

Abstract
Preeclampsia is a placental disease characterized by hypertension and proteinuria in pregnant women, and it is associated with a high maternal and neonatal morbidity. However, circulating biomarkers that are able to predict the prognosis of preeclampsia are lacking. Thirty-eight women were included in the current study. They consisted of 19 patients with preeclampsia (13 with severe preeclampsia and 6 with non-severe preeclampsia) and 19 gestational age-matched women with normal pregnancies as controls. We measured circulating factors that are associated with the coagulation pathway (including fibrinogen, fibronectin, factor VIII, antithrombin, protein S and protein C), endothelial activation (such as soluble endoglin and CD146), and the release of total and platelet-derived microparticles. These markers enabled us to discriminate the preeclampsia condition from a normal pregnancy but were not sufficient to distinguish severe from non-severe preeclampsia. We then used a microarray to study the transcriptional signature of blood samples. Preeclampsia patients exhibited a specific transcriptional program distinct from that of the control group of women. Interestingly, we also identified a severity-related transcriptional signature. Functional annotation of the upmodulated signature in severe preeclampsia highlighted two main functions related to “ribosome” and “complement”. Finally, we identified 8 genes that were specifically upmodulated in severe preeclampsia compared with non-severe preeclampsia and the normotensive controls. Among these genes, we identified VSIG4 as a potential diagnostic marker of severe preeclampsia. The determination of this gene may improve the prognostic assessment of severe preeclampsia.
Introduction
Preeclampsia (PE) is a placental disease characterized by the onset of hypertension and proteinuria after 20 weeks of gestation [1]. PE occurs in 2-8% of pregnancies [2] and is a leading cause of feto-maternal morbidity and mortality [1]. The clinical presentations of PE are heterogeneous, which makes effective treatment difficult. Two nonexclusive theories account for the pathophysiology of PE. The first emphasizes the vascular aspect of the pathophysiology, including placental hypoxia, enhanced platelet aggregation and endothelial dysfunction. Endothelial dysfunction is believed to persist after delivery, thus explaining maternal and neonatal complications. The second theory focuses on immunological dysfunction, whereby fetal tissue is involved in the onset and duration of the clinical presentation [3,4]. It is likely that PE encompasses a combination of genetic, immune and biological aspects, making it a multifactorial disease. This characteristic explains the difficulty in identifying specific biomarkers and designing efficient therapies. Indeed, numerous circulating factors have been investigated, but none have been identified as consistent biomarkers [5–8]. After promising initial results, recent advances have shown that angiogenic factors, such as soluble endoglin (sEndoglin), soluble vascular endothelial growth factor receptor (sVEGF-R, also known as sFLT1) and placental growth factor (PlGF) are not predictive of early PE [9–11]. Microparticles have been associated with recurrent miscarriages and PE and have been evaluated for their diagnostic properties [12,13]. An approach combining the clinical and biological features of PE has a better predictive value, likely reflecting the multifactorial origin of PE [14].
The development of new high-throughput methods enables the identification of novel biomarkers and increases the probability of their use in clinical practice [15]. Many studies have applied microarray analyses of placental biopsies to determine potential biomarkers of PE [16]. The work has revealed gene expression differences in placentas from PE women compared with normotensive (NT) women [17–21]. These changes have a relative predictive value because dysregulated gene expression in the early placenta (samples were obtained from the chorionic villous) is observed 6 months before the development of PE [19]. The functional annotation of the modulated genes in PE shows an enrichment in inflammatory immune and metabolic categories [17]. Some of the gene products have been identified as potential biomarkers of PE, including leptin [17], endoglin pathway genes [22], platelet-derived growth factor (PDGF) [21], VEGF and its soluble receptor [23], and metalloprotease (MMP)-12 [24]. However, the invasive sampling approach precludes its routine use. Three studies have focused on the analysis of the transcriptome in blood samples taken from a limited number of patients with PE. These studies demonstrated the modulation of some genes, but only a few were shared, confirming that larger cohorts of patients are required to verify these results [25,26].
In the current study, we showed that circulating factors associated with the coagulation pathway, endothelial activation and microparticle release were modulated in PE compared with NT controls, but these factors did not discriminate severe and non-severe PE. We therefore used a microarray approach to further distinguish severe and non-severe PE. Overall, the pattern of gene expression was distinct between PE and control women, and the functional annotation of the upmodulated signature in severe PE highlighted functions related to “ribosome” and “complement”. A subanalysis showed that 8 genes, including the VSIG4 gene, were specifically upmodulated in severe PE. This gene may serve as a potential diagnostic marker of severe PE and improve the prognostic assessment of severe PE.
Materials and Methods
Patients
The study was approved by the Ethics Committee "CPP Sud Méditerranée 1" (n° 2010-A00633-36, on 05/11/2010). Written informed consent was obtained from each pregnant woman. Thirty-eight women were included in the study: 19 patients with PE, including 6 women with non-severe PE and 13 women with severe PE, and 19 NT women selected according to age, weight, smoking status, race, gestational age at the inclusion and blood pH (Table 1 ). NT women had no history of medical illness or use of medication and received routing prenatal care. The diagnosis of PE was based on a blood pressure of ≥ 140/90 mmHg taken twice, uricemia, and a positive proteinuria either on urine stick or an urine sample, occurring after 20 gestational weeks in previously normotensive women (Table 2 ). Following the ACOG definition [27], severe PE was defined by the presence of one or more of the following conditions: a blood pressure higher than 160/110 mmHg, a proteinuria higher than 5000 mg/24h, a multisystem disorder, maternal cerebral symptoms (seizures, stroke) or an intrauterine growth restriction below the 3rd percentile. Women with multiple gestations, fetal congenital malformations/chromosomal abnormalities, recent infection, antiphospholipid antibodies, trauma, drug or alcohol abuse during pregnancy, preexisting hypertension, thrombophilia with a history of PE or receiving anticoagulant or anti-aggregation therapy were excluded from the study.
