

Abstract
Background
We previously demonstrated angiotensin converting enzymes (ACE) over-expression in a dextransodium sulfate colitis model; ACE inhibitor (ACE-I) treatment reduced colitis severity in this model. However, ACE-I has not been tested in more immunologically relevant colitis models.
Aim
We hypothesized that ACE-I would decrease disease severity in an IL-10 knockout (−/−) colitis model.
Methods
Colitis was induced by giving 10-week old IL-10−/− mice piroxicam (P.O.) for 14 days. The ACE-I enalaprilat was given transanally at a dose of 6.25 mg/kg for 21 days. Prednisolone (PSL) with or without enalaprilat were used as therapeutic, comparative groups. All groups were compared to a placebo treated group. Outcome measures were clinical course, histology, abundance of pro-inflammatory cytokines/chemokines, and epithelial barrier function.
Results
Enalaprilat exhibited better survival (91 %) versus other treatment groups (PSL: 85.7 %, PSL + ACE-I: 71.4 %, placebo: 66.6 %). The ACE-I and PSL + ACE-I groups showed significantly better histological scores versus placebo mice. ACE-I and the PSL groups significantly reduced several pro-inflammatory cytokines versus placebo mice. FITC-dextran permeability was reduced in the ACE-I and PSL + ACE-I groups. Blood pressure was not affected in ACE-I treated mice compared to placebo mice.
Conclusions
ACE-I was effective in reducing severity of colitis in an IL-10−/− model. The addition of prednisolone minimally augmented this effect. The findings suggest that appropriately dosed ACE-I with or without steroids may be a new therapeutic agent for colitis.
Keywords: Colitis, IL-10 deficient, Angiotensin, Steroid, Renin angiotensin system
Introduction
Inflammatory bowel disease (IBD) affects approximately 1 million people in the United States and 2 million people in Europe [1]. While a number of medical strategies are available, many of these have substantial side-effects including immune suppression; thus, newer approaches are greatly needed [2].
Although angiotensin converting enzymes (ACE) are commonly thought to control blood pressure, the expression of ACE and its associated receptors have been identified within the gastrointestinal mucosa [3], and their abundance has been shown to increase in states of intestinal injury and colitis in both animal models and humans [4–6]. Thus, ACE may be associated with the pathogenesis of IBD. Recently ACE has been shown to play a critical role in the gastrointestinal tract [5, 6]; whereby, ACE appears to modulate intestinal epithelial cell (IEC) growth. Based on these findings, our laboratory and others have shown that the use of ACE-inhibitors (ACE-I) suppressed intestinal epithelial cell (IEC) apoptosis and decreased expression of TNF-α [6]. We subsequently demonstrated that the use of ACE-I reduced the severity of colitis using a dextran sodium sulfate (DSS)-induced colitis model [6]. These findings are similar to other reports which demonstrated reduced colitis using a trinitrobenzene sulfonic acid (TNBS)-induced colitis model in angiotensinogen (Atg) knockout mice [7], as well as use of the ACE-I captopril to reduce the degree of fibrosis in this same colitis model [8].
DSS disrupts intestinal epithelial barrier function, most likely by increasing the abundance of medium chain fatty acids, promoting increased cellular exposure to normal mucosal microflora, and a progressive loss of IECs due to apoptosis [9, 10]. While DSS is perhaps the most commonly used agent to induce a colitis model, it has experimental deficiencies, since the model leads to a chemical injury to the gastrointestinal tract, which is opposed to a predominately immunologically-driven model. The use of the TNBS model also may have similar drawbacks and lacks a robust immunologic component. In particular, many have felt that a chemically-induced injury to the bowel fails to fully resemble the histologic manifestations of human IBD, and a shift toward examining drug actions in more immunologically-driven models has been increasingly desired. Thus, we tested whether ACE-I could prevent the severity of colitis in an IL-10 knockout (−/−) mouse model of colitis, which develop colitis in an immunologically-relevant mechanism. The spontaneous development of colitis in the proximal colon [11] will occur in IL-10 deficient mice only sporadically after several months [12]. Additionally, the severity of colitis in this model depends on both environmental conditions and genetic factors [13, 14]. Spontaneous development of colitis, as it happens in IL-10−/− mice, is an inefficient model to study potential colitis therapies. Sporadic development of colitis produces diseased animals of different ages. Since the severity of the disease in this model depends on environmental conditions, animals which develop colitis at different times will not develop the same disease. To address these problems, Berg et al. [15] reported that piroxicam (a non-steroidal anti-inflammatory drug, NSAID) treatment induced severe and consistent formation of colitis in IL-10−/− C57/BL6 mice within a few weeks post-treatment. The addition of piroxicam to the IL-10−/− mouse model allows for the study of animals with a predictable disease time course, with similar age, and under similar environmental influences, making it more desirable for treatment studies than the IL-10−/− mice alone. While the complete mechanism leading to the development of colitis in the presence of piroxicam is not fully understood, this model is very similar to human IBD in terms of cytokine expression, clinical features of weight loss and gastrointestinal bleeding, and, in particular, its histological features [16]. Further, Hale et al. [12] demonstrated that this model was also associated with a significant pro-apoptotic action, which more closely resembled human IBD than the DSS model.
In the present study, we tested the efficacy of the ACE-I enalaprilat against colitis, and evaluated it against the efficacy of a corticosteroid, the standard of care for colitis, alone or a combination of ACE-I and steroid. We hypothesized that ACE-I would decrease the severity of colitis in an IL-10−/− model to a similar degree as that achieved with steroid use. We elected to use enalaprilat given via a transanal route. This unique approach capitalized on the fact that enalaprilat is poorly absorbed via the gastrointestinal tract (as it is normally given intravenously) and is able to act directly on the increased ACE activity, which is most abundant on the surface of the gastrointestinal mucosa [17]. We demonstrated significant reduction in colitis severity with ACE-I treatment, which was superior in many aspects compared to steroid treatment. This suggests the use of ACE-I may be a novel clinical approach for the treatment of patients with IBD.
