

Abstract
SR proteins are essential splicing factors whose biological function is regulated through phosphorylation of their C-terminal RS domains. Prior studies have shown that cytoplasmic–nuclear translocalization of the SR protein SRSF1 is regulated by multisite phosphorylation of a long Arg-Ser repeat in the N-terminus of the RS domain while subnuclear localization is controlled by phosphorylation of a shorter Arg-Ser repeat along with several Ser-Pro dipeptides in the C-terminus of the RS domain. To better understand how these two kinases partition Arg-Ser versus Ser-Pro specificities, we monitored the phosphorylation of SRSF1 by CLK1 and SRPK1. Although SRPK1 initially binds at the center of the RS domain phosphorylating in an orderly, N-terminal direction, CLK1 makes widespread contacts in the RS domain and generates multiple enzyme–substrate complexes that induce a random addition mechanism. While SRPK1 rapidly phosphorylates N-terminal serines, SRPK1 and CLK1 display similar activities toward Arg-Ser repeats in the C-terminus, suggesting that these kinases may not separate function in a strict linear manner along the RS domain. CLK1 induces a unique gel shift in SRSF1 that is not the result of enhanced Arg-Ser phosphorylation but rather is the direct result of the phosphorylation of several Ser-Pro dipeptides. These prolines are important for binding and phosphorylation of the SR protein by CLK1 but not for the SRPK1-dependent reaction. The data establish a new view of SR protein regulation in which SRPK1 and CLK1 partition activities based on Ser-Pro versus Arg-Ser placement rather than on N- and C-terminal preferences along the RS domain.
Keywords: kinase, kinetics, phosphorylation, splicing, SR protein
Introduction
SR proteins are essential splicing factors that bind to precursor mRNA guiding the proper assembly of the spliceosome, a macromolecular complex that removes intronic sequences and joins exons to form mature mRNA for protein translation. SR proteins contain one or two RNA recognition motifs (RRMs) that bind precursor mRNA and a C-terminal domain rich in Arg-Ser dipeptide repeats (RS domains).1 Two families of protein kinases [SR-specific protein kinases (SRPKs) and Cdc2-like kinases (CLKs)] phosphorylate the RS domain and regulate SR protein function.2 SRPKs that are in the cytoplasm phosphorylate SR proteins, a key modification that facilitates binding to a transportin protein (Tr-SR) and entry of the splicing factor into the nucleus.3,4 The function of SRPKs in the nucleus is not well understood although association of U1 small nuclear ribonucleoprotein with SRPK1 and U4/U6-U5 tri-small nuclear ribonucleoprotein with SRPK2 suggests a selective role for these kinase forms during splicing.5 In comparison, the CLKs are predominantly localized in the nucleus of over-expressed cells where they control subnuclear partitioning of SR proteins and also regulate alternative splicing.6,7
Currently, there are several forms of CLK (CLK1-4) that possess homologous kinase cores and long N-terminal sequences with unknown function.8 Although it is not clear how phosphorylation controls splicing, many reports have now established that the phosphorylation levels of RS domains in SR proteins can actively regulate alternative gene splicing by controlling protein–protein or protein–RNA interactions in the spliceosome.9–13
Phospho-mapping and immunohistochemical analyses suggest that cytoplasmic SRPK1 phosphorylates an N-terminal stretch of Arg-Ser repeats [(RS)8NS(RS)2, termed “RS1”] in the RS domain of the SR protein SRSF1 (Fig. 1).14,15 RS1 modification then enhances interactions with Tr-SR that shuttles the SR protein into speckles, membrane-free storage compartments in the nucleus.4,15,16 The C-terminal half of the RS domain (RS2) contains a region with several Ser-Pro dipeptides [SPRRSRGSPRYSP] and an additional, shorter Arg-Ser repeat [(SR)3] (Fig. 1). Although these residues are not essential for translocation of SRSF1,15 they can be phosphorylat-ed by nuclear CLK1, a process that leads to a hyper-phosphorylation phenotype and a gel shift on SDS-PAGE.14 The latter modifications lead to dispersion of SRSF1 from the speckles along with changes in alternative splicing.6,17–19 These studies support a model for cellular regulation in which cytoplasmic SRPK1 directs the SR protein to the nucleus by phosphorylating RS1 and then CLK1 alters its subnuclear position by phosphorylating RS2 (Fig. 1). However, the question remains as to how these kinases function when both are present in the nucleus. Since SRPK1-2 are integral parts of the spliceosome, they are likely to have increased proximal access to the RS domains of SRSF1 and other SR proteins5 and may effectively compete with CLKs. Under these conditions, the simple, linear view of RS1/RS2 partitioning of phosphorylation derived from translocation studies may not provide a complete picture of RS domain phosphorylation specificities. Furthermore, the RS domains of SR proteins differ considerably in length and Arg-Ser and Ser-Pro arrangements. Establishing general rules for SRPK/CLK specificity toward SRSF1 may help to understand how other SR proteins are regulated through multisite phosphorylation.
Fig. 1.

Regiospecific phosphorylation model controlling SRSF1 nuclear–cytoplasmic localization. Cytoplasmic SRPK1 phosphorylates RS1 in the RS domain allowing nuclear translocation of SRSF1. Nuclear CLK1 then phosphorylates the serines in RS2.