Table 1. Clinical characteristics of patients.
| Controls (n = 19) | Non severe PE (n = 6) | Severe PE (n = 13) | p | |
|---|---|---|---|---|
| Maternal age (years) | 28 [26-32] | 36 [33-37] | 30 [27-34] | 0.17 |
| Weight (kg) | 70 [64-83] | 77 [72-81] | 78 [66-86] | 0.60 |
| Smoking | 1 (5) | 1 (17) | 1 (8) | 1 |
| Previous PE | 0 | 0 | 3 (23) | 0.52 |
| Caucasian race | 9 (47) | 4 (67) | 9 (69) | 1 |
| Gestity / Parity | G2/P1 [1/0 - 3/2] | G3/P1 [2/0 - 4/2] | 1 | |
| Gestational age at inclusion | 36 [33-37] | 34 [32-36] | 34 [31-35] | 0.79 |
| Gestational age at birth | 40 [39-40]** | 36 [35-38] | 34 [31-36] | 0.12 |
| Birth weight (g) | 3240 [2990-3620]** | 1805 [1495-1930] | 1900 [1060-2120] | 0.92 |
| Blood pH at birth | 7.32 [7.25-7.34] | 7.29 [7.21-7.30] | 7.33 [7.28-7.36] | 0.12 |
The values were expressed as median and interquartile range (in brackets) or absolute count and percentages (in parentheses). The p values correspond to the comparison between non-severe and severe PE. The asterisks correspond to the comparison between controls and PE patients independently of the severity of the disease. *p < 0.01; **p < 0.001.
Table 2. Biological characteristics of patients.
| Controls (n = 19) | Non severe PE (n = 6) | Severe PE (n = 13) | p-value | |
|---|---|---|---|---|
| Proteinuria (g/24h) | 0** | 0.38 [0.20-0.38] | 1.00 [0.53-3.00] | 0.02 |
| Uricemia (µmol/L) | 277 [196-292]* | 365 [289-384] | 336 [286-382] | 0.78 |
| Platelets (109/L) | 236 [212-259] | 209 [181-258] | 259 [229-347] | 0.22 |
| Neutrophils (109/L) | 8.2 [7.0-10.3] | 6.7 [5.3-7.0] | 9.2 [5.3-13.3] | 0.10 |
| Lymphocytes (109/L) | 2.2 [1.6-2.4] | 2.2 [1.7-2.5] | 2.1 [1.3-2.9] | 0.86 |
| Monocytes (109/L) | 0.6 [0.6-0.8] | 0.5 [0.4-0.5] | 0.6 [0.5-0.7] | 0.28 |
| ASAT (UI/L) | NA | 24 [22-27] | 21 [19-24] | 0.15 |
| ALAT (UI/L) | NA | 17 [11-21] | 16 [14-19] | 0.90 |
| LDH (UI/L) | NA | 457 [310-506] | 386 [204-540] | 1 |
The values were expressed as median and interquartile range (in brackets). As inclusion criteria for proteinuria was based on a positive urinary stick or an urine sample, one patient from the non-severe group displayed a low value on the 24h collection time proteinuria (0.13g/24h). The asterisks correspond to the comparison between controls and PE independently of the severity of the disease. The p values correspond to the comparison between non-severe and severe PE. *p < 0.01; **p < 0.001. ASAT: aspartate amino transferase; ALAT: alanine amino transferase ; LDH: lactate dehydrogenase. NA: not available.
Circulating angiogenic factors
The angiogenic factors sFLT1, sEndoglin and PlGF were measured in citrate plasma using commercially available ELISA kits (R&D Systems, Lille, France). Soluble CD146 (sCD146) was measured using an ELISA kit from Biocytex (Marseille, France).
Microparticles
Total (Annexin V+) and platelet-derived (CD41+) microparticles were measured by flow cytometry as previously described [28]. Briefly, whole blood was drawn into sodium citrated tubes and centrifuged within 2 hours of collection at 1,500 x g for 15 min. Residual platelets were removed by centrifugation at 13,000 x g for 2 min, and plasma containing microparticles was stored at -80°C until analysis. After thawing, 30 µL of the plasma samples was incubated for 30 min with Annexin V-FITC and CD41-PE or isotype controls (IgG1-PE) obtained from Beckman Coulter (Villepinte, France). Microparticles were measured on an FC500 flow cytometer from Beckman Coulter using calibrated Megamix beads (Biocytex). Flow Count Beads (Beckman Coulter) were added to each sample to express the microparticle counts as absolute numbers.
Microarrays
Venous blood samples (2.5 mL) were collected at the time of study inclusion in PAXgene Blood RNA tubes (PreAnalytiX, Hombrechtikon, Switzerland) and were kept at room temperature for 2 hours before freezing. RNA was extracted using the PAXgene Blood RNA kit (Qiagen, Courtaboeuf, France) with a DNase I step included to eliminate DNA contaminants according to the manufacturer’s recommendations. The quantity and quality of the RNA were assessed using a Nanodrop (Thermo Science, Orsay, France) and a 2100 Bioanalyseur (Agilent Technologies, Massy, France), respectively. The microarray study was performed using microarray chips that included 45,000 probes (1 microarray for each patient, 4X44K Whole Human Genome microarray G4112F) and the One Color Microarray Based Gene Expression Analysis based on the Agilent Technologies procedures recently described [29]. In brief, 400 ng of RNA was labeled with cyanine-3 CTP using the quick amp labeling kit (Agilent Technologies, Massy, France). The hybridization was performed for 17 hours at 65°C using the gene expression hybridization kit (Agilent Technologies, Massy, France). The microarray slides were scanned with a G2505C DNA Microarray scanner at a resolution of 5 µm. The scanned images were analyzed using the Feature Extraction 10.5.1 software.