Materials and Methods
Animals
Healthy 10-week-old (body weight around 19–25 g) IL-10−/− mice with a C57BL/6 background (originally obtained from Jackson Laboratories, but now bred at University of Michigan) were used for this study. Mice were bred under high level specific pathogen-free conditions, and were fed ad libitum with rodent chow powder and water. All experiments were approved by the University Committee on Use and Care of Animals at the University of Michigan (UCUCA protocol 08773-3).
Induction of Colitis
Colitis was induced in IL-10 deficient mice by feeding NSAIDs (160 ppm piroxicam [Sigma-Aldrich, St Louis, MO]) in a powdered diet for 2 weeks. Drug treatment commenced at the start of the NSAID treatment and continued for 3 weeks.
Drug Treatment
NSAID-primed mice were distributed into five study groups (N = 7–12 mice per group): placebo control (piroxicam-primed only with transanal saline and oral PBS); enalaprilat (6.25 mg/kg/every 12 h, transanal and oral PBS); oral predonisolone (PSL; 5 mg/kg and transanal saline); and oral PSL + transanal enalaprilat. An additional naïve group was studied which consisted of age-matched IL-10−/− mice that did not receive piroxicam or drug therapy. All drugs were started at the beginning of study concurrent the NSAID, and were delivered for 21 days, every 12 h. Enalaprilat or PBS control (0.2 ml volume) was administered via the transanal route using a blunt plastic gavage needle. To maintain the drug within the entire colon lumen, mice were held in a sitting position, and the needle was held in place for 30 s after transanal injection.
Hemodynamic Assessment
The most common complication of ACE-I is hypotension. Thus, we evaluated the hemodynamic status of all mice. Systemic blood pressure via tail-cuff measurements were performed noninvasively using the CODA non-invasive blood pressure system (Kent Scientific Co, Torrington, WY) 2 h after drug administration on the final day of the experiment to allow for full systemic absorption.
Colonic mucosa and small bowel mesenteric blood flow were also measured just before harvesting tissue using laser Doppler perfusion imaging (LDPI, Perimed Inc., North Royalton, OH), as previously reported [6]. Colonic mesentery was not possible due to the very foreshortened colon mesentery of mice. Briefly, a median laparotomy was performed and the mesentery exposed to the laser Doppler beam. A 670-nm helium–neon laser beam was placed 12 cm above the mesentery. The surface of the mesentery and then the colonic mucosa (after opening the lumen up longitudinally) were sequentially scanned. Scanning areas of 8 × 10 mm and 20 × 20 mm in distal mucosal colon and mesentery, respectively, were used. The perfusion response was presented in arbitrary perfusion units (PU). As the PU values are not absolute blood flow, the magnitude of the change in blood flow perfusion was calculated as the ratio between maximum peak and baseline perfusions. Maximum, minimum, and mean percent perfusions were normalized to total pixel area. At the end of the measurements the mice were killed via CO2 asphyxiation.
Evaluation of Colitis
The clinical score was calculated using the Cooper scoring method, as previously described [18]. Clinical score was based on a measure of stool stiffness, body weight change and presence of hematochezia daily to estimate colitis severity [18]. Hematochezia was tested using Hemoccult® cards and developer (Beckman Coulter, Brea, CA). We defined a clinical score over 7 or mouse death as severe colitis. Histology was scored in a blinded fashion from H&E stained sections [19] using a modification of previously published methods [20] (Table 1). This scoring modification accounted for the greater degree of lamina propria inflammatory infiltration in the IL-10−/− model versus other chemically-induced models.
Table 1.
Histological score
| Mucosal ulceration, 0–3 | Lamina propria mononuclear infiltrate, 0–5 |
|---|---|
| 0, normal | 0, none |
| 1, surface epithelial inflammation | 1, a few cells |
| 2, erosions | 2, more cells, less aggregate |
| 3, ulcerations | 3, one small aggregate |
| 4, a few aggregates | |
| 5, large area aggregate | |
|
| |
| Epithelial hyperplasia, 0–3 | Lamina propria neutrophil infiltrate, 0–2 |
|
| |
| 0, normal | 0, normal |
| 1, mild | 1, slightly increased |
| 2, moderate | 2, markedly increased |
| 3, pseudopolyps | |
Ten fields from each mouse were scored. Means and standard deviation (SD) of the ten scores are reported. A maximum score of 13 represents the worst colitis
Collection of Tissue
After mice were euthanized, the colon was completely removed and placed in RPMI cell culture medium, and fecal contents were gently flushed out [6]. Sections (5 mm) were taken from the proximal colon, as this location represented the maximal degree of colitis formation, and were placed in 10 % neutral buffered formalin. Remaining colonic epithelium was mechanically stripped and subsequently isolated for RNA and mucosal protein as described previously [6].
Real Time-Polymerase Chain Reaction (RT-PCR)
Mucosal scrapings underwent RNA extraction and purification by RNeasy Micro Kit (Qiagen, Hilden, Germany). All primers for selected gene sequences were selected by Primer-BLAST software. Real-time PCR (RT-PCR) was performed using a Rotor-Gene 6000 (Qiagen, Hilden, Germany) and 18S RNA was used as an internal control for normalization. Fold changes of target genes were calculated using comparative quantification to 18S RNA.
Barrier Function Assessment
Methods of assessing the in vitro passage across the intestinal wall were similar to those previously described [21]. Briefly, an Using chamber was utilized with freshly isolated mid-colonic tissue using standard techniques [22]. The histological results in middle colon were similar to the proximal colon, so to conserve tissue, we used mid-colonic tissue for barrier function analysis. Intestinal tissues with an exposed surface area of 0.031 cm2 were incubated in 5 ml of preheated 37 °C Krebs buffer [140 mM NaCl, 1.2 mM MgCl2, 1.2 mM CaCl2, 10 mM KHCO3, 0.2 mM KH2PO4, and 1.2 mM K2HPO4] on each side (serosal and mucosal), and pH was adjusted to 7.4. Each chamber was continuously oxygenated with O2/CO2 (95/5 %) and stirred by gas flow in the chambers. Transepithelial resistance was measured using previously described techniques. Additionally, a measure of intercellular permeability was performed with FITC-dextran (Sigma-Aldrich; 150 μl) as previously described [23]. FITC-dextran was added to the chamber on the mucosal side after balancing unlabeled dextran on serosal and mucosal sides. After 30 min equilibration, 500 μl of Krebs buffer of the serosal side was collected at 60, 90, and 120 min after injection to evaluate colon permeability. Fluorescence was measured using an M5 Microplate Reader (Molecular Dynamics) at an excitation wavelength of 492 nm and an emission wavelength of 515 nm. Permeability was expressed as the mucosal-to-serosal clearance of FITC-dextran-4000.
