001003204.x. [DOI] [PubMed] [Google Scholar]
- 6.Trulock EP. Lung transplantation. Am J Respir Crit Care Med. 1997;155:789–818. doi: 10.1164/ajrccm.155.3.9117010. [DOI] [PubMed] [Google Scholar]
- 7.Kingsley CI, Nadig SN, Wood KJ. Transplantation tolerance: lessons from experimental rodent models. Transpl Int. 2007;20:828–841. doi: 10.1111/j.1432-2277.2007.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lechler RI, Sykes M, Thomson AW, Turka LA. Organ transplantation—how much of the promise has been realized? Nat Med. 2005;11:605–613. doi: 10.1038/nm1251. [DOI] [PubMed] [Google Scholar]
- 9.Li XC, Strom TB, Turka LA, Wells AD. T cell death and transplantation tolerance. Immunity. 2001;14:407–416. doi: 10.1016/s1074-7613(01)00121-2. [DOI] [PubMed] [Google Scholar]
- 10.Long E, Wood KJ. Regulatory T cells in transplantation: transferring mouse studies to the clinic. Transplantation. 2009;88:1050–1056. doi: 10.1097/TP.0b013e3181bb7913. [DOI] [PubMed] [Google Scholar]
- 11.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 12.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 13.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 14.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 15.Sánchez-Fueyo A, Weber M, Domenig C, Strom TB, Zheng XX. Tracking the immunoregulatory mechanisms active during allograft tolerance. J Immunol. 2002;168:2274–2281. doi: 10.4049/jimmunol.168.5.2274. [DOI] [PubMed] [Google Scholar]
- 16.Taylor PA, Noelle RJ, Blazar BR. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via co-stimulatory blockade. J Exp Med. 2001;193:1311–1318. doi: 10.1084/jem.193.11.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng XX, Sanchez-Fueyo A, Domenig C, Strom TB. The balance of deletion and regulation in allograft tolerance. Immunol Rev. 2003;196:75–84. doi: 10.1046/j.1600-065x.2003.00089.x. [DOI] [PubMed] [Google Scholar]
- 18.Starzl TE, Murase N, Abu-Elmagd K, Gray EA, Shapiro R, Eghtesad B, Corry RJ, Jordan ML, Fontes P, Gayowski T, et al. Tolerogenic immunosuppression for organ transplantation. Lancet. 2003;361:1502–1510. doi: 10.1016/s0140-6736(03)13175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claas FH. Towards clinical transplantation tolerance. Lancet. 2003;361:1489–1490. doi: 10.1016/S0140-6736(03)13220-5. [DOI] [PubMed] [Google Scholar]
- 20.Belperio JA, Keane MP, Burdick MD, Lynch JP, III, Zisman DA, Xue YY, Li K, Ardehali A, Ross DJ, Strieter RM. Role of CXCL9/CXCR3 chemokine biology during pathogenesis of acute lung allograft rejection. J Immunol. 2003;171:4844–4852. doi: 10.4049/jimmunol.171.9.4844. [DOI] [PubMed] [Google Scholar]
- 21.Medoff BD, Seung E, Wain JC, Means TK, Campanella GS, Islam SA, Thomas SY, Ginns LC, Grabie N, Lichtman AH, et al. BLT1-mediated T cell trafficking is critical for rejection and obliterative bronchiolitis after lung transplantation. J Exp Med. 2005;202:97–110. doi: 10.1084/jem.20042481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabinowich H, Zeevi A, Paradis IL, Yousem SA, Dauber JH, Kormos R, Hardesty RL, Griffith BP, Duquesnoy RJ. Proliferative responses of bronchoalveolar lavage lymphocytes from heart-lung transplant patients. Transplantation. 1990;49:115–121. doi: 10.1097/00007890-199001000-00026. [DOI] [PubMed] [Google Scholar]
- 23.Luster AD. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 24.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 25.Luster AD, Tager AM. T-cell trafficking in asthma: lipid mediators grease the way. Nat Rev Immunol. 2004;4:711–724. doi: 10.1038/nri1438. [DOI] [PubMed] [Google Scholar]
- 26.Medoff BD, Thomas SY, Luster AD. T cell trafficking in allergic asthma: the ins and outs. Annu Rev Immunol. 2008;26:205–232. doi: 10.1146/annurev.immunol.26.021607.090312. [DOI] [PubMed] [Google Scholar]
- 27.el-Sawy T, Fahmy NM, Fairchild RL. Chemokines: directing leukocyte infiltration into allografts. Curr Opin Immunol. 2002;14:562–568. doi: 10.1016/s0952-7915(02)00382-5. [DOI] [PubMed] [Google Scholar]