Microarray analysis
Analysis of the microarray data was performed, and the figures were created using the R (v.2.13) and Bioconductor software suite as recently described [30]. The raw data were filtered and normalized using the Agi4x44PreProcess library. Two microarrays were discarded because they did not pass the quality control check. Unsupervised and supervised analyses were performed using hierarchical clustering, principal component analysis (made4 library) [31] and the Linear Models for Microarray Analysis (limma library) [32]. We considered genes to be differentially expressed when the adjusted p value was below 0.05, and the absolute fold change (FC) was higher than 1.5. The average linkage was based on the Pearson correlation distance. Functional enrichment analysis was performed on selected genes with the DAVID bioinformatics tool using Gene Ontology (GO) pathways. Keywords were selected when the Benjamini-Hochberg-corrected p-value for enrichment was less than 0.01. The data were generated in compliance with the MIAME guidelines and were deposited in the National Center for Biotechnology Information's Gene Expression Omnibus (accession number: GSE48424).
Quantitative real-time RT-PCR
Potential severity-associated genes identified by the microarray were selected and validated by quantitative real-time RT-PCR (qRT-PCR) as previously described [29]. Reverse transcription of 100 ng of RNA was performed with the M-MLV-RT kit (Invitrogen, Life Technologies, Saint-Aubin, France). cDNA was obtained using oligo(dT) primers and the M-MLV reverse transcriptase, and quantitative PCR was performed using the SYBR Green Fast Master Mix (Roche Diagnostics, Meylan, France) and an ABI7900 Fast Real-Time PCR machine (Life Technologies). The primers were designed using Primer3 [33] and their sequences are presented in Table S1 . The results were normalized using the housekeeping gene β-actin and were expressed as FC = 2-∆∆Ct, where ∆∆Ct = (CtTarget-CtActin)PE - (CtTarget-CtActin)NT control as previously described [34].
Statistical analysis
Quantitative parameters were expressed as the median and inter-quartile range. Qualitative parameters were expressed as absolute counts and percentages. Differences between groups were assessed with the nonparametric Wilcoxon rank sum test. Differences were considered as significant when the p value was lower than 0.05.
Results
Characterization of PE patients
We included 19 PE women matched with 19 NT women in the study. The diagnosis of PE was based on changes in blood pressure, proteinuria and increased uricemia (Table 2 ). We first analyzed the clinical and biological features known to be associated with PE. The maternal age, weight and gestational age at inclusion were similar between the two groups of patients. As expected, the gestational age at birth and the birth weight were significantly (p < 0.001) lower in PE women than in NT controls (Table 1 ). Among the biological parameters, we did not find any differences in circulating leukocytes and platelets (Table 2 ). The PE patients were divided into 2 subgroups according to the severity of the disease (6 and 13 patients with non-severe and severe PE, respectively). A comparison of the two groups revealed that proteinuria was higher in the severe PE than in the non-severe PE patients (1.00 [0.53-3.00] g/24h vs. 0.38 [0.20-0.38]; p = 0.02), but that other clinical (Table 1 ) and biological (Table 2 ) parameters were similar between these patients.
Measurement of potential biomarkers in PE patients
We measured biological parameters associated with either the vascular or inflammatory hypothesis and biomarkers previously identified in placentas from PE patients in circulating blood. The circulating levels or the activity of molecules involved in coagulation or fibrinolysis, such as fibrinogen, fibronectin, factor VIII, antithrombin, protein S and protein C, were similar in the PE patients and NT controls. The severity of PE did not affect the levels or activity of these molecules with the exception of protein C, which was significantly higher (p < 0.02) in severe PE than in non-severe PE but without clinical significance (111 [106-116] % in severe PE vs. 93 [81-99] % in non-severe PE). We also found that potential biomarkers, such as sEndoglin and sCD146, were not significantly modulated in PE patients. In contrast, PlGF was significantly downmodulated in PE patients (63 [50-88] pg/mL vs. 225 [104-282]; p < 0.001), independently of the degree of severity. Finally, microparticle levels (total and platelet-derived microparticles) were increased in PE patients compared with NT controls, but the levels were similar between the severe and non-severe PE patients (Table 3 ). Taken together, our results demonstrate that PlGF is the only diagnostic biomarker that is significantly modulated between PE patients and NT controls. However, none of these biomarkers is able to assess the severity of PE.
Table 3. Potential circulating biomarkers of PE patients.
| NT controls | non-severe PE | severe PE | p value | |
|---|---|---|---|---|
| Fibrinogen (g/L) | 4.82 [4.21-5.86] | 4.94 [4.87-5.12] | 4.42 [4.27-5.34] | 0.52 |
| Fibronectin (g/L) | NA | 0.54 [0.36-0.73] | 0.61 [0.59-0.71] | 0.46 |
| Factor VIII activity (%) | 2.0 [1.6-2.2] | 2.4 [1.6-3.0] | 2.4 [1.8-2.5] | 1 |
| Antithrombin (%) | 94 [88-99] | 82 [81-86] | 91 [86-93] | 0.27 |
| Protein S (%) | 48 [40-63] | 66 [60-69] | 52 [42-62] | 0.12 |
| Protein C (%) | 98 [90-123] | 93 [81-99] | 111 [106-116] | 0.02 |
| sEndoglin (ng/mL) | 4.8 [3.7-7.9]* | 25.5 [14.0-31.2] | 20.8 [11.2-30.8] | 0.79 |
| sCD146 (ng/mL) | 159 [154-191] | 182 [177-187] | 149 [140-169] | 0.25 |
| sFLT1 (pg/mL) | 2815 [1640-21980]* | 13010 [8633-18257] | 12367 [10871-13280] | 0.85 |
| PlGF (pg/mL) | 225 [104-282]** | 61 [54-73] | 75 [44-91] | 0.52 |
| Total MP (number/µL) | 595 [360-755]* | 979 [599-1205] | 1316 [812-1415] | 0.29 |
| Platelet-derived MP (number/µL) | 515 [267-761]* | 706 [474-1152] | 947 [863-1352] | 0.19 |
The values were expressed as median and interquartile range (in brackets). The p values correspond to the comparison between non-severe and severe PE. The asterisks correspond to the comparison between controls and PE independently of the severity of the disease. *p < 0.05; **p < 0.001. MP: microparticles; sFLT1: soluble vascular endothelial growth factor receptor; PlGF: Placental Growth Factor.