Much is currently known about the mechanism of catalysis of SRPKs from the studies on SRPK1 and its substrate SRSF1. In single turnover experiments, SRPK1 rapidly adds about 10–12 phosphates to RS1.20 The active site of the kinase initially binds to the center of the RS domain at the C-terminal end of RS1 and then phosphorylates the serines in a strict N-terminal direction.21 However, it has recently been shown that SRPK1 can also modify the shorter repeat at the C-terminal end in RS2 although this reaction is about 10-fold less efficient and, as stated previously, unnecessary for nuclear translocation.15,22 This secondary reaction requires dissociation and re-positioning of the kinase. X-ray studies show that the directional mechanism is supported by an electronegative docking groove in the large lobe of the kinase domain that sequentially feeds Arg-Ser dipeptides into the active site.23 Mutations in this groove disrupt the directional mechanism leading to random and less efficient phosphorylation rates with no change in overall regiospecificity.22 SRPK1 is constitutively active since it does not require phosphorylation of a loop segment near the active site termed the “activation loop”.15 For some kinases, loop phosphorylation is essential for an efficient phosphoryl transfer step.24 For SRPK1, rapid quench flow studies reveal that the phosphoryl transfer and Arg-Ser translocation steps from the docking groove to the active site are highly efficient whereas the release of the product ADP limits transit time at each dipeptide subsite.25 SRPK1 facilitates ADP release rates through two short sequences external to the kinase domain that act cooperatively as a nucleotide release factor.26 A combination of mass spectrometric and substrate peptide analyses have shown that although SRSF1 contains several serines that flank prolines rather than arginines in the RS domain, these residues are not modified by SRPK1.19,22
Although the CLK family of kinases catalyzes multisite SR protein phosphorylation, they appear to use a very different mechanism than the SRPKs. Unlike SRPK1 that catalyzes a highly ordered phosphorylation mechanism, CLK1 randomly phosphorylates the RS domain of SRSF1.27 In keeping with this observation, X-ray structural studies show that CLK1 lacks a docking groove that could support a feeding mechanism akin to that for SRPK1.28 Nonetheless, previous reports indicate that CLK1 may bind SRSF1 with high affinity.14,19 In addition to phosphorylating Arg-Ser dipeptides, CLK1 modifies Ser-Pro dipeptides in the RS domain.14,19
In biochemical studies, SRSF1 that is pre-phosphorylated by SRPK1 can be then fully phosphorylated by the addition of CLK1, suggesting a cooperative interplay consistent with the translocation model catalyzed by partitioned kinases (Fig. 1).14 The X-ray structures of several CLKs have been solved and reveal an active conformation without activation loop phosphorylation, similar to that for SRPK1.28 While these structures provide a beginning framework for future investigations, they lack a bound SR protein or fragment from which a phosphorylation mechanism can be inferred.28
To provide insights into SR protein substrate specificities, we investigated the phosphorylation mechanism of mouse CLK1 and its SR protein substrate SRSF1 and compared it to that for SRPK1. We found that both kinases interact with the RS domain in SRSF1 using very different mechanisms. While SRPK1 binds in the center of the RS domain and phosphorylates in a directional manner, CLK1 has more flexibility in docking to various parts of the RS domain, thereby initiating phosphorylation at different spots through multiple forms of the bound substrate. Although SRPK1 has a preference for phosphorylating the long Arg-Ser stretch in the RS1 segment, it also phosphorylates the shorter Arg-Ser stretch in RS2 at about the same rate as CLK1, suggesting that SRPK1 is not prohibited from modifying RS2 serines and may compete at these sites with CLK1 in the nucleus. Unlike SRPK1, CLK1 induces a unique structural form of SRSF1 observed by SDS-PAGE that is exclusively the result of Ser-Pro rather than Arg-Ser phosphorylation. The data establish a new way to look at how splicing kinases modify their SR protein targets. Rather than restricting their activities to separate termini of the RS domain as suggested in Fig. 1 for SRSF1, SRPK1 and CLK1 may incorporate overlapping specificities defined largely by the presence of Arg-Ser versus Ser-Pro repeats. This new principle may be very important when considering phosphorylation-dependent changes in other SR proteins that differ considerably in RS domain size and amino acid composition.
Results
CLK1 and SRPK1 use different rate-limiting steps
We showed previously that the phosphoryl transfer step for SRPK1 is fast and does not limit SRSF1 phosphorylation.25 To investigate CLK1, we initially performed steady-state kinetic experiments. We found that the Km for SRSF1 (KSR) is 94 ± 10 nM for CLK1, which compares to a value of 100 nM reported for SRPK1.20 In contrast, the Km for ATP (KATP) at 60 ± 18 μM is 5-fold higher than that for SRPK1.25 Most notably, the kcat for CLK1 is 0.16 ± 0.02 s−1, a value about 6-fold lower than that for SRPK1.25,26 To investigate whether lower turnover is due to a change in rate-limiting step, we measured the values of kcat, kcat/KSR and kcat/KATP with varying sucrose concentrations (Fig. 2a). No effects of solvent viscosity on kcat were detected when either ATP or SRSF1 was varied consistent with a slow phosphoryl transfer step. However, while increasing sucrose levels lowered kcat/KATP within expected bounds, the effects on kcat/KSR were outside the theoretical slope limits from 0 to 1.29 These findings suggest that added viscosogen may affect SRSF1 conformation interfering with substrate association. To address this, we performed pre-steady-state kinetic experiments. A complex of CLK1 and SRSF1 was mixed with ATP in the rapid quench machine, and a linear production of phospho-SRSF1 with no evidence for an enzyme-stoichiometric “burst” was observed (Fig. 2b). Taken together, the data indicate that, while the phosphoryl transfer step for SRPK1 is fast, this step limits maximum turnover for CLK1.
Fig. 2.

Kinetic analysis of SRSF1 phosphorylation by CLK1. (a) Viscometric experiments. The steady-state kinetic parameters kcat with varying ATP (○) or SRSF1 (△) and kcat/Km with varying ATP (●) or SRSF1 (▲) are plotted as relative values in the absence and presence of varying sucrose concentrations versus relative solvent viscosity. Broken lines represent theoretical slope values of 0 and 1. (b) Rapid quench flow experiment. Final concentrations of CLK1, SRSF1 and 32P-ATP are 0.25 μM, 0.5 μM and 100 μM. Production of phosphoproduct normalized to the total CLK1 concentration ([P]/[E]) is fit to a linear function with a slope of 0.08 s−1. (c) Competition experiments. Fixed amounts of SR(ΔRRM1) (50 nM) and 32P-ATP (50 μM) are added to CLK1 (10 nM) or SRPK1 (1 nM) with varying amounts of SRSF1 (competitor), and the reactions are stopped and run on SDS-PAGE. The relative velocities for SR(ΔRRM1) phosphorylation are plotted as a function of total SRSF1, and the appKI values are listed in Table 1.
CLK1 forms a highly stable complex with SRSF1
The steady-state kinetic data suggest that SRSF1 may bind with high affinity to CLK1. However, since Km values oftentimes do not reflect true dissociation constants, we used a competition assay to assess substrate affinity.20 The phosphorylation of a fixed amount of SRSF1 lacking one of its RRMs [SR(ΔRRM1); Fig. 3a] is monitored with increasing concentrations of wild-type SRSF1. SR(ΔRRM1) is used as a fixed, control substrate so that it can be separated from SRSF1 by SDS-PAGE (Fig. 2c). With the use of competition method, a decline in SR(ΔRRM1) phosphorylation velocity is detected along with an increase in SRSF1 phosphorylation (Fig. 2c). Since approximately 50% of the velocity for SR(ΔRRM1) phosphorylation using 10 nM CLK1 is lost within 30 nM SRSF1, we used Eq. (1) for a tight binding inhibitor to define an appKI of 20 nM for SRSF1 (Fig. 2c and Table 1). The true KI is likely to be lower, owing to competition with SR(ΔRRM1). Indeed, the phosphorylation of SRSF1 increases linearly with SRSF1, suggesting an upward perturbation in Km. Using Eq. (2) and the Km for SR(ΔRRM1) (20 nM), we estimate a true KI of about 6 nM for SRSF1 (Table 1). For comparison, we performed similar competition experiments using SRPK1 and obtained an appKI of 145 nM (Fig. 2c and Table 1). Owing to the higher activity of SRPK1 and weaker apparent affinity for SRSF1, we could use much lower concentrations for the assay (1 nM) such that fitting to a quadratic function was unnecessary. Using Eq. (2) and a Km of 34 nM for SR(ΔRRM1) measured in separate experiments, we were able to establish a true KI of 60 nM for SRSF1 to SRPK1 (Table 1). These analyses indicate that CLK1 binds with high affinity to SRSF1, exhibiting a KI that is an order of magnitude lower than that for SRPK1.