- 28.Agostini C, Calabrese F, Rea F, Facco M, Tosoni A, Loy M, Binotto G, Valente M, Trentin L, Semenzato G. Cxcr3 and its ligand CXCL10 are expressed by inflammatory cells infiltrating lung allografts and mediate chemotaxis of T cells at sites of rejection. Am J Pathol. 2001;158:1703–1711. doi: 10.1016/S0002-9440(10)64126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancock WW. Chemokine receptor-dependent alloresponses. Immunol Rev. 2003;196:37–50. doi: 10.1046/j.1600-065x.2003.00084.x. [DOI] [PubMed] [Google Scholar]
- 30.Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD. Donor-derived IP-10 initiates development of acute allograft rejection. J Exp Med. 2001;193:975–980. doi: 10.1084/jem.193.8.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Kaptanoglu L, Haddad W, Ivancic D, Alnadjim Z, Hurst S, Tishler D, Luster AD, Barrett TA, Fryer J. Donor T cell activation initiates small bowel allograft rejection through an IFN-gamma-inducible protein-10-dependent mechanism. J Immunol. 2002;168:3205–3212. doi: 10.4049/jimmunol.168.7.3205. [DOI] [PubMed] [Google Scholar]
- 32.Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, Gerard C. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miura M, Morita K, Kobayashi H, Hamilton TA, Burdick MD, Strieter RM, Fairchild RL. Monokine induced by IFN-gamma is a dominant factor directing T cells into murine cardiac allografts during acute rejection. J Immunol. 2001;167:3494–3504. doi: 10.4049/jimmunol.167.6.3494. [DOI] [PubMed] [Google Scholar]
- 34.Medoff BD, Wain JC, Seung E, Jackobek R, Means TK, Ginns LC, Farber JM, Luster AD. CXCR3 and its ligands in a murine model of obliterative bronchiolitis: regulation and function. J Immunol. 2006;176:7087–7095. doi: 10.4049/jimmunol.176.11.7087. [DOI] [PubMed] [Google Scholar]
- 35.Kwun J, Hazinedaroglu SM, Schadde E, Kayaoglu HA, Fechner J, Hu HZ, Roenneburg D, Torrealba J, Shiao L, Hong X, et al. Unaltered graft survival and intragraft lymphocytes infiltration in the cardiac allograft of Cxcr3-/- mouse recipients. Am J Transplant. 2008;8:1593–1603. doi: 10.1111/j.1600-6143.2008.02250.x. [DOI] [PubMed] [Google Scholar]
- 36.Zerwes HG, Li J, Kovarik J, Streiff M, Hofmann M, Roth L, Luyten M, Pally C, Loewe RP, Wieczorek G, et al. The chemokine receptor Cxcr3 is not essential for acute cardiac allograft rejection in mice and rats. Am J Transplant. 2008;8:1604–1613. doi: 10.1111/j.1600-6143.2008.02309.x. [DOI] [PubMed] [Google Scholar]
- 37.Thomas SY, Hou R, Boyson JE, Means TK, Hess C, Olson DP, Strominger JL, Brenner MB, Gumperz JE, Wilson SB, Luster AD. CD1d-restricted NKT cells express a chemokine receptor profile indicative of Th1-type inflammatory homing cells. J Immunol. 2003;171:2571–2580. doi: 10.4049/jimmunol.171.5.2571. [DOI] [PubMed] [Google Scholar]
- 38.Eksteen B, Miles A, Curbishley SM, Tselepis C, Grant AJ, Walker LS, Adams DH. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J Immunol. 2006;177:593–603. doi: 10.4049/jimmunol.177.1.593. [DOI] [PubMed] [Google Scholar]
- 39.Vanbervliet B, Bendriss-Vermare N, Massacrier C, Homey B, de Bouteiller O, Brière F, Trinchieri G, Caux C. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J Exp Med. 2003;198:823–830. doi: 10.1084/jem.20020437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grabie N, Delfs MW, Westrich JR, Love VA, Stavrakis G, Ahmad F, Seidman CE, Seidman JG, Lichtman AH. IL-12 is required for differentiation of pathogenic CD8+ T cell effectors that cause myocarditis. J Clin Invest. 2003;111:671–680. doi: 10.1172/JCI16867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medoff BD, Sauty A, Tager AM, Maclean JA, Smith RN, Mathew A, Dufour JH, Luster AD. IFN-gamma-inducible protein 10 (CXCL10) contributes to airway hyperreactivity and airway inflammation in a mouse model of asthma. J Immunol. 2002;168:5278–5286. doi: 10.4049/jimmunol.168.10.5278. [DOI] [PubMed] [Google Scholar]
- 43.Belperio JA, Keane MP, Burdick MD, Lynch JP, III, Xue YY, Li K, Ross DJ, Strieter RM. Critical role for CXCR3 chemokine biology in the pathogenesis of bronchiolitis obliterans syndrome. J Immunol. 2002;169:1037–1049. doi: 10.4049/jimmunol.169.2.1037. [DOI] [PubMed] [Google Scholar]