Transcriptional signature of PE patients
Because the use of single biomarkers was not sufficient to assess the degree of PE severity, we used a whole-genome microarray approach to define the peripheral blood transcriptional signature of PE. The principal component analysis showed that the overall gene expression of PE women and control patients was organized in two different groups. Note that the variance in gene expression appeared to be higher in the PE group than the control group (Figure 1A ), suggesting a relative heterogeneity of PE patients. Supervised analysis using the SAM algorithm identified 239 probes (184 genes) that were modulated in PE patients compared with control women (Figure 1B ). Among the probes modulated in PE, 161 probes (150 genes) were downmodulated and 78 (45 genes) were upmodulated (Table S2 ). The functional annotation of the upmodulated genes identified specific enriched functions such as "regulation of molecular function", “cell communication”, “intracellular signaling cascade”, “cell cycle”, “cell differentiation” and “gene expression”. Many genes were also annotated with keywords linked to key cellular functions, such as “Regulation of immune process”, “membrane organization”, “hemopoiesis”, and “cell death” (Table 4 ). Taken together, these results show that PE patients exhibit a specific transcriptional program compared with NT controls.
Figure 1. Preeclampsia related transcriptional signature.
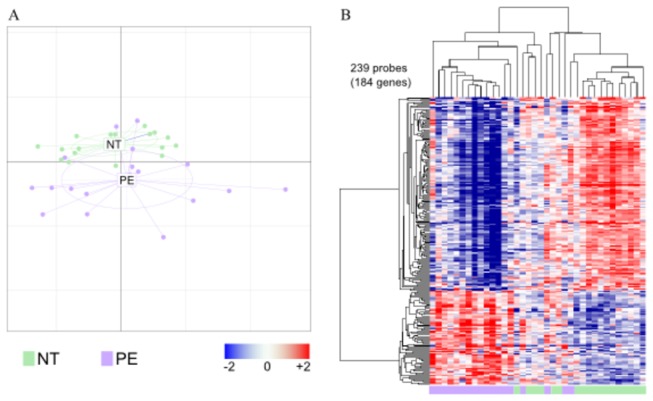
RNA was extracted from whole blood and microarrays were performed. A, The impact of the PE disease was analyzed by principal component analysis. The distance along each axis represents the degree of variance in gene expression. B, Supervised analysis using linear models identified 239 probes (184 genes) modulated in preeclampsia (PE, in purple) patients compared with normotensive (NT, in green) patients. These modulated probes are represented as a heatmap with probes in rows and samples in columns. Dendrograms show the results of the hierarchical clustering of the probes (left) and samples (top). Gene expression is represented by a color gradient ranging from blue (downmodulated genes) to red (upmodulated genes).
Table 4. Functional annotation of upmodulated genes in PE.
| GO ID | GO term | Number of associated genes | p value |
|---|---|---|---|
| 0065009 | regulation of molecular function | 19 | 1.7 × 10-3 |
| 0007154 | cell communication | 59 | 3.2 × 10-3 |
| 0007049 | cell cycle | 17 | 2.8 × 10-3 |
| 0030154 | cell differentiation | 21 | 5.9 × 10-2 |
| 0030029 | actin-filament based process | 8 | 8.3 × 10-3 |
| 0050896 | response to stimulus | 29 | 6.6 × 10-1 |
| 0042060 | wound healing | 5 | 2.1 × 10-2 |
| 0009893 | positive regulation of metabolic process | 25 | 1.1 × 10-8 |
| 0008219 | cell death | 17 | 2.2 × 10-4 |
| 0006950 | response to stress | 20 | 1.3 × 10-1 |
| 0016044 | membrane organization | 12 | 8.4 × 10-4 |
| 0007242 | intracellular signalling cascade | 35 | 1.4 × 10-6 |
| 0007165 | signal transduction | 56 | 1.7 × 10-3 |
| 0002682 | regulation of immune system process | 7 | 4.5 × 10-2 |
| 0030097 | hemopoiesis | 8 | 2.0 × 10-3 |
| 0010467 | gene expression | 60 | 6.5 × 10-9 |
The genes that were specifically upmodulated in PE compared to NT controls were annotated using GO database. The number of modulated genes associated with a particular term is indicated. The p value was obtained after Benjamini-Hochberg correction.
Transcriptional signature of severe PE
As the variability of gene expression patterns was higher in PE patients than NT controls, we analyzed the modulation of gene expression according to PE severity to identify severity-associated biomarkers. In a supervised analysis comparing severe and non-severe PE, we found 116 probes (69 genes) that were upmodulated in severe PE (Table S3 ). Functional annotation of this signature highlighted three main functions related to “intracellular transport”, “translation” and “immune response” (Figure 2A ). The interactome map of the modulated genes associated with the “translation” cluster showed that the upregulation of many genes encoding proteins of the large ribosome subunit (RPL17, RPL23, RPL26, RPL27, RPL31 and RPL34) and involved in ribosome biogenesis (SNRPG, NPM1, EEF1B2) were up-modulated in women with severe PE (Figure 2B ). The “immune response” cluster included four genes related to the complement system (C1QA, C1QB, C4BPA, VSIG4) (Figure 2C ).
Figure 2. Functional annotation of genes modulated in severe PE.