Fig. 3.

Role of RRMs for CLK1-induced phosphorylation of SRSF1. (a) Deletion constructs. (b) CLK1 competition experiments. Fixed amounts of SRSF1 (50 nM) and 32P-ATP (50 μM) are added to CLK1 (10 nM) with varying amounts of the competitors SR(ΔRRM1), SR(ΔRRM1) and SR(ΔRRM1-2); the reactions are stopped and run on SDS-PAGE. The relative velocities for SRSF1 phosphorylation are plotted as a function of total competitor, and the appKI values are listed in Table 1. (c) SRPK1 competition experiments. Fixed amounts of SRSF1 (50 nM) are added to SRPK1 (1 nM) with varying amounts of the competitors SR(ΔRRM1), SR(ΔRRM1) and SR(ΔRRM1-2); the reactions are stopped and run on SDS-PAGE. The relative velocities for SRSF1 phosphorylation are plotted as a function of total competitor, and the appKI values are listed in Table 1. (d) Bar graph. KI values from Table 1 are plotted for various RRM deletions relative to the RS domain construct SR(ΔRRM1-2) for CLK1 and SRPK1. Values above 1 reflect reductions in binding affinity upon addition of the RRMs, whereas values below 1 reflect increases in affinity.
Table 1.
Dissociation constants for SRSF1 and mutant constructs to CLK1 and SRPK1
| Substrate |
appKI (nM)
|
KI (nM)
|
||
|---|---|---|---|---|
| CLK1 | SRPK1 | CLK1 | SRPK1 | |
| SRSF1 | 20 ± 5 | 150 ± 30 | 6 ± 2 | 60 ± 10 |
| SR(ΔRRM1) | 13 ± 4 | 30 ± 10 | 8 ± 2 | 21 ± 9 |
| SR(ΔRRM2) | 200 ± 40 | 500 ± 200 | 130 ± 30 | 300 ± 100 |
| SR(ΔRRM1-2) | 40 ± 5 | 110 ± 20 | 26 ± 3 | 70 ± 10 |
| SR(RRM1-2) | 230 ± 30 | 260 ± 40 | 150 ± 20 | 170 ± 30 |
| SR(RRM2) | 190 ± 20 | 4400 ± 700 | 120 ± 20 | 2900 ± 500 |
| SR(1–226) | 8 ± 2 | 5 ± 1 | ||
| SR(1–219) | 26 ± 5 | 17 ± 3 | ||
| SR(188–235) | 180 ± 20 | 170 ± 10 | 120 ± 10 | 110 ± 7 |
| SR(188–228) | 370 ± 40 | 280 ± 50 | 240 ± 30 | 190 ± 30 |
| SR(188–220) | 440 ± 40 | 520 ± 40 | 290 ± 20 | 350 ± 200 |
| SR(P200A) | 24 ± 4 | 90 ± 20 | 7 ± 1 | 35 ± 9 |
| SR(P228A) | 28 ± 5 | 105 ± 4 | 8 ± 1 | 43 ± 2 |
| SR(P235A) | 260 ± 60 | 140 ± 30 | 80 ± 20 | 60 ± 10 |
| SR(P239A) | 110 ± 20 | 110 ± 7 | 30 ± 5 | 45 ± 3 |
| SR(P228,235A) | 240 ± 50 | 100 ± 10 | 70 ± 10 | 41 ± 6 |
| SR(200,228,235A) | 440 ± 70 | 80 ± 8 | 130 ± 20 | 33 ± 3 |
| SR(P235,239A) | 90 ± 30 | 80 ± 10 | 26 ± 7 | 34 ± 4 |
Differential effects of RRMs on SRSF1 binding to CLK1 and SRPK1
To better understand why CLK1 binds with higher affinity to SRSF1 than does SRPK1, we used the competition assay to determine whether the RRMs in SRSF1 facilitate enhanced binding. In these experiments, the initial velocity for SRSF1 phosphorylation is measured as a function of three deletion mutants lacking RRM1 [SR(ΔRRM1)], RRM2 [SR(ΔRRM2)] and both RRM1 and RRM2 [SR(ΔRRM1-2)] (Fig. 3a and b). While deletion of RRM1 had no effect, removal of both RRMs increased the KI by about 4-fold to CLK1 (Fig. 3b and Table 1). In comparison, while deletion of RRM1 reduced the KI by 3-fold, removal of both RRMs had no effect on binding to SRPK1 (Fig. 3c and Table 1). Despite this differential effect, both CLK1 and SRPK1 prefer RRM2 over RRM1 directly N-terminal to the RS domain. The KI values for SR(ΔRRM2) to CLK1 and SRPK1 are 24- and 6-fold higher than that for SRSF1 (Fig. 3b and c and Table 1). By comparing changes in KI relative to SR(ΔRRM1-2), we can show that addition of RRM2 to the RS domain enhances binding affinity by similar amounts to both kinases (Fig. 3d). The stabilizing effects of RRM2 are clearly unique as adding RRM1 in the flanking position to the RS domain significantly reduces binding affinity to both kinases. Interestingly, the addition of RRM1 to the RRM2 RS domain construct has no significant effect on binding to CLK1 but instead reduces binding to SRPK1 by about 3-fold (Fig. 3d). Thus, the 10-fold weaker binding affinity of SRSF1 to SRPK1 compared to CLK1 can be explained largely by a combination of an intrinsic, 3-fold weaker binding of the RS domain and a further 3-fold weakening by the addition of the isolated RRM1 to the RRM2 RS domain construct.