Immunofluorescence Staining
Immunofluorescence staining was also used to determine the integrity of the tight junction and adherens junction in each treatment group using previously described methods [24]. Briefly, tissue was frozen and sectioned [24]. Primary antibodies (dilution) anti rabbit ZO-1 (1/100) and anti-mouse occludin (6/1000) (Invitrogen, Carlsbad, CA) were applied overnight at 4 °C. Slides were incubated with secondary antibody (goat anti rabbit IgG 488 or goat anti mouse IgG 555) for 2 h and counterstained with DAPI for detection of nucleus. Images were visualized on a Nikon A1 confocal microscope (Nikon Instruments Inc, Tokyo, Japan) under 20× magnification.
Statistical Analysis
For comparison of two groups unpaired T test was used. For multiple group comparisons, ANOVA was performed with a post hoc Tukey Multiple Comparison analysis. These statistical methods were used to analyze for all data except clinical scores. Data are expressed as mean ± SD.
Data for clinical scores analysis were analyzed using a repeated measures mixed model. We analyzed the clinical score for each mouse at each timepoint relative to the other mice. An autoregressive correlation structure was used to account for the relationship of measures on the same mouse over time since the clinical scores in the days closest to the current day would be more similar than scores that were much earlier or later. The model is able to estimate an average clinical score for each group at each day. Differences between groups were tested using pairwise comparisons within the model. p values less than 0.05 were considered statistically significant.
Results
Hemodynamic Status
Based on our previous work with ACE-I in a DSS colitis model, we chose a dose of 6.25 mg/kg/every 12 h given via transanal instillation. This dosing was predicted to have poor systemic exposure due to poor enteral absorption [25]. Thus, this dose was actually ten times higher than the typical dose given to rodents to treat high blood pressure. Our first aim was to determine if an ACE-I at the selected dose affected hemodynamic measures in each treatment group. Both systolic and diastolic blood pressures were not statistically different between study groups (Fig. 1a, b). As another measure of systemic perfusion, mesenteric and distal colonic blood flow measurements were performed. These also showed no statistically significant difference between any of the study groups (Fig. 1c, d). Taken together, our data suggest that use of transanal ACE-I should be safely tolerated.
Fig. 1.

Hemodynamic status does not change with treatment. Systolic blood pressure (a) and diastolic blood pressure (b) were accessed from the tail with an external blood pressure machine. Both systolic and diastolic blood pressures were not statistically different between study groups. Due to stress on the animal, BP was taken only once per animal at the end of the study. Laser Doppler measurement of mesenteric blood flow in the small bowel (c) and distal colon (d). No statistical differences were seen with blood flow in either area. A minimum of five mice per study group were analyzed
Enalaprilat Led to Improved Survival and Better Clinical Outcomes
We first assessed whether treated mice had better survival rates versus the placebo group (Fig. 2a). The placebo group exhibited the worst survival rate at 66.7 %. All treatment groups exhibited a statistically better survival rate compared to placebo group (p < 0.01). The PSL group had an intermediate survival rate of 85.7 %. As well, the PSL + ACE-I group showed an intermediate survival rate of 71.4 %. The ACE-I group exhibited a significantly better survival rate (90.9 %) compared to all other treatment groups. Percent body weight change demonstrated a decline as the colitis progressed. On average, the percent decline in body weight was: IL10−/− naïve, 5.9 %; placebo, −8.5 %; ACE-I, PSL, −12.5 %, and PSL + ACE-I, −12.0 %. All colitis groups differed significantly from the IL10−/− naïve group (p < 0.05). The ACE-I group weight loss was less than the PSL or PSL + ACE-I groups, but differences were not statistically significant (Fig. 2b).
Fig. 2.

Enalaprilat led to increased survival and a decrease in clinical severity. a A Kaplan–Meier survival curve through the 21-day study period is shown. Note that the ACE-I group exhibited the best survival at day 21 (p < 0.01 using one-way ANOVA with a Tukey’s multiple comparison test). All treatment groups exhibited statistically better survival than placebo. The PSL + ACE-I group also had statistically better survival (p < 0.01). A minimum of N = 7 mice were used per study group. b Body weight was measured daily, and a percent body weight change was calculated. The key is the same as Fig. 2a. Note the positive weight change in the IL-10−/− naïve group. Weight change was compared between groups using a one-way ANOVA with Tukey’s post hoc analysis. The following comparisons were found to be statistically different (p < 0.05): IL10−/− naïve over all groups; ACE-I less change than PSL; and ACE-I less change than PSL + ACE-I. c Cooper clinical score was calculated for each study day and shown for all treatment groups. Clinical scores were compared between groups on each day and statistically different scores were observed between groups at days 14–21. Note the best clinical score (lowest number) was in the ACE-I group, and worst clinical score was in the PSL group. There was no statistical significance in one-way ANOVA with Tukey’s post hoc analysis. A few animals in some groups had severe colitis, thus driving the scores up. Thus we observed the proportion of animals that had severe colitis (see d). A minimum of N = 7 per study group. d The proportion of mice exhibiting severe colitis (clinical score over 7 or death) was compared on each day to determine if the score for a few mice skewed the overall clinical score for the group. One-way ANOVA with Tukey’s post hoc analysis was performed and demonstrated that the PSL group had the highest proportion of animals with severe colitis, which was statistically worse than the placebo group (p < 0.01). The ACE-I group fared significantly better than the placebo group (p < 0.01) Data for days 14 and 21 are shown. *p < 0.01 for PSL versus all other groups; §p < 0.01 for placebo versus ACE-I group. This graph does not have standard deviation because it shows percentage of mice having severe colitis in each group
The clinical severity score was based on a compilation of stool stiffness, body weight change and presence of hematochezia. A definitively positive hemoccult stool test along with diarrhea indicated severe colitis. Differences in clinical scores between groups were tested on day 21 after colitis induction (Fig. 2c). Mice dying at or before this time period were scored as a 12, or a maximum score for this analysis. As a trend, the clinical score progressively rose in all groups; however, between 14 and 21 days, the ACE-I group exhibited a significantly better score compared to either the placebo group (p < 0.05) or PSL (p < 0.01 or PSL + ACE-I groups (p < 0.01). Mice treated only with PSL had the worst clinical severity score. These data indicate that all groups had worsening disease over the time course, but the end disease was best with ACE-I treatment.