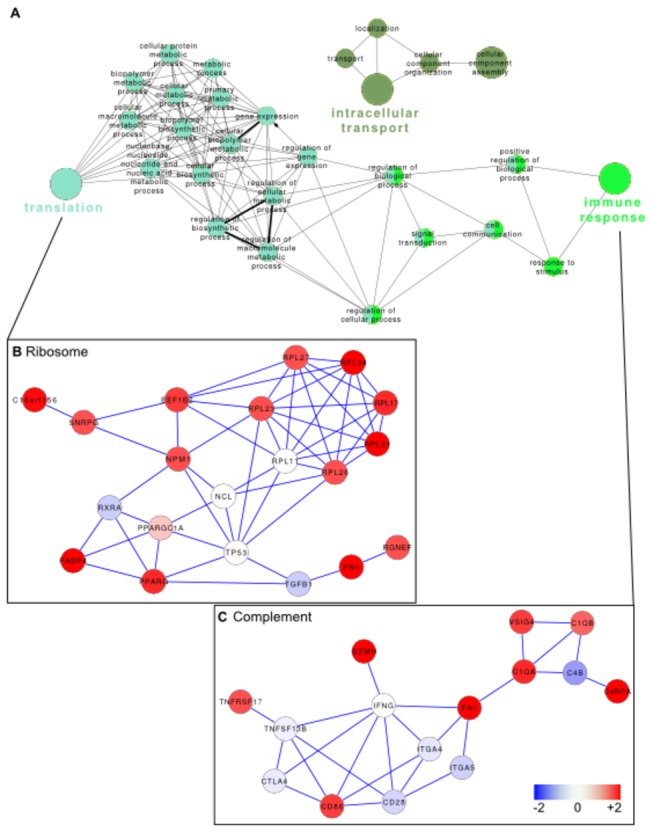
The genes that were modulated in severe PE compared with non-severe PE were analyzed using GO terms in Cytoscape. A, The network represents the relationship between the significant GO terms that were associated with the identified genes. B, An interactome map was built from the genes associated with the terms of the “ribosome” cluster, and gene expression in severe PE is color-coded from blue (downmodulated) to red (upmodulated). C, An interactome map was built from the genes associated with the terms of the “complement” cluster, and gene expression in severe PE is color-coded from blue (downmodulated) to red (upmodulated).
We then selected 8 genes that were specifically upmodulated in severe PE compared with NT controls but that were not modulated in non-severe PE (Figure 3 ). Among these genes, C1QB, C4BPA and VSIG4 genes were related to the complement system and RPL17, RPL26 and RPL34 to the ribosome pathway. Interestingly, we also identified the gene encoding the estrogen receptor (GPER) in this signature. The specific modulation of RPL26, RPL34, MACROD2 and VSIG4 genes was also confirmed by qRT-PCR (Table 5 ). The severity ratio was higher for VISG4 (severe/non-severe ratio = 8.8) than other genes. Taken together, our approach identified potential diagnostic biomarkers of PE severity.
Figure 3. Expression of potential biomarkers of severity of PE.
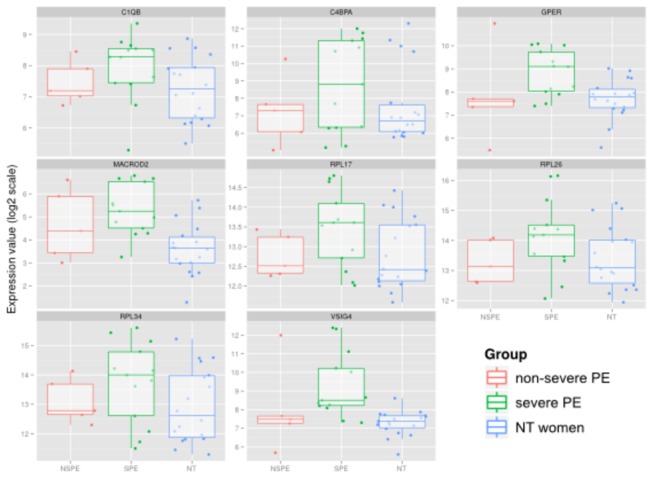
Eight genes were selected as potential biomarkers of the severity of PE. Their expression in the microarray is represented as box and whiskers, according to the disease status and severity of PE: normotensive (NT) patient (blue), non-severe PE (red) and severe PE (green). Individual results are shown with box and whisker representation.
Table 5. Analysis of severity-associated biomarkers by qRT-PCR.
| Microarray ratio |
qRT-PCR ratio |
|||
|---|---|---|---|---|
| non-severe | severe | non-severe | severe | |
| RPL26 | 1.0 | 2.1 | 2.6 | 13.2 |
| RPL34 | 1.1 | 2.6 | 1.9 | 6.2 |
| MACROD2 | 1.6 | 3.1 | 3.7 | 5.8 |
| VSIG4 | 1.1 | 2.2 | 0.9 | 7.9 |
The selected biomarkers identified in microarray were analyzed by qRT-PCR. The results were expressed relative to NT controls. The Spearman correlation coefficient was 0.6
Discussion
The objective of our study was to identify new circulating biomarkers of PE severity. To date, the most relevant serum biomarker identifying severe PE is the variation in plasma sEndoglin levels during the pregnancy trimesters [35]. A ratio between soluble FLT1 and PlGF in the first trimester may predict an evolution toward PE, but this ratio is not related to severity [36].
We chose to study circulating cells because this method is non-invasive and may be easily applied to the general population. First, we selected different biomarker candidates from the literature [9,11]. Most of them were modulated in PE independently of the degree of severity of the disease. The measurement of circulating microparticles was also evaluated. Microparticles are associated with thrombotic conditions, and their increase has been documented in women with recurrent miscarriage, PE, intrauterine growth retardation and gestational hypotension [37,38]. We found that total microparticles and platelet-derived microparticles, known to be highly thrombogenic, were clearly increased in PE compared with NT women. Again, their measurement could not discriminate severe PE from non-severe PE, thereby excluding microparticles as a potential marker of PE severity.
Microarray analysis may be an excellent technology to study a systemic disorder such as PE. Indeed, it allows the large-scale screening of biomarkers, and a peripheral transcriptional signature of PE has been reported in a limited number of patients [25]. A transcriptional signature associated with PE severity has also been described in a small cohort of patients [26]. Nevertheless, the comparison of these two microarray analyses is difficult because different microarrays or various variable filtering or statistical methods were used. In our study, we included a larger cohort of patients and matched controls. Despite a relative heterogeneity in the PE patients, we identified a disease-related signature in which 184 genes were significantly modulated, and the functional categories were related to cell proliferation, differentiation and apoptosis. Genes from the cell proliferation and apoptosis categories were also found in PE patients from Rajakumar's study [25]. Importantly, a transcriptional signature of severe PE was also identified in the current study. This signature included the overexpression of the ribosomal genes RPL26, RPL34, RPS15A and RPS26, along with genes encoding proteins involved in ribosome biogenesis, suggesting that severe PE is characterized by a selective modulation of the translational machinery. It also included GPER that encodes an oestrogen receptor. The more interesting effect of GPER in the context of PE is its effect on the cardiovascular system and regulation of blood pressure [39]. PE has been recently reported to increase the long-term risk of cardiovascular disease with increased maternal mortality, and this risk factor may be related to estrogen levels [40,41].