RRM2 makes direct contacts with CLK1
While replacement of RRM2 with RRM1 in SR(ΔRRM2) results in large decreases in binding affinity to CLK1, deletion of RRM1 has no effect on binding of SRSF1 (Table 1). Such findings suggest that RRM2, unlike RRM1, could make productive contacts with the kinase. To investigate whether RRM2 interacts with CLK1, we evaluated whether two proteins containing the two RRMs [SR(RRM1-2)] or only RRM2 [SR(RRM2)] could affect the phosphorylation of SRSF1 (Fig. 4a). For these experiments, we used a filter-binding assay since we showed that neither deletion protein is phosphorylated by CLK1 (data not shown). We found that both SR(RRM1-2) and SR(RRM2) inhibited CLK1 with similar affinities (Fig. 4b and Table 1), suggesting that RRM2 within the RRM1–RRM2 domain pair is most important for interactions with CLK1. To determine how these constructs bind to SRPK1, we performed competition experiments and found that, while SRPK1 binds with similar affinity to SR(RRM1-2) as CLK1, it binds very poorly to SR(RRM2) (Fig. 4b and Table 1). The KI for SR(RRM1-2) is almost 20-fold lower than that of SR(RRM2) to SRPK1 (Fig. 4c). Consistent with these experiments, GST (glutathione S-transferase)-RRM2 was found to interact with CLK1 in a pull-down assay (Fig. 4d). In control experiments, CLK1 did not interact with free GST bound to the beads. In comparison, prior studies showed that GST-RRM2 did not pull down SRPK1 in line with the poor binding affinity observed in the competition experiments.30 These findings indicate that, whereas CLK1 interacts with RRM2, SRPK1 binds poorly to the isolated RRM2 and instead requires cooperative interactions with both RRMs for efficient interactions.
Fig. 4.
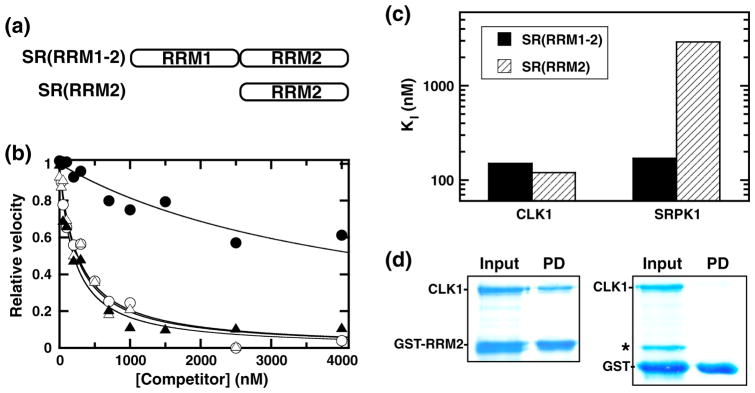
Binding of RRM constructs to CLK1. (a) Deletion constructs. (b) Competition experiments. Fixed amounts of SRSF1 (50 nM) are mixed either with CLK1 (10 nM) and varying amounts of SR(RRM1-2) (△) and SR(RRM2) (▲) or with SRPK1 (1 nM) and varying amounts of SR(RRM1-2) (○) and SR(RRM2) (●). The relative velocities for SRSF1 phosphorylation monitored using a filter-binding assay are plotted as a function of total competitor, and the appKI values are listed in Table 1. (c) Bar graph displaying KI values for SR(RRM1-2) and SR(RRM2) using CLK1 and SRPK1. (d) Pull-down of CLK1 by GST-RRM2. Input contains CLK1 with GST-RRM2 or free GST. Pull-down (PD) contains proteins on G-agarose beads after washing. Starred band represents an impurity, not pulled down by the beads.
CLK1 makes extensive interactions with the RS domain
In prior mapping experiments, we showed that CLK1 phosphorylates a larger number of serines in the RS domain of SRSF1 than does SRPK1.14,22 This broader substrate specificity suggests that CLK1 could make more extensive contacts with the RS domain, possibly explaining the higher affinity of SRSF1 for this kinase compared to SRPK1 (Fig. 2c). We initially made two deletion forms of SRSF1 that lack C-terminal residues in the RS domain (Fig. 5a). SR(1–226) was found to bind with high affinity to CLK1 similar to that for the full-length substrate (Fig. 5b and Table 1). Thus, while CLK1 phosphorylates residues both within and outside the long, N-terminal Arg-Ser repeat,27 those residues outside this sequence appear to play no role in controlling the overall binding affinity of the full-length substrate. Removal of additional residues in SR(1–219) reduces binding affinity by about 3-fold compared to SRSF1, suggesting that further deletion of positive charges begins to weaken affinity (Fig. 5b). These data show that C-terminal residues in the RS domain appear dispensable for high-affinity binding of the SR protein when the RRMs are present.
Fig. 5.

Residue contacts guiding RS domain interactions with CLK1. (a) Deletion constructs. (b) Effects of C-terminal deletion on CLK1 binding in the presence of RRMs. Phosphorylation of SR(ΔRRM1) (50 nM) using CLK1 (10 nM) was measured as a function of SR(1–219) and SR(1–226), and the appKI values are listed in Table 1. (c) Effects of C-terminal deletion on CLK1 binding in the absence of RRMs. The phosphorylation of SRSF1 (50 nM) using CLK1 (4 nM) was measured as a function of SR(ΔRRM1-2), SR(188–235), SR(188–228) and SR(188–200). The appKI for SRSF1 is 26 ± 4 nM and those for SR(188–235), SR(188–228) and SR(188–200) are listed in Table 1. (d) Effects of C-terminal deletion on SRPK1 binding in the absence of RRMs. The phosphorylation of SR(ΔRRM1) (50 nM) using SRPK1 (2 nM) was measured as a function of SR(ΔRRM1-2), SR(188–235), SR(188–228) and SR(188–200); the appKI values are listed in Table 1. (e) Bar graph showing KI values for deletion constructs lacking RRMs.
To evaluate whether C-terminal residues in the RS domain make stable contacts with CLK1, we measured the binding affinities of deletion constructs lacking the RRMs. Removal of the RRMs eliminates the possibility of compensatory binding attractions that might otherwise mask the effects of deletions in the RS domain and allows a better analysis of potential contacts with RS domain residues. We made a series of C-terminal deletions in the parent substrate SR(ΔRRM1-2) and measured their abilities to bind to CLK1 (Fig. 5a and c). Overall, the KI values for SR(188–235), SR(188–228) and SR(188–220) increased 5-, 9- and 11-fold compared to that for SR(ΔRRM1-2) (Table 1). In comparison, more modest changes are observed for SRPK1. While SR(188–235) and SR(ΔRRM1-2) bind with similar affinities, the KI values for SR(188–228) and SR(188–220) are only 3- and 5-fold higher than that for SR(ΔRRM1-2) with SRPK1 (Fig. 5d and Table 1). The large affinity change for SR(188–235) compared to SR(ΔRRM1-2) for CLK1 indicates that residues between 236 and 248 make contacts with CLK1 (Fig. 5e). In comparison, SRPK1 binds SR(188–235) and SR(ΔRRM1-2) with similar affinities, suggesting that these C-terminal residues play no significant role in stabilizing contacts between the RS domain and SRPK1 (Fig. 5e). Overall, these findings suggest that, while SRPK1 principally makes direct contacts with the long Arg-Ser stretch in the N-terminal region, CLK1 makes more widespread contacts within the RS domain.