Since the mice in all groups exhibited worsening colitis over the course of the experiment, we examined the cause for the increased severity in the treatment groups. We hypothesized that the number of mice with severe colitis would be fewer in the treatment groups, and that the scores for those mice would skew the group’s clinical severity scores. Thus, we determined the proportion of mice in each group with severe colitis, as defined by a clinical score >7 or death. Significant differences in the number of mice with severe colitis in each group were observed at days 14 and 21 (Fig. 2d). The PSL group had the highest proportion of animals with severe colitis, and this was statistically worse than the placebo group (p < 0.01). The ACE-I group fared significantly better than the placebo group (p < 0.01). The ACE-I + PSL group had an intermediate number of mice with severe colitis.
Enalaprilat Reduced the Histological Severity of Colitis
Since the lamina propria has a greater degree of inflammatory infiltration in the IL-10−/− model versus other chemically-induced models, we used a scoring system which was modified from previously published methods [20] (Table 1 and Supplemental Figure). Histologic assessment was performed for the proximal colon, because in the IL-10−/− model, this area represents the predominate area of colitis formation [11]. The histologic scoring consisted of a measurement of mucosal ulceration, epithelial hyperplasia, neutrophil infiltrate, and mononuclear infiltrate. Neutrophil infiltrates were widely dispersed in the placebo group, along with moderate degrees of mucosal epithelial ulceration. The placebo group had a significantly higher histological score compared to the naïve group. The ACE-I and the PSL + ACE-I groups showed remarkable preservation of the epithelial integrity and a lack of inflammatory infiltration, and exhibited markedly lower scores than the placebo group (Fig. 3). This decline in histologic score was statistically significant in the ACE-I group. Interestingly, the PSL group had only a marginal improvement in the histology score.
Fig. 3.

ACE-I treatment significantly decreased the histological colitis score. a Representative histological sections from proximal colon were blindly scored using our scoring criteria for colitis (see Fig. 3; Table 1). b Histologic scores for all treatment groups are shown. ACE-I and PSL + ACE-I exhibited statistically better histology than placebo or PSL groups (p < 0.05). A minimum of N = 7 per study group, and a minimum of ten microscopic fields were scored for each animal. These images were taken ×10 magnifications
Cytokine and Chemokine Expression via RT-PCR
Inflammatory cytokines were then measured as a marker of the pro-inflammatory response in this colitis model [26]. As a trend, drug treatment led to a reduction in the abundance of several of the pro-inflammatory cytokines and chemokine. TNF-α and IFN-γ decreased in all treatment groups compared to placebo (Table 2; Fig. 4). However, overall, the PSL group exhibited the greatest decrease in these cytokines compared to the other treatment groups. IL1β and IL6 were also significantly decreased in the PSL group. In addition, the PSL + ACE-I group demonstrated significant declines in pro-inflammatory cytokine expression. In particular, the PSL + ACE-I group showed a significantly decreased level compared to the placebo group for TNF-α and IFN-γ. The ACE-I group exhibited reductions in the expression of several pro-inflammatory cytokines, however, the declines were not statistically significant. Thus, the use of corticosteroids, alone or in combination with ACE-I, appears to have the greatest benefit on reduction of pro-inflammatory cytokine expression.
Table 2.
Cytokine and chemokine expression
| Study groups | TNF-α | IFN-γ | IL1β | IL6 | IL8 | MIP2 | KC |
|---|---|---|---|---|---|---|---|
| IL-10−/− naïve | 4.7 ± 2.3 | 0.7 ± 0.4 | 3.0 ± 1.0 | 1.6 ± 1.3 | 1.6 ± 0.6 | 12.4 ± 11.4 | 9.5 ± 3.9 |
| Placebo | 14.1 ± 4.3 | 5.2 ± 1.3* | 7.0 ± 1.5 | 25.0 ± 11.5 | 2.7 ± 0.7 | 22.7 ± 4.9 | 20.8 ± 5.4 |
| ACE-I | 11.2 ± 2.3 | 3.1 ± 0.7 | 6.4 ± 2.1 | 10.0 ± 3.0 | 1.8 ± 0.5 | 12.0 ± 4.5 | 15.6 ± 5.5 |
| PSL | 3.9 ± 0.7 | 2.1 ± 1.1 | 1.1 ± 0.1** | 1.0 ± 0.3** | 2.1 ± 0.7 | 16.8 ± 5.3 | 8.0 ± 2.9 |
| PSL + ACE-I | 3.2 ± 0.9** | 0.8 ± 1.6** | 6.0 ± 5.2 | 1.3 ± 0.3 | 2.1 ± 0.5 | 13.3 ± 4.0 | 6.4 ± 1.0 |
Data are divided by 103. Results are expressed as mean ± SEM. Fold changes of target genes relative to 18S RNA. No statistical significance between all the other groups
p < 0.05 for comparing with naïve group
p < 0.05 for comparing with placebo group
Fig. 4.