We also found that some genes belonging to the complement pathway were upmodulated in severe PE compared with non-severe PE. There is already accumulating evidence for a role of complement in the pathogenesis of several complications of pregnancy, including PE, intrauterine fetal death and recurrent spontaneous abortion [42,43]. The activation of complement is necessary to maintain immune homeostasis during pregnancy, but excessive or inappropriate activation of complement contributes to the pathogenesis of PE [44]. Our data suggested that molecules of the classical complement pathway were altered in PE, especially C1q and C4bp. In addition, the alteration of complement may be involved in renal lesions of PE as suggested by the aggravation of these lesions in genetic defects of complement regulatory proteins and the deposition of C1q in glomerular capillaries [45]. The deposition of C4BP along the glomerular capillary wall has also been found in PE patients [46].
Interestingly, the VSIG4 gene was specifically upmodulated in severe PE. This result is consistent with that obtained by Sun et al. who found that the VSIG4 gene was slightly overexpressed in peripheral blood mononuclear cells from patients with PE [26]. VSIG4 is a member of the B7 family that is only present on resident placental macrophages [47] and is involved in T-cell anergy [48]. VSIG4 belongs to the complement pathway due to its ability to bind C3b, C3i [49] and C1q (interactome databases). Because the VSIG4 gene is the only gene found to be modulated in PE in the transcriptional studies published to date [25,26], it represents a superior candidate biomarker to assess PE severity. Given the large variation in expression value and the overlap between the three groups in our dataset, this biomarker requires validation in an independent cohort of patients. Finally, as PAXgene tubes were used in this study, the transcriptome we analyzed comes from heterogeneous origins (mainly leukocytes). Therefore, despite interesting pathophysiological hypothesis were raised, they should be confirmed by further studies.
In the present study, we evaluated a large panel of PE biomarkers. We confirmed the diagnostic value of most of them, but they were unable to predict the severity of the disease. We completed the analysis with a transcriptional study that identified several circulating biomarkers of PE severity. One of them, VSIG4, is a potential candidate that may help physicians to manage PE patients.
Supporting Information
Nucleotide sequences of oligonucleotide primers. Oligonucleotide primers used for qRT-PCR validation.
(DOC)
Genes modulated in pre-eclampsia (severe or non severe, versus control women). List of the modulated genes in pre-eclampsia with probe IDs, gene symbols and fold-changes.
(XLS)
Genes modulated in severe pre-eclampsia versus non-severe pre-eclampsia. List of the modulated genes in severe pre-eclampsia with probe IDs, gene symbols and fold-changes. both methods was 0.6.
(XLS)
Acknowledgments
We thank Jean-François Cocallemen for data management and Dr Christian Capo for his careful reading of the manuscript.
Funding Statement
This work was funded in part by a grant from Assistance Publique – Hôpitaux de Marseille: Appel d'Offre Promotion 2010 (ref. 2010-04). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding received for this study.
References
- 1. Villar J, Say L, Gulmezoglu AM et al. (2003) Eclampsia and pre-eclampsia: a worldwide health problem for 2000 years. In: Critchley H. Pre-eclampsia. London: RCOG; pp. 189–207. [Google Scholar]
- 2. WHOICS Pregnancy of HD of (1988) Geographic variation in the incidence of hypertension in pregnancy. Am J Obstet Gynecol 158: 80–83. doi: 10.1016/0002-9378(88)90782-X. PubMed: 2962500. [DOI] [PubMed] [Google Scholar]
- 3. Redman CW, Sargent IL (2010) Immunology of pre-eclampsia. Am J Reprod Immunol 63: 534–543. doi: 10.1111/j.1600-0897.2010.00831.x. PubMed: 20331588. [DOI] [PubMed] [Google Scholar]
- 4. Eiland E, Nzerue C, Faulkner M (2012) Preeclampsia. J Pregnancy 2012: ID 586578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carty DM, Delles C, Dominiczak AF (2008) Novel biomarkers for predicting preeclampsia. Trends Cardiovasc Med 18: 186–194. doi: 10.1016/j.tcm.2008.07.002. PubMed: 18790389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Romero R, Nien JK, Espinoza J, Todem D, Fu W et al. (2008) A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J Matern Fetal Neonatal Med 21: 9–23. doi: 10.1080/14767050701830480. PubMed: 18175241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grill S, Rusterholz C, Zanetti-Dällenbach R, Tercanli S, Holzgreve W et al. (2009) Potential markers of preeclampsia--a review. Reprod Biol Endocrinol 7: 70. doi: 10.1186/1477-7827-7-70. PubMed: 19602262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hawfield A, Freedman BI (2009) Pre-eclampsia: the pivotal role of the placenta in its pathophysiology and markers for early detection. Ther. Adv - Cardiovasc Dis 3: 65–73. doi: 10.1177/1753944708097114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McElrath TF, Lim KH, Pare E, Rich-Edwards J, Pucci D et al. (2012) Longitudinal evaluation of predictive value for preeclampsia of circulating angiogenic factors through pregnancy. Am J Obstet Gynecol 207: 407: e1–e7. PubMed: 22981320. [DOI] [PubMed] [Google Scholar]
- 10. Levine RJ, Lam C, Qian C, Yu KF, Maynard SE et al. (2006) Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 355: 992–1005. doi: 10.1056/NEJMoa055352. PubMed: 16957146. [DOI] [PubMed] [Google Scholar]