- 44.Campanella GS, Medoff BD, Manice LA, Colvin RA, Luster AD. Development of a novel chemokine-mediated in vivo T cell recruitment assay. J Immunol Methods. 2008;331:127–139. doi: 10.1016/j.jim.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 46.Nelson PJ, Krensky AM. Chemokines, chemokine receptors, and allograft rejection. Immunity. 2001;14:377–386. doi: 10.1016/s1074-7613(01)00118-2. [DOI] [PubMed] [Google Scholar]
- 47.Kurts C, Sutherland RM, Davey G, Li M, Lew AM, Blanas E, Carbone FR, Miller JF, Heath WR. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc Natl Acad Sci USA. 1999;96:12703–12707. doi: 10.1073/pnas.96.22.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hänninen A, Nurmela R, Maksimow M, Heino J, Jalkanen S, Kurts C. Islet beta-cell-specific T cells can use different homing mechanisms to infiltrate and destroy pancreatic islets. Am J Pathol. 2007;170:240–250. doi: 10.2353/ajpath.2007.060142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melter M, Exeni A, Reinders MEJ, Fang JC, McMahon G, Ganz P, Hancock WW, Briscoe DM. Expression of the chemokine receptor CXCR3 and its ligand IP-10 during human cardiac allograft rejection. Circulation. 2001;104:2558–2564. doi: 10.1161/hc4601.098010. [DOI] [PubMed] [Google Scholar]
- 50.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uppaluri R, Sheehan KC, Wang L, Bui JD, Brotman JJ, Lu B, Gerard C, Hancock WW, Schreiber RD. Prolongation of cardiac and islet allograft survival by a blocking hamster anti-mouse CXCR3 monoclonal antibody. Transplantation. 2008;86:137–147. doi: 10.1097/TP.0b013e31817b8e4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas SY, Banerji A, Medoff BD, Lilly CM, Luster AD. Multiple chemokine receptors, including CCR6 and CXCR3, regulate antigen-induced T cell homing to the human asthmatic airway. J Immunol. 2007;179:1901–1912. doi: 10.4049/jimmunol.179.3.1901. [DOI] [PubMed] [Google Scholar]
- 53.Sauty A, Colvin RA, Wagner L, Rochat S, Spertini F, Luster AD. CXCR3 internalization following T cell-endothelial cell contact: preferential role of IFN-inducible T cell alpha chemoattractant (CXCL11) J Immunol. 2001;167:7084–7093. doi: 10.4049/jimmunol.167.12.7084. [DOI] [PubMed] [Google Scholar]
- 54.Menning A, Höpken UE, Siegmund K, Lipp M, Hamann A, Huehn J. Distinctive role of CCR7 in migration and functional activity of naive- and effector/memory-like Treg subsets. Eur J Immunol. 2007;37:1575–1583. doi: 10.1002/eji.200737201. [DOI] [PubMed] [Google Scholar]
- 55.Schnickel GT, Bastani S, Hsieh GR, Shefizadeh A, Bhatia R, Fishbein MC, Belperio J, Ardehali A. Combined CXCR3/CCR5 blockade attenuates acute and chronic rejection. J Immunol. 2008;180:4714–4721. doi: 10.4049/jimmunol.180.7.4714. [DOI] [PubMed] [Google Scholar]
- 56.Kang SM, Tang Q, Bluestone JA. CD4+CD25+ regulatory T cells in transplantation: progress, challenges and prospects. Am J Transplant. 2007;7:1457–1463. doi: 10.1111/j.1600-6143.2007.01829.x. [DOI] [PubMed] [Google Scholar]
- 57.Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol. 2007;7:875–888. doi: 10.1038/nri2189. [DOI] [PubMed] [Google Scholar]
- 58.McLachlan JB, Catron DM, Moon JJ, Jenkins MK. Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity. 2009;30:277–288. doi: 10.1016/j.immuni.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest. 2004;114:1398–1403. doi: 10.1172/JCI23238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172:6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- 61.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 63.Chappert P, Leboeuf M, Rameau P, Lalfer M, Desbois S, Liblau RS, Danos O, Davoust JM, Gross DA. Antigen-specific Treg impair CD8(+) T-cell priming by blocking early T-cell expansion. Eur J Immunol. 2010;40:339–350. doi: 10.1002/eji.200839107. [DOI] [PubMed] [Google Scholar]
- 64.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 66.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O′Brien S, Blank R, Lamb E, Natarajan S, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.