Cytokine and chemokine expression during colitis treatment. Expression for pro-inflammatory cytokines and chemokines was obtained using RT-PCR. Results are expressed as mean ± SEM. Fold changes of target genes relative to 18S RNA. *p < 0.05 for comparing with naïve group; #p < 0.05 for comparing with placebo group. No statistical significance was observed between the other groups
Barrier Function Was Preserved with ACE-I Treatment
Epithelial barrier function loss is a major mediator of colitis formation and worsens with the severity of the colitis. Figure 5a shows TER measurements from each group. TER measurements did not decline with the development of colitis and were found not to differ significantly between the study groups. FITC dextran permeability was used as another measure of epithelial barrier function. FITC dextran permeability (Fig. 5b) was significantly increased in the placebo group and thus was felt to represent a better measure of loss of epithelial barrier function in the IL-10−/− colitis model. ACE-I resulted in a significant (p < 0.05) preservation of barrier function compared to the placebo group. As well, the PSL + ACE-I group also led to a significant reduction in permeability (p < 0.01) as compared to placebo. The permeability levels in the ACE-I and PSL + ACE-I groups were not significantly different from the naïve group of mice. Also, the PSL group failed to show any reduction in permeability compared to the placebo group.
Fig. 5.
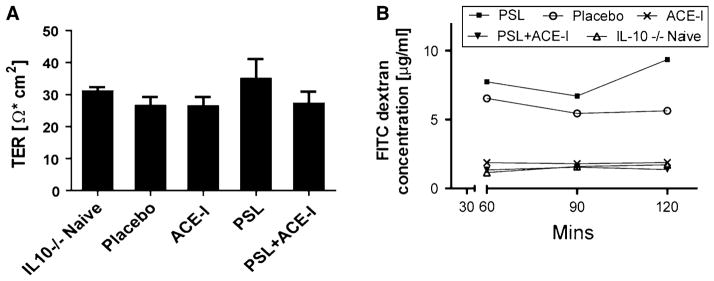
Intestinal permeability was improved with ACE-I treatment. a Transepithelial resistance (TER) was measured with Ussing chambers, and the TER taken after a 120 min incubation period is shown. No statistical difference was found between the naïve mice and any other group. b Barrier function was also measured by FITC-dextran permeability from the mucosal to the serosal side starting at 60 min, and repeated at 30 min intervals up to 120 min. Permeability was significantly increased in the placebo group. Permeability was not improved with PSL treatment; however, it was reduced in the ACE-I group (p < 0.05) and the PSL + ACE-I (p < 0.01) group compared to the placebo group. A minimum of N = 5 per study group
Tight Junctions Were Preserved with ACE-I Treatment
Liu et al. [27] demonstrated that Lactobacillus plantarum prevented the development of colitis in IL-10-deficient mice by blocking changes in the expression of tight junction proteins, tight junction structure and intestinal permeability. Thus, a more in-depth investigation of barrier function was performed by examining the distribution and density of tight junction proteins. Immunofluorescence staining was performed for ZO-1 and occludin and each of these showed a marked loss in the placebo group and in the PSL group. As well, an intracellular internalization of occludin was noted in both of these study groups. These histologic findings are consistent with other models of colitis [28]. The use of ACE-I as well as PSL + ACE-I resulted in a preservation of each junctional protein and a prevention of the internalization of occludin (Fig. 6). Although it is difficult to precisely quantify, it appeared that the ACE-I group maintained tight junction intensity and distribution closest to the naïve group. Thus, the greatest action of ACE-I treatment appears to be preservation of barrier function in this colitis model.
Fig. 6.

Tight junctions are preserved with ACE-I treatment. Immunofluorescence staining was performed on each animal to localize the tight junction proteins ZO-1 (red), occludin (green) and Merge (ZO-1/Occludin/DAPI). Note a loss of tight junction structure in the placebo group which also occurred in the PSL alone group. A preservation of tight junction staining was seen in both ACE-I and the PSL + ACE-I groups. Representative images are shown. A minimum of N = 3 per study group. Scale bar is 10 μm, and these images were taken at ×100 magnifications
Discussion
Treatment of IBD is associated with substantial morbidity in patients with both Crohn’s disease and ulcerative colitis [29]. Early microarray work by our laboratory demonstrated an increase in ACE expression during times of intestinal epithelial cell remodeling [30], and outside of the GI tract, a rich cardiovascular literature suggests that ACE may mediate pro-inflammatory and pro-apoptotic signaling [31, 32]. Thus, our group and others have demonstrated that blockade of ACE and its downstream signaling pathway, including the AT1a-receptor, can very effectively decrease the severity of colitis in either a DSS or TNBS model. However, these chemically-induced colitis models lack a strong phenotypic resemblance to human IBD which puts into question whether blockade of ACE would have a beneficial clinical role.
Although several authors have used the DSS colitis model to investigate Renin-Angiotensin System (RAS) blockade and colitis [26, 33–36], the data presented here are the first which demonstrate RAS blockade efficacy using an immune-based model. In the present study we utilized an IL-10−/− colitis immune-based model and demonstrated that ACE-I treatment was as good, or far superior, to that of conventional steroid treatment.
A recent paper [37] suggested that ACE-2 acts as a protective protein during DSS-induced inflammation. Since enalaprilat is an ACE-2/AT1aR inhibitor, we are not affecting ACE-2 directly with therapy; however, our data do not rule out ACE-2 as a player in our model. Recent work in a related protein to ACE, ACE2 shows that the loss of ACE2 (ACE2 KO mice) results in a strong predisposition to colitis using dextran sodium sulfate [37]. This work is quite consistent with our present findings in that ACE2 behaves in an anti-inflammatory fashion and balances the pro-inflammatory actions of ACE [38]. IL-10 deficient mice develop an immunologically-mediated colitis that more closely matches the human condition than DSS-colitis models [39]. This model has proved itself to be a valuable tool to study IBD and has been widely used to examine ulcerative colitis therapies [40, 41]. IL-10 is an anti-inflammation cytokine that is a potent suppressor of macrophage activation in vitro and in vivo. IL-10 inhibits the production of inflammatory cytokines such as IL-1, IL-6, and TNF-α [42]. Deletion of IL-10 severely inhibits the inflammatory response [43]. Due to the decreased inflammatory response, IL-10-deficient mice developed spontaneous colitis in the proximal colon under pathogen-free conditions; however, disease development takes several months [12]. We utilized the method of Berg et al. [15] where NSAIDs accelerated the formation of severe colitis in a nicely timed and consistent fashion, allowing for the testing of new therapies. In the present study we tested the efficacy of ACE-I, and compared this to a known standard of care prednisolone or a combination of these two agents.