- 11. Kleinrouweler CE, Wiegerinck MM, Ris-Stalpers C, Bossuyt PM, van der Post JA et al. (2012) Accuracy of circulating placental growth factor, vascular endothelial growth factor, soluble fms-like tyrosine kinase 1 and soluble endoglin in the prediction of pre-eclampsia: a systematic review and meta-analysis. BJOG 119: 778–787. doi: 10.1111/j.1471-0528.2012.03311.x. PubMed: 22433027. [DOI] [PubMed] [Google Scholar]
- 12. Germain SJ, Sacks GP, Sooranna SR, Soorana SR, Sargent IL et al. (2007) Systemic inflammatory priming in normal pregnancy and preeclampsia: the role of circulating syncytiotrophoblast microparticles. J Immunol 1950 178: 5949–5956. PubMed: 17442979. [DOI] [PubMed] [Google Scholar]
- 13. Marques FK, Campos FMF, Sousa LP, Teixeira-Carvalho A, Dusse LMS et al. (2013) Association of microparticles and preeclampsia. Mol Biol Rep 40: 4553–4559. doi: 10.1007/s11033-013-2536-0. PubMed: 23645085. [DOI] [PubMed] [Google Scholar]
- 14. Parra-Cordero M, Rodrigo R, Barja P, Bosco C, Rencoret G et al. (2013) Prediction of early and late pre-eclampsia from maternal characteristics, uterine artery Doppler and markers of vasculogenesis during first trimester of pregnancy. Ultrasound Obstet Gynecol 41: 538–544. PubMed: 22807133. [DOI] [PubMed] [Google Scholar]
- 15. Drucker E, Krapfenbauer K (2013) Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J 4: 7. doi: 10.1186/1878-5085-4-7. PubMed: 23442211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Founds SA, Shi H, Conley YP, Jeyabalan A, Roberts JM et al. (2012) Variations in Discovery-Based Preeclampsia Candidate Genes. Clin. Transl Sci 5: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reimer T, Koczan D, Gerber B, Richter D, Thiesen HJ et al. (2002) Microarray analysis of differentially expressed genes in placental tissue of pre-eclampsia: up-regulation of obesity-related genes. Mol Hum Reprod 8: 674–680. doi: 10.1093/molehr/8.7.674. PubMed: 12087083. [DOI] [PubMed] [Google Scholar]
- 18. Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H et al. (2007) Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and late onset severe pre-eclampsia. Placenta 28: 487–497. doi: 10.1016/j.placenta.2006.05.010. PubMed: 16860862. [DOI] [PubMed] [Google Scholar]
- 19. Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A, Hogge WA et al. (2009) Altered global gene expression in first trimester placentas of women destined to develop preeclampsia. Placenta 30: 15–24. doi: 10.1016/j.placenta.2008.09.018. PubMed: 19027158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Enquobahrie DA, Meller M, Rice K, Psaty BM, Siscovick DS et al. (2008) Differential placental gene expression in preeclampsia. Am J Obstet Gynecol 199: 566: e1–11. PubMed: 18533121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sitras V, Paulssen RH, Grønaas H, Leirvik J, Hanssen TA et al. (2009) Differential placental gene expression in severe preeclampsia. Placenta 30: 424–433. doi: 10.1016/j.placenta.2009.01.012. PubMed: 19249095. [DOI] [PubMed] [Google Scholar]
- 22. Bell MJ, Roberts JM, Founds SA, Jeyabalan A, Terhorst L et al. (2013) Variation in endoglin pathway genes is associated with preeclampsia: a case-control candidate gene association study. BMC Pregnancy Childbirth 13: 82. doi: 10.1186/1471-2393-13-82. PubMed: 23548068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rolfo A, Giuffrida D, Nuzzo AM, Pierobon D, Cardaropoli S et al. (2013) Pro-inflammatory profile of preeclamptic placental mesenchymal stromal cells: new insights into the etiopathogenesis of preeclampsia. PLOS ONE 8: e59403. doi: 10.1371/journal.pone.0059403. PubMed: 23527185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Founds SA, Terhorst LA, Conrad KP, Hogge WA, Jeyabalan A et al. (2011) Gene expression in first trimester preeclampsia placenta. Biol Res Nurs 13: 134–139. doi: 10.1177/1099800410385448. PubMed: 21044967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rajakumar A, Chu T, Handley DE, Bunce KD, Burke B et al. (2011) Maternal gene expression profiling during pregnancy and preeclampsia in human peripheral blood mononuclear cells. Placenta 32: 70–78. doi: 10.1016/j.placenta.2010.10.004. PubMed: 21075447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun C-J, Zhang L, Zhang W-Y (2009) Gene expression profiling of maternal blood in early onset severe preeclampsia: identification of novel biomarkers. J Perinat Med 37: 609–16. PubMed: 19681734. [DOI] [PubMed] [Google Scholar]
- 27. ACOG Committee on Obstetric Practice (2002) Practice bulletin 33: diagnosis and management of preeclampsia and eclampsia. Obstet Gynecol 99: 159-167. doi: 10.1016/S0029-7844(01)01747-1. PubMed: 16175681. [DOI] [PubMed] [Google Scholar]
- 28. Robert S, Poncelet P, Lacroix R, Arnaud L, Giraudo L et al. (2009) Standardization of platelet-derived microparticle counting using calibrated beads and a Cytomics FC500 routine flow cytometer: a first step towards multicenter studies? J Thromb Haemost 7: 190–197. doi: 10.1111/j.1538-7836.2008.03200.x. PubMed: 18983485. [DOI] [PubMed] [Google Scholar]
- 29. Thuny F, Textoris J, Amara AB, Filali AE, Capo C et al. (2012) The gene expression analysis of blood reveals S100A11 and AQP9 as potential biomarkers of infective endocarditis. PLOS ONE 7: e31490. doi: 10.1371/journal.pone.0031490. PubMed: 22319637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moal V, Textoris J, Ben Amara A, Mehraj V, Berland Y et al. (2013) Chronic hepatitis E virus infection is specifically associated with an interferon-related transcriptional program. J Infect Dis 207: 125–132. PubMed: 23072754. [DOI] [PubMed] [Google Scholar]