CLK1 binding and phosphorylation of SRSF1 is assisted by prolines in the RS domain
In addition to long Arg-Ser dipeptide repeats, RS domains in SR proteins commonly contain multiple prolines. Since CLK1 is a cyclin-like kinase and expected to phosphorylate serines next to prolines,14,19 we wished to determine the potential role of several prolines in the RS domain of SRSF1. For these studies, we made single and multiple alanine mutations at four prolines in the RS domain (Fig. 6a). In single turnover experiments, we found that most of the single mutants had no or only minor effects on total phosphorylation levels within the assay time course (Fig. 6b and c). While most of the mutants obey single exponential kinetics, one progress curve is best fit to a biphasic model. For SR(P239A), although the first 5 serines are rapidly modified similar to the wild-type substrate, the remaining serines are only slowly phosphorylated. While these findings suggest that Pro239 is important for rapid phosphorylation of the full RS domain, it appears that the prolines can work together to regulate SR protein phosphorylation. For example, while SR(P239A) is slowly phosphorylated, the inclusion of a second mutation at Pro235 [SR(P235,239A)] leads to a restoration of rapid RS domain phosphorylation (Fig. 6b). In separate single turnover experiments, we showed that SRPK1 phosphorylated all mutants at the same rate and to the same extent as wild-type SRSF1 (data not shown). Overall, the kinetic data indicate that Pro239 plays a significant role in positioning the RS domain for efficient phosphorylation by CLK1.
Fig. 6.

Effects of proline mutations on SRSF1 binding and phosphorylation. (a) Mutations in RS domain prolines. Residues phosphorylated by SRPK1 are highlighted with brackets. (b) Single turnover kinetics. CLK1 (0.5 μM) and 100 μM 32P-ATP are added to 0.2 μM SRSF1 (●), SR(P200A) (○), SR(P228A) (▲), SR(P235A) (△), SR(P239A) (■), SR(P228,235A) (□), SR(P200,228,235A) (▼) and SR(P235,239A) (▽). The data are fit to a single exponential to obtain rate constants and amplitudes of 0.60 ± .08 min−1 and 18 ± 0.6 for SRSF1, 0.44 ± .05 min−1 and 16 ± 0.6 for SR(P200A), 0.34 ± .06 min−1 and 15 ± 0.6 for SR(P228A), 0.58 ± .04 min−1 and 19 ± 0.4 for SR(P235A), 0.41 ± .02 min−1 and 20 ± 0.4 for SR(P228,235A), 0.25 ± .02 min−1 and 13 ± 0.39 for SR(P200,228,235A) and 0.26 ± .02 min−1 and 17 ± 0.34 for SR(P235,239A), respectively. SR(P239A) was fit to a double exponential with amplitudes of 5 ± 0.4 and 15 ± 0.4 sites and rate constants of 1.1 ± .25 and 0.034 ± 0.002 min−1, respectively. (c) Bar graph showing phosphoryl contents of mutants. Data are taken from the total amplitudes in (b). (d and e) Competition experiments for mutants using SRPK1 (d) and CLK1 (e). Fixed amounts of SR(ΔRRM1) (50 nM) are mixed with either CLK1 (10 nM) or SRPK1 (1 nM) and varying amounts of mutant SR proteins. Mutants are labeled as in (b), and the appKI and KI values are displayed in Table 1. (f) Bar graph showing KI values for the mutants relative to SRSF1 for CLK1 and SRPK1.
Since the kinetic data suggest that prolines serve a role in serine phosphorylation, we wished to determine whether they also affect the stability of the CLK1–SR protein complex. Although none of the proline mutations impacted binding to SRPK1 in competition assays, some had large effects on CLK1 binding (Fig. 6d and e). While SR(P200A) and SR(P228A) had no effect, SR(P235A) and SR(P239A) reduced binding to CLK1 by 12- and 5-fold (Fig. 6f and Table 1).
Additional proline mutations within the SR(P235A) construct [SR(P228, 235A) and SR(P200, 228, 235A)] had either no or only a 2-fold additional effect on binding (Table 1). These findings suggest that Pro235 and Pro239 in the RS domain play pivotal roles in controlling the stability of the CLK1–SRSF1 complex. Combining the Pro235 and Pro239 mutations into one construct [SR(P235,239A)] did not reduce binding affinity beyond SR(P239A), suggesting that the prolines can function in a complex manner to control interactions with CLK1 (Fig. 6f). Overall, the single turnover and competition binding experiments reveal that, while rapid phosphorylation of the RS domain is largely dependent on Pro239, high-affinity SR protein binding relies on Pro235 and Pro239.
Multisite phosphorylation of SRSF1 by SRPK1 and CLK1
In a previous study, we showed that CLK1 phosphorylates SRSF1 to a greater extent than SRPK1, inducing a hyper-phosphorylated state that can be readily detected by a gel shift on SDS-PAGE.14 In Fig. 7a, the phosphorylation of SRSF1 in single turnover experiments using SRPK1 and CLK1 is shown. SRPK1 initially phosphorylates about 11 serines in the first minute and then modifies several additional serines over a longer time frame until an endpoint of about 15 phosphates is reached.
Fig. 7.

Gel-shift analyses of Ser-Pro mutants of SRSF1. (a) Progress curves for SRSF1 phosphorylation by SRPK1 (●) or CLK1 (▲). SRPK1 (1 μM) or CLK1 (0.5 μM) is preequilibrated with 0.2 μM SRSF1 and reacted with 100 μM 32P-ATP. The data for SRPK1 are fit to a double exponential with amplitudes of 11 ± 1 and 4 ± 1 sites and rate constants of 5 ± 4 and 0.3 ± 0.1 min−1. The data for CLK1 are fit to a single exponential with an amplitude and rate constant of 18 ± 0.5 sites and 0.65 ± 0.04 min−1. (b) Mutations in Ser-Pro dipeptides of RS domain. Residues 227–248 of the RS domain are displayed. (c) Single turnover kinetics. CLK1 (0.5 μM) and SR protein (0.2 μM) are mixed with 100 μM 32P-ATP, and the total number of sites is plotted as a function of time. Data are fit to single exponential functions to obtain amplitudes and rate constants of 18 ± 0.4 and 0.44 ± 0.04 min−1 for SR(S227A) (○), 17 ± 0.85 and 0.62 ± 0.10 min−1 for SR(S234A) (△), 18 ± 0.50 and 0.61 ± 0.071 min−1 for SR(S238A) (□) and 13 ± 0.40 and 0.34 ± 0.022 min−1 for SR(S227,234,238A) (■), respectively. (d) Kinetic analyses of gel shift. CLK1 (0.5 μM) and SRSF1 or SR(S227,234,238A) (TM) are mixed with 100 μM 32P-ATP, and the reaction is stopped at various time points and run on a 10% SDS-PAGE. (e) Effects of phosphorylation on mobility of single mutants. CLK1 (0.5 μM) and SR protein (0.2 μM) are mixed with 100 μM 32P-ATP, and the reaction is stopped after 30 min and run on an 10% SDS-PAGE.