Based on our previous data showing that ACE is over-expressed in the damaged colonic mucosa, we elected to deliver the ACE-I via a transanal route, which would permit the most direct targeting of this area of disease. The advantage of giving enalaprilat transanally is that it is acting directly on the colonic mucosa which has the highest activity of ACE, particularly during periods of colitis [6]. The present use of enalaprilat is in contrast to enalapril which is normally given via the gastrointestinal tract to treat hypertension. Enalapril is the inactive form of enalaprilat and is well-absorbed through the gastrointestinal mucosa where endogenous esterases act to convert it to the active enalaprilat form. Enalaprilat is poorly absorbed via the gastrointestinal mucosa [25], which allowed us to dose this at over 10-fold higher than normal anti-hypertensive levels without systemic side-effects.
The mechanisms by which blockade of the renin angiotensin system (RAS) are not fully understood. However, ACE-I has been shown to have a variety of anti-inflammatory effects [40, 44]. A series of pro-inflammatory and growth signaling actions occur after ACE mediated conversion of angiotensin I to angiotensin II [45]. Downstream signals from this receptor include an up-regulation of MAdCAM-1 under colonic inflammatory conditions via NF-κB translocation into the nucleus and subsequent translocation [35]. ACE-I has been shown to decrease the inflammatory process in hypertensive rats; and its action appears to be due to a prevention of NF-κB activation [46].
Although our previous study demonstrated that AT1aR antagonist markedly decrease pro-inflammatory cytokines (TNF-α, IL1β, IL-6) in a DSS model, the results in this manuscript for cytokine levels were not statistically decreased. There are several differences between our DSS model and our IL-10 KO model. First, the DSS model is a chemical induction of colitis; whereas, the IL-10 KO model is an immunological model. Thus, the cytokine levels in the immunological model may be subjected to different controls when presented with the ACE-I therapy. Also, the mice in the DSS model are 5-week-old mice. The IL10-KO mice do not present with colitis until later, so we used 10-week-old mice. The age difference may account for differences in cytokine levels as well. Also, being an immunological model lends itself to abnormal control of the immune system, regardless if there is colitis or not. Since the mice only presented with colitis around day 14 of the study (7 days from the end of the study), there may not have been enough time to detect changes in the treated populations.
Although steroids represent a major therapeutic standard for colitis patients, in the present study oral prednisolone was not as effective as ACE-I treatment for the clinical score, histologic score, and the prevention of epithelial barrier function loss. There are two possible explanations for the ineffective nature of the steroid in our study. First, Okada et al. [41] showed that NSAID-induced colitis in IL-10 mice could be resistant to steroid treatment. However, their data are controversial as other studies have shown efficacy for steroids in this model [47]. Second, actions of prednisolone and ACE-I in this present study appeared to be different. We noted that prednisolone had an equal or greater effect on reducing pro-inflammatory cytokine expression, whereas the ACE-I groups were more effective in preventing histologic and barrier function injury. Clearly, ACE-I had a very strong benefit in protecting the barrier function of the epithelium; and this action was far superior to that seen with prednisolone. This may be how the use of angiotensin II blockade mediates its action in an indomethacin intestinal injury model [48]. It is also possible that the use of ACE-I may be used chronically in IBD patients to maintain barrier function, and thus prevent recurrent episodes of acute inflammation. Our data argue that, although the prednisolone alone did not have much efficacy in reducing injury and restoring barrier function, prednisolone could lower inflammatory cytokines, and thus was not without benefit. Even though our data did not show a large benefit to using prednisolone in conjunction with ACE-I, it is certainly possible that use of ACE-I along with a corticosteroid may prove more beneficial than either drug alone since steroid treatment is efficacious in the clinic. Therefore, addition of an ACE-I to NSAID therapy may allow for lower steroid dosing, reducing harsh side effects seen with steroid use.
Future work is needed to optimize ACE-I dosing to avoid potential adverse effects, including the risk of ischemic colitis or hypotension. Although very rarely described, ischemic colitis or angioedema have been reported in the literature during ACE-I treatment [49, 50]. Risk factors for the development of ischemic colitis in patients on ACE inhibitors include elderly age, underlying renal disease, and cardiovascular disease [51]. Nevertheless, the safety record for ACE-I usage is strong and may thus have a strong appeal for clinical applications.
In conclusion, we demonstrated that the use of ACE-I was effective in an immunologically-driven colitis model. This suggests that the clinical application of an ACE-I may have a potential therapeutic role as either single therapy for IBD or in combination with other immunosuppressive agents. Future work will be needed to better understand the mechanisms which lead to its anti-inflammatory effect.
Supplementary Material
Acknowledgments
We thank Nathan Opaleski and Pele Browner for their excellent technical assistance. The authors received Grant Support NIH-R01 AI-44076-13, NIH UL1-RR024986 and NIH 1 R43 DK088495-01.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10620-013-2825-4) contains supplementary material, which is available to authorized users.
Conflict of interest None.
Contributor Information
Ryo Sueyoshi, Section of Pediatric Surgery, Department of Surgery, Mott Children’s Hospital, University of Michigan, 1540 E. Hospital Dr., SPC 4211, Ann Arbor, MI 48109-4211, USA.
Kathleen M. Woods Ignatoski, Section of Pediatric Surgery, Department of Surgery, Mott Children’s Hospital, University of Michigan, 1540 E. Hospital Dr., SPC 4211, Ann Arbor, MI 48109-4211, USA
Stephanie Daignault, Biostatistics, Comprehensive Cancer Center, University of Michigan Health System, Ann Arbor, MI, USA.
Manabu Okawada, Section of Pediatric Surgery, Department of Surgery, Mott Children’s Hospital, University of Michigan, 1540 E. Hospital Dr., SPC 4211, Ann Arbor, MI 48109-4211, USA.