- 31. Culhane AC, Thioulouse J, Perrière G, Higgins DG (2005) MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics 21: 2789–2790. doi: 10.1093/bioinformatics/bti394. PubMed: 15797915. [DOI] [PubMed] [Google Scholar]
- 32. Smyth GK (2005) Limma: linear models for microarray data. In: Gentleman R, Carey V, Irizarry R, Huber W. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; pp. 397–420. [Google Scholar]
- 33. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R et al. (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35: W71–W74. doi: 10.1093/nar/gkm093. PubMed: 17485472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Desnues B, Raoult D, Mege JL (2005) IL-16 is critical for Tropheryma whipplei replication in Whipple’s disease. J Immunol 175: 4575–4582. PubMed: 16177102. [DOI] [PubMed] [Google Scholar]
- 35. Myatt L, Clifton R, Roberts J, Spong C, Wapner R et al. (2013) Can changes in angiogenic biomarkers between the first and second trimesters of pregnancy predict development of pre-eclampsia in a low-risk nulliparous patient population? BJOG (in press). doi: 10.1111/1471-0528.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schnettler WT, Dukhovny D, Wenger J, Salahuddin S, Ralston SJ et al. (2013) Cost and resource implications with serum angiogenic factor estimation in the triage of pre-eclampsia. BJOG (2013). doi: 10.1111/1471-0528.12259. PubMed: 23647884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bretelle F, Sabatier F, Desprez D, Camoin L, Grunebaum L et al. (2003) Circulating microparticles: a marker of procoagulant state in normal pregnancy and pregnancy complicated by preeclampsia or intrauterine growth restriction. Thromb Haemost 89: 486–492. PubMed: 12624632. [PubMed] [Google Scholar]
- 38. Shetty S, Patil R, Ghosh K (2013) Role of microparticles in recurrent miscarriages and other adverse pregnancies: a review. Eur J Obstet Gynecol Reprod Biol 169: 123–9. doi: 10.1016/j.ejogrb.2013.02.011. PubMed: 23490540. [DOI] [PubMed] [Google Scholar]
- 39. Prossnitz ER, Barton M (2009) Signaling, physiological functions and clinical relevance of the G protein-coupled estrogen receptor GPER. Prostaglandins Other Lipid Mediat 89: 89–97. doi: 10.1016/j.prostaglandins.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brown MC, Best KE, Pearce MS, Waugh J, Robson SC et al. (2013) Cardiovascular disease risk in women with pre-eclampsia: systematic review and meta-analysis. Eur J Epidemiol 28: 1–19. doi: 10.1007/s10654-013-9820-0. PubMed: 23397514. [DOI] [PubMed] [Google Scholar]
- 41. Van Rijn BB, Nijdam ME, Bruinse HW, Roest M, Uiterwaal CS et al. (2013) Cardiovascular disease risk factors in women with a history of early-onset preeclampsia. Obstet Gynecol 121: 1040–1048. doi: 10.1097/AOG.0b013e31828ea3b5. PubMed: 23635741. [DOI] [PubMed] [Google Scholar]
- 42. Soto E, Romero R, Vaisbuch E, Erez O, Mazaki-Tovi S et al. (2010) Fragment Bb: evidence for activation of the alternative pathway of the complement system in pregnant women with acute pyelonephritis. J Matern Fetal Neonatal Med 23: 1085–1090. doi: 10.3109/14767051003649870. PubMed: 20218820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lynch AM, Eckel RH, Murphy JR, Gibbs RS, West NA et al. (2012) Prepregnancy obesity and complement system activation in early pregnancy and the subsequent development of preeclampsia. Am J Obstet Gynecol 206: 428: e1–e8. PubMed: 22542119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Denny KJ, Coulthard LG, Finnell RH, Callaway LK, Taylor SM et al. (2013) Elevated complement factor C5a in maternal and umbilical cord plasma in preeclampsia. J Reprod Immunol 97: 211–216. doi: 10.1016/j.jri.2012.11.006. PubMed: 23415845. [DOI] [PubMed] [Google Scholar]
- 45. Java A, Atkinson J, Salmon J (2013) Defective complement inhibitory function predisposes to renal disease. Annu Rev Med 64: 307–324. doi: 10.1146/annurev-med-072211-110606. PubMed: 23121180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Joyama S, Yoshida T, Koshikawa M, Sawai K, Yokoi H et al. (2001) C4d and C4bp deposition along the glomerular capillary walls in a patient with preeclampsia. Am J Kidney Dis 37: E6. doi: 10.1016/S0272-6386(01)90003-4. PubMed: 11136195. [DOI] [PubMed] [Google Scholar]
- 47. Nagamatsu T, Schust DJ (2010) The immunomodulatory roles of macrophages at the maternal-fetal interface. Reprod Sci 17: 209–218. doi: 10.1177/1933719109349962. PubMed: 20065301. [DOI] [PubMed] [Google Scholar]
- 48. Vogt L, Schmitz N, Kurrer MO, Bauer M, Hinton HI et al. (2006) VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J Clin Invest 116: 2817–2826. doi: 10.1172/JCI25673. PubMed: 17016562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. He JQ, Wiesmann C, van Lookeren Campagne M (2008) A role of macrophage complement receptor CRIg in immune clearance and inflammation. Mol Immunol 45: 4041–4047. doi: 10.1016/j.molimm.2008.07.011. PubMed: 18752851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nucleotide sequences of oligonucleotide primers. Oligonucleotide primers used for qRT-PCR validation.
(DOC)
Genes modulated in pre-eclampsia (severe or non severe, versus control women). List of the modulated genes in pre-eclampsia with probe IDs, gene symbols and fold-changes.
(XLS)
Genes modulated in severe pre-eclampsia versus non-severe pre-eclampsia. List of the modulated genes in severe pre-eclampsia with probe IDs, gene symbols and fold-changes. both methods was 0.6.
(XLS)