In comparison, CLK1 modifies a larger number of sites (18 serines) although the rate of this reaction is slower than the first kinetic phase for SRPK1. Prior mapping studies showed that the initial phase for SRPK1 represents phosphorylation in the N-terminus of the RS domain between residues 205 and 225 whereas the second phase includes phosphorylation after residue 229 (Fig. 5a).22 Interestingly, while SRPK1 is considerably faster than CLK1 in the initial phase, the rate constant for the slower phase is similar to that for CLK1 (0.3 versus 0.6 min−1). These data indicate that SRPK1 and CLK1 have similar phosphorylation activities toward C-terminal residues in the RS domain of SRSF1.
SRSF1 hyper-phosphorylation is the result of Ser-Pro phosphorylation
Phosphorylation kinetics indicate that SRPK1 can modify 15 serines in the RS domain whereas CLK1 can modify about 18 serines in a similar time frame. Since we showed previously that SRPK1 is directed at Arg-Ser repeats,22 we wondered whether the difference in total phosphoryl contents in these experiments are the result of CLK1-induced Ser-Pro phosphorylation. To address this question, we made several serine-to-alanine mutations in the RS domain of SRSF1 (Fig. 7b). We made single alanine mutations at Ser227, Ser234 and Ser238 as well as a triple mutant to assess Ser-Pro phosphorylation. In single turnover experiments, we found that the single mutants had only small effects as expected, whereas the overall phosphoryl content of the triple mutant [SR(S227,234,238A)] is reduced by a level consistent with the removal of three serines (Fig. 7c). The phosphorylation level of the triple mutant after 15 min of incubation with CLK1 is reduced to about 13 sites, a value close to that for the wild-type SR protein after phosphorylation with SRPK1 (Fig. 7a and c). The minor difference between this mutant and SRPK1-phosphorylated SRSF1 could reflect a small effect of Ser-Pro mutations on Arg-Ser phosphorylation. Overall, none of the mutations affected the rate constant for multisite phosphorylation. These findings indicate that CLK1 not only phosphorylates Ser-Pro dipeptides as previously suggested6 but also does not require these modifications for multisite phosphorylation of other serines in the RS domain of SRSF1.
To determine whether any of the mutations in the Ser-Pro dipeptides affect the gel-shift phenomenon, we performed SDS-PAGE analyses. As shown in Fig. 6d, CLK1 readily induced a gel shift in SRSF1 that occurs within the first 2 min after approximately 5–10 serines are modified. In comparison, the triple mutant does not undergo a gel shift even after 10 min when the reaction has reached an apparent endpoint (Fig. 7d). To determine whether phosphorylation at all three serines is necessary to attain the gel shift, we compared the mobilities of the single mutants to that for wild-type SR protein and the triple mutant at the end of the phosphorylation reaction. In general, the single mutants display intermediate mobilities on SDS-PAGE compared to SRSF1 and the triple mutant (Fig. 7e). These findings suggest that phosphorylation of all three serines in this region may be required for the observed gel shift. To assess whether this deficiency is due to poor interactions of the SR(S227,234,238A) with CLK1, we performed competition studies and found that the triple mutant observed no defects in binding compared to SRSF1 (data not shown). Overall, these data indicate that the gel-shifted form of SRSF1 is induced by CLK1-dependent phosphorylation of three Ser-Pro dipeptides in the RS domain. Also, it appears that SRPK1 and CLK1 have overlapping substrate specificities with CLK1 being distinctive for its ability to uniquely modify Ser-Pro dipeptides.
Discussion
CLK1 and SRPK1 use different strategies for SR protein phosphorylation
Through detailed kinetic and structural studies, the mechanism of SRSF1 phosphorylation by SRPK1 is now well understood.2 An electronegative docking groove in the large lobe of this kinase firmly orients a long Arg-Ser repeat so that the C-terminal end of the stretch initially resides in the active site (Fig. 8a). As the reaction proceeds, phosphoserines are expelled and new dipeptide repeats are fed from the docking groove into the active site. Thus, the juxtaposition of a docking groove and active site along with neighboring RRMs enforce a highly ordered mechanism for RS domain processing. In comparison, CLK1 lacks a similar docking groove and has been shown to catalyze a random phosphorylation mechanism.27 Despite this apparent handicap, CLK1 binds SRSF1 with better affinity than SRPK1, a result of a combination of enhanced RS domain as well as RRM2 contacts (Figs. 2–4). While SRPK1 only requires N-terminal Arg-Ser repeats for high-affinity binding, CLK1 utilizes these along with additional contacts in the short, C-terminal Arg-Ser stretch to initially bind the RS domain (Fig. 5). Given these new discoveries, we propose that CLK1, lacking the constraints of a docking groove, is likely to initially bind the RS domain in several modes, generating multiple, distinct enzyme–substrate complexes (Fig. 8b). While SRPK1 imposes a strict zone of initiation in the center of the RS domain owing to the docking groove, CLK1 initiates at two or more positions creating an observed, random phosphorylation mechanism.
Fig. 8.

Model for CLK1- and SRPK1-dependent phosphorylation of SRSF1. (a) SRPK1 uses a docking groove to orient the RS domain and induce C-to-N-terminal phosphorylation. (b) CLK1 likely allows several bound forms of the RS domain to induce random phosphorylation. (c) Model for interplay of SRPK1 and CLK1 activities on SRSF1. Black regions in the RS domain are rich in Arg-Ser repeats and divided into N- and C-terminal repeats (RS-N and RS-C). Red region is rich in Ser-Pro dipeptides.
How CLK1 accommodates the RS domain of SRSF1 in various binding modes is not yet understood. RS domains are expected to be highly disordered and capable of sampling numerous conformations in solution.31 While CLK1 is likely to use this flexibility to access different binding modes, the new data suggest that some prolines may provide critical switch points for specific conformations. We found that, while none of the prolines in the RS domain are important for SRPK1, Pro235 and Pro239 are important for CLK1 binding (Fig. 6). Prolines are unique residues owing to their ability to adopt cis and trans conformers, greatly affecting protein structure. We propose that the prolines in SRSF1 may be important for arranging the RS domain in more than one binding conformation on the surface of CLK1 (Fig. 8b). Furthermore, since all SR proteins contain multiple prolines in their RS domains, these residues could serve a general function. Why CLK1 would use several attachment points along the RS domain to bind SRSF1 is not well understood. It is possible that this kinase, lacking a docking groove, may have evolved alternate features to accommodate diverse RS domains. For example, CLKs have long N-terminal extensions with isolated Arg-Ser dipeptides that, based on yeast two-hybrid studies, are necessary for CLK1 binding to SRSF1.6 It is possible that these Arg-Ser dipeptides, while not substrates, may interact with the RS domains in SR proteins and provide both high affinity and elasticity for the random phosphorylation mechanism. Overall, while the unique structure of CLK1 appears to offer flexibility in targeting the RS domain, the loss of a rigid apposition of docking groove and active site results in a less efficient kinase. While the phosphoryl transfer step for SRPK1 is fast and does not limit SR protein turnover, that for CLK1 is about 2 orders of magnitude lower and is fully rate limiting (Fig. 1).