Daniel H. Teitelbaum, Email: dttlbm@med.umich.edu, dttlbm@umich.edu, Section of Pediatric Surgery, Department of Surgery, Mott Children’s Hospital, University of Michigan, 1540 E. Hospital Dr., SPC 4211, Ann Arbor, MI 48109-4211, USA
References
- 1.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 2.Mayer L. Evolving paradigms in the pathogenesis of IBD. J Gastroenterol. 2010;45:9–16. doi: 10.1007/s00535-009-0138-3. [DOI] [PubMed] [Google Scholar]
- 3.Duggan KA, Mendelsohn FA, Levens NR. Angiotensin receptors and angiotensin I-converting enzyme in rat intestine. Am J Physiol. 1989;257:G504–G510. doi: 10.1152/ajpgi.1989.257.4.G504. [DOI] [PubMed] [Google Scholar]
- 4.Jaszewski R, Tolia V, Ehrinpreis M, et al. Increased colonic mucosal angiotensin I and II concentrations in Crohn’s colitis. Gastroenterology. 1990;98:1543–1548. doi: 10.1016/0016-5085(90)91088-n. [DOI] [PubMed] [Google Scholar]
- 5.Wildhaber B, Yang H, Haxhija E, et al. Intestinal intraepithelial lymphocyte derived angiotensin converting enzyme modulates epithelial cell apoptosis. Apoptosis. 2005;10:1305–1315. doi: 10.1007/s10495-005-2138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spencer A, Yang H, Haxhija E, et al. Reduced severity of a mouse colitis model with angiotensin converting enzyme inhibition. Dig Dis Sci. 2007;52:1060–1070. doi: 10.1007/s10620-006-9124-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inokuchi Y, Morohashi T, Kawana I, et al. Amelioration of 2, 4, 6-trinitrobenzene sulphonic acid induced colitis in angiotensinogen gene knockout mice. Gut. 2005;54:349–356. doi: 10.1136/gut.2003.036343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wengrower D, Zannineli G, Pappo O, et al. Prevention of fibrosis in experimental colitis by captopril: the role of TGF-beta1. Inflamm Bowel Dis. 2004;10:536–545. doi: 10.1097/00054725-200409000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation 1. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 10.Laroui H, Ingersoll SA, Liu HC, et al. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS ONE. 2012;7:e32084. doi: 10.1371/journal.pone.0032084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kühn R, Löhler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 12.Hale LP, Gottfried MR, Swidsinski A. Piroxicam treatment of IL-10-deficient mice enhances colonic epithelial apoptosis and mucosal exposure to intestinal bacteria. Inflamm Bowel Dis. 2005;11:1060–1069. doi: 10.1097/01.mib.0000187582.90423.bc. [DOI] [PubMed] [Google Scholar]
- 13.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson NJ, Hudak SA, Lesley RE, et al. IL-12, but not IFN-gamma, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J Immunol. 1998;161:3143–3149. [PubMed] [Google Scholar]
- 15.Berg DJ, Zhang J, Weinstock JV, et al. Rapid development of colitis in NSAID-treated IL-10–deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 16.Rennick DM, Fort MM, Davidson NJ. Studies with IL-10−/− mice: an overview. J Leukoc Biol. 1997;61:389–396. doi: 10.1002/jlb.61.4.389. [DOI] [PubMed] [Google Scholar]
- 17.Haxhija E, Yang H, Spencer A, et al. Modulation of mouse intestinal epithelial cell turnover in the absence of angiotensin converting enzyme. Amer J Physiol Gastroint Liver Physiol. 2008;295:G88–G98. doi: 10.1152/ajpgi.00589.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper HS. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 19.Saverymuttu SH. Indium 111-granulocyte scanning in the assessment of disease extent and disease activity in inflammatory bowel disease. A comparison with colonoscopy, histology, and fecal indium 111-granulocyte excretion. Gastroenterology. 1986;90:1121–1128. doi: 10.1016/0016-5085(86)90376-8. [DOI] [PubMed] [Google Scholar]
- 20.Madsen K, Cornish A, Soper P, et al. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology. 2001;121:580–591. doi: 10.1053/gast.2001.27224. [DOI] [PubMed] [Google Scholar]
- 21.Lennernös H, Nylander S, Ungell AL. Jejunal permeability: a comparison between the using chamber technique and the single-pass perfusion in humans. Pharm Res. 1997;14:667–671. doi: 10.1023/a:1012121632357. [DOI] [PubMed] [Google Scholar]
- 22.Yang H, Finaly R, Teitelbaum DH. Alteration in epithelial permeability and ion transport in a mouse model of total parenteral nutrition. Crit Care Med. 2003;31:1118–1125. doi: 10.1097/01.CCM.0000053523.73064.8A. [DOI] [PubMed] [Google Scholar]
- 23.Krug SM, Amasheh S, Richter JF, et al. Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol Biol Cell. 2009;20:3713–3724. doi: 10.1091/mbc.E09-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng Y, Sun X, Yang H, Teitelbaum D. Dissociation of E-cadherin and beta-catenin in a mouse model of total parenteral nutrition: a mechanism for the loss of epithelial cell proliferation and villus atrophy. J Physiol (London) 2009;587:641–654. doi: 10.1113/jphysiol.2008.162719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahlin P, Kristl J, Kristl A, et al. Investigation of polymeric nanoparticles as carriers of enalaprilat for oral administration. Int J Pharm. 2002;239:113–120. doi: 10.1016/s0378-5173(02)00076-5. [DOI] [PubMed] [Google Scholar]
- 26.Okawada M, Koga H, Larsen S, et al. Use of enterally delivered angiotensin II type Ia receptor antagonists to reduce the severity of colitis. Dig Dis Sci. 2011;56:2553–2565. doi: 10.1007/s10620-011-1651-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Zhang P, Ma Y, et al. Lactobacillus plantarum prevents the development of colitis in IL-10-deficient mouse by reducing the intestinal permeability. Mol Biol Rep. 2011;38:1353–1361. doi: 10.1007/s11033-010-0237-5. [DOI] [PubMed] [Google Scholar]