CLK1 induces a unique structural form of SRSF1
In detailed kinetic studies, SRPK1 was found to phosphorylate a total of 15 serines whereas CLK1 phosphorylates 18 serines in a similar time frame (Fig. 7a). SRPK1 initially phosphorylates about 10–12 serines in the N-terminus of the RS domain and then phosphorylates the remaining serines in a shorter C-terminal Arg-Ser repeat.22 Interestingly, these later phosphorylation events occur at approximately the same rate for both CLK1 and SRPK1, suggesting that neither kinase displays a preference for this region. CLK1 induces a gel shift in SRSF1 on SDS-PAGE.14 Based on prior cellular studies, it was proposed that this phenotype is the result of additional phosphorylation sites introduced by CLK1 in the RS2 segment of the RS domain (Fig. 1). We now show that this gel-shifted form is not the result of large, bulk increases in serine phosphorylation but rather is due to the phosphorylation of only 3 Ser-Pro dipeptides in the RS domain (Fig. 7). Thus, while both SRPK1 and CLK1 phosphorylate the Arg-Ser repeats in SRSF1, CLK1 uniquely phosphorylates Ser-Pro dipeptides. Since CLK1 adds only 3 additional phosphates above the large basal phosphorylation of 15 sites catalyzed by SRPK1, we propose that the observed gel shift is the likely result of a structural change in the RS domain. A recent study showed that SRPK1 phosphorylation promotes an interaction between the RS domain and RRMs in SRSF1.32 In comparison, CLK1 hyper-phosphorylation promotes a ternary interaction of SRSF1 with U1 and pre-mRNA establishing the 5′ splice site. Such changes in binding partners are likely to be induced by changes in the conformation of the RS domain that regulates RRM contacts in the spliceosome. The studies presented herein suggest that this molecular trigger may have its origins in Ser-Pro phosphorylation, which is uniquely catalyzed by CLK1.
Developing a new model for SRPK–CLK substrate specificities
There is now ample evidence that SRPK1-directed phosphorylation of N-terminal sequences in the RS domain promotes translocation of SRSF1 from the cytoplasm to the nucleus.15,33 Both phosphorylation kinetics and phospho-mapping studies support a model where SRPK1 rapidly modifies a stretch of 10–12 serines in this region, thus providing a biochemical connection to this cellular mechanism.14,22 Once in the nucleus, it is postulated that CLK1 phosphorylates the remaining C-terminal sequences including the shorter Arg-Ser stretch and Ser-Pro dipeptides (Fig. 1). The studies presented herein now suggest a new model that may have a broader application within the SR protein family. SRPK1 is known to transition into the nucleus at various stages of the cell cycle and in response to cellular signals.9,34 Furthermore, SRPKs have been found associated with the spliceosome where they may serve a basal function in nuclear splicing.5
These findings indicate that SRPKs and CLKs can occupy the same compartments in the cell and have access to the same SR protein pool. Our observations that SRPK1 and CLK1 can target the same Arg-Ser repeats in the C-terminus of SRSF1 at similar rates raises the possibility that their activities in the nucleus may be differentiated based entirely on Arg-Ser versus Ser-Pro phosphorylation patterns (Fig. 8b) rather than on regiospecific grounds where SRPK1 targets only N-terminal Arg-Ser repeats and CLK1 targets all remaining serines in the C-terminal portion (Fig. 1). This new activity could be extended to all SR proteins since Ser-Pro dipeptides are a common feature of RS domains. Finally, although RS2 phosphorylation is not necessary for cytoplasmic–nuclear shuttling of SRSF1,15 SRPK1 may still phosphorylate Arg-Ser repeats in this segment in the cytoplasm, leaving Ser-Pro phosphorylation under the province of nuclear CLKs.
Conclusions
The kinetic data presented herein provide several new insights into the phosphorylation mechanism of the SR protein SRSF1 by SRPK1 and CLK1. First, despite lacking a docking groove, CLK1 binds SRSF1 with very high affinity. Deletion analyses reveal that CLK1 binds the RS domain better than SRPK1 even though CLK1 lacks a docking groove for the recognition of Arg-Ser repeats. CLK1 also appears to use the neighboring RRMs to enhance substrate binding with RRM2, making direct contacts with the kinase. Second, while SRPK1 makes strong interactions with mostly N-terminal Arg-Ser repeats, CLK1 appears to make more widespread contacts both with the N-terminal as well as the shorter, C-terminal Arg-Ser stretch. The ability to make multi-dentate contacts without a well-defined docking groove is likely to explain why CLK1 catalyzes random phosphorylation of the SRSF1 RS domain while SRPK1 moves in a unidirectional manner. These observations suggest that, unlike SRPK1, CLK1 is likely to form several distinct enzyme–substrate complexes for phosphorylation initiation at more than one position in the RS domain. Third, prolines are essential for high-affinity SR protein binding and phosphorylation of SRSF1. We propose that these prolines play a role in generating the multiple, bound forms of the RS domain that guide the random phosphorylation mechanism. Fourth, we showed that Ser-Pro phosphorylation is under the control of the CLK family of protein kinases and that these modifications induce a unique form of SRSF1 and possibly other SR proteins that plays highly specialized functions in splicing.
Materials and Methods
Materials
ATP, 3-(N-morpholino)propanesulfonic acid (Mops), Tris, MgCl2, NaCl, ethylenediaminetetraacetic acid, glycerol, sucrose, acetic acid, lysozyme, DNase, RNase, Phenix imaging film, bovine serum albumin (BSA), Whatman P81-grade filter paper and liquid scintillant were obtained from Fisher Scientific. [γ-32P]ATP was obtained from NEN Products, a division of Perkin-Elmer Life Sciences. Protease inhibitor cocktail was obtained from Roche.