- 28.Clayburgh DR, Barrett TA, Tang Y, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manz M, Vavricka SR, Wanner R, et al. Therapy of steroid-resistant inflammatory bowel disease. Digestion. 2012;1:11–15. doi: 10.1159/000341952. [DOI] [PubMed] [Google Scholar]
- 30.Wildhaber B, Yang H, Coran A, Teitelbaum D. Gene alteration of intestinal intraepithelial lymphocytes in response to massive small bowel resection. Pediatr Surg Int. 2003;19:310–315. doi: 10.1007/s00383-003-1001-x. [DOI] [PubMed] [Google Scholar]
- 31.Papp M, Li X, Zhuang J, et al. Angiotensin receptor subtype AT(1) mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol. 2002;282:L713–L718. doi: 10.1152/ajplung.00103.2001. [DOI] [PubMed] [Google Scholar]
- 32.Uhal BD, Gidea C, Bargout R, et al. Captopril inhibits apoptosis in human lung epithelial cells: a potential antifibrotic mechanism. Am J Physiol. 1998;275:L1013–L1017. doi: 10.1152/ajplung.1998.275.5.L1013. [DOI] [PubMed] [Google Scholar]
- 33.Haxhija EQ, Yang H, Spencer AU, et al. Modulation of mouse intestinal epithelial cell turnover in the absence of angiotensin converting enzyme. Am J Physiol Gastroint Liver Physiol. 2008;295:G88–G98. doi: 10.1152/ajpgi.00589.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koga H, Yang H, Adler J, et al. Transanal delivery of angiotensin converting enzyme inhibitor prevents colonic fibrosis in a mouse colitis model: development of a unique mode of treatment. Surgery. 2008;144:259–268. doi: 10.1016/j.surg.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizushima T, Sasaki M, Ando T, et al. Blockage of angiotensin II type 1 receptor regulates TNF-α-induced MAdCAM-1 expression via inhibition of NF-κB translocation to the nucleus and ameliorates colitis. Am J Physiol Gastroint Liver Physiol. 2010;298:G255–G266. doi: 10.1152/ajpgi.00264.2009. [DOI] [PubMed] [Google Scholar]
- 36.Koga H, Haxhija EQ, Teitelbaum DH. The role of angiotensin II type 1a receptor on intestinal epithelial cells following small bowel resection in a mouse model. Pediatr Surg Int. 2008;24:1279–1286. doi: 10.1007/s00383-008-2277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hashimoto T, Perlot T, Rehman A, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487:477–481. doi: 10.1038/nature11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. 2008;93:543–548. doi: 10.1113/expphysiol.2007.040048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikenoue Y, Tagami T, Murata M. Development and validation of a novel IL-10 deficient cell transfer model for colitis. Int Immunopharmacol. 2005;5:993–1006. doi: 10.1016/j.intimp.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 40.Pang T, Benicky J, Wang J, et al. Telmisartan ameliorates lipo-polysaccharide-induced innate immune response through peroxisome proliferator-activated receptor-gamma activation in human monocytes. J Hypertens. 2012;30:87–96. doi: 10.1097/HJH.0b013e32834dde5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okada Y, Maeda N, Takakura S, et al. Preventive and therapeutic effects of tacrolimus in an interleukin-10-deficient mouse model of colitis. Inflamm Res. 2011;60:1049–1059. doi: 10.1007/s00011-011-0366-x. [DOI] [PubMed] [Google Scholar]
- 42.Fiorentino D, Zlotnik A, Mosmann T, et al. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 43.Ito K, Takaishi H, Jin Y, et al. Staphylococcal enterotoxin B stimulates expansion of autoreactive T cells that induce apoptosis in intestinal epithelial cells: regulation of autoreactive responses by IL-10. J Immunol. 2000;164:2994–3001. doi: 10.4049/jimmunol.164.6.2994. [DOI] [PubMed] [Google Scholar]
- 44.Nemati F, Rahbar-Roshandel N, Hosseini F, et al. Anti-Inflammatory effects of anti-hypertensive agents: influence on interleukin-1β secretion by peripheral blood polymorphonuclear leukocytes from patients with essential hypertension. Clin Exp Hypertens. 2011;33:66–76. doi: 10.3109/10641963.2010.496521. [DOI] [PubMed] [Google Scholar]
- 45.Phillips MI, Kagiyama S. Angiotensin II as a pro-inflammatory mediator. Curr Opin Investig Drugs. 2002;3:569–577. [PubMed] [Google Scholar]
- 46.Miguel-Carrasco J, Zambrano S, Blanca A, et al. Captopril reduces cardiac inflammatory markers in spontaneously hypertensive rats by inactivation of NF-kB. J Inflamm. 2010;7:21. doi: 10.1186/1476-9255-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishiyori A, Nagakura Y, Ichikawa K. Piroxicam accelerates development of colitis in T-cell receptor α chain-deficient mice. Eur J Pharmacol. 2009;615:241–245. doi: 10.1016/j.ejphar.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 48.Okuda T, Yoshida N, Takagi T, et al. CV-11974, angiotensin II type I receptor antagonist, reduces the severity of indomethacin-induced rat enteritis. Dig Dis Sci. 2008;53:657–663. doi: 10.1007/s10620-007-9931-0. [DOI] [PubMed] [Google Scholar]
- 49.Makins R. Gastrointestinal side effects of drugs. Expert Opin Drug Saf. 2003;2:421–429. doi: 10.1517/14740338.2.4.421. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt TD, McGrath KM. Angiotensin-converting enzyme inhibitor angioedema of the intestine: a case report and review of the literature. Am J Med Sci. 2002;324:106–108. doi: 10.1097/00000441-200208000-00011. [DOI] [PubMed] [Google Scholar]
- 51.Higgins PDR, Davis KJ, Laine L. The epidemiology of ischaemic colitis. Aliment Pharmacol Ther. 2004;19:729–738. doi: 10.1111/j.1365-2036.2004.01903.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.