Expression and purification of recombinant proteins
SRPK1, CLK1 and SRSF1 were expressed from a pET19b vector containing a 10× His tag at the N-terminus.14 All mutations in SRSF1 were generated by single or sequential polymerase chain reactions using the QuikChange™ mutagenesis kit and relevant primers (Stratagene, La Jolla, CA). The construction of the plasmids for the deletion mutants was described in a previous report.20,22 The plasmids for wild-type and mutant forms of SRSF1, SRPK1 and CLK1 were transformed into the BL21 (DE3) Escherichia coli strain; the cells were then grown at 37 °C in LB broth supplemented with 100 μg/mL ampicillin, and protein expression was induced with 1 μg/mL IPTG at room temperature for 5 h for SRSF1 constructs, 1 μg/mL IPTG at room temperature for 12 h for SRPK1 and 2.5 μg/mL IPTG (16 h) for CLK1. All SRPK1 constructs were purified by Ni-resin affinity chromatography using a published procedure.35 All SRSF1 constructs were refolded and purified using a previously published protocol.21 For CLK1, 2 L of pelleted cells was lysed in 50 mL lysis buffer [50 mM phosphate-buffered saline and 500 mM NaCl (pH 7.8)] with DNase, RNase, lysozyme, inhibitor cocktail (Roche) and 1 mM PMSF added. The soluble fraction was applied to a 3-mL Ni2+–Sepharose column, and the bound protein was washed with 40 mM imidazole and then eluted with 300 mM imidazole. A buffer exchange [50 mM Mops (pH 7.6), 500 mM NaCl, 1 mM DTT, 1 mM ethylenediaminetetraacetic acid and 20% glycerol] was then performed with a 50,000-cutoff centrifugal filter (Millipore), and concentration measurements were taken using a Bradford assay.
Phosphorylation reactions and manual mixing
The phosphorylation of wild-type and mutant forms of SRSF1 by SRPK1 and CLK1 were carried out in the presence of 100 mM Mops (pH 7.4), 10 mM free Mg2+ and 5 mg/mL BSA, at 23 °C according to previously published procedures.21 For single turnover experiments, reactions were carried out with 1 μM enzyme, 0.2 μM SRSF1 and 100 μM [γ-32P]ATP (4000–8000 cpm/pmol) unless otherwise stated. For competition experiments, reactions were carried out with 1–2 nM SRPK1 and 10 nM CLK1 with 5 mM and 20 mM ATP [γ-32P]ATP (4000–8000 cpm/pmol), respectively. Competition reactions were carried out using fixed amounts of SR(μRRM1) (50 nM) or SRSF1 (50 nM) as substrates and varying concentrations of the competitors (substrate inhibitors). All reactions were carried out in a total reaction volume of 10 μL and then were quenched with 10 μL SDS-PAGE loading buffer. Phosphorylated SRSF1 was separated from unreacted ATP by loading the quenched reaction on an SDS-PAGE gel (10% or 16%) and running at 170 V for 1 h. Protein bands corresponding to phosphorylated SRSF1 were cut from the dried SDS-PAGE gel and quantitated on the 32P channel in liquid scintillant. The total amount of phosphoproduct was then determined by considering the specific activity (cpm/min) of the reaction mixture and the background retention of 32P-ATP in the absence of enzyme.
Rapid quench flow experiments
Pre-steady-state kinetic measurements were made using a KinTek Corporation Model RGF-3 quench flow apparatus following a previously published procedure.25 The apparatus consists of three syringes driven by a stepping motor. Typical experiments were performed by mixing equal volumes of the enzyme–SRSF1 complex in one reaction loop and [γ-32P]ATP (5000–15,000 cpm/pmol) in the second reaction loop all in the presence of 100 mM Mops (pH 7.4), 10 mM free Mg2+ and 5 mg/mL BSA, with the reaction quenched with 30% acetic acid in the third syringe. Phosphorylated SRSF1 was separated from unreacted ATP using a filter-binding assay where a portion of each quenched reaction (50 μL) was spotted onto a phosphocellulose filter disk and was washed 3 times with 0.5% phosphoric acid. The filter disks were rinsed with acetone, dried and counted on the 32P channel in liquid scintillant. The total amount of phosphoproduct was then determined by considering the specific activity (cpm/min) of the reaction mixture and the background retention of 32P-ATP in the absence of enzyme. Full retention of the phosphorylated product on the filters was confirmed by running quenched reaction samples on SDS-PAGE and counting the bands. Control experiments lacking enzyme–SRSF1 complex were run to define a background correction.
Viscosity studies
The steady-state phosphorylation of SRSF1 was monitored using the filter-binding assay as described above in the presence of 0–30% sucrose. The relative solvent viscosity (ηrel) of the buffer (100 mM Mops, pH 7.4) containing 0–30% sucrose was measured using an Ostwald viscometer and a previously published protocol.36 ηrel values of 1.44, 1.83, 2.32 and 3.43 were measured for buffer containing 10%, 20%, 25% and 30% sucrose at 23 °C.
Pull-down assays
For pull-down experiments, GST-RRM2 (10 μM) or free GST (10 μM) was incubated with His-CLK1 (2.5 μM) for 30 min in the binding buffer [0.1% NP40, 20 mM Tris (pH 7.5) and 75 mM NaCl]. The mixtures were then incubated with 15 μL glutathione-modified agarose resin for 30 min. The resin was washed 4 times with 40 μL binding buffer, and the bound proteins were eluted by treating with SDS quench buffer for 5 min at 90 °C. Bound protein was resolved by 16% SDS-PAGE and visualized by Instant Blue Coomassie stain.
Data analysis
The counts per minute were corrected for background and converted to concentrations of phosphoproduct using the specific activity of ATP. In single turnover experiments, the time-dependent production of phosphoproduct was fit to either single or double exponential functions. The initial velocity data were fit to the Michaelis–Menten equation to obtain Km and Vmax. The Vmax values were converted to kcat using the total enzyme concentration determined from a Bradford assay (kcat = Vmax/Etot). The relative initial velocities for the competition data were fit to Eq. (1):
| (1) |
where vi/vo is the relative initial velocity (ratio of v in the presence and absence of inhibitor), Eo is the total enzyme concentration, Io is the total substrate inhibitor concentration and appKI is the apparent dissociation constant for the substrate inhibitor. The KI is calculated from appKI using Eq. (2):
| (2) |
where KI is the true dissociation constant for the substrate inhibitor, Km is the Michaelis constant for the fixed substrate and [S] is the fixed substrate concentration.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants GM67969 and GM98528 to J.A.A. R.M.P. was supported by NIH under the Ruth L. Kirschstein National Research Service Award (GM090484). C-T.M. and J.C.H. were supported by an NIH training grant (GM07752).
Abbreviations used
- BSA
bovine serum albumin
- CLK
Cdc2-like kinase
- RRM
RNA recognition motif
- SRPK
SR-specific protein kinase
- NIH
National Institutes of Health
References
- 1.Stojdl DF, Bell JC. SR protein kinases: the splice of life. Biochem Cell Biol. 1999;77:293–298. [PubMed] [Google Scholar]
- 2.Ghosh G, Adams JA. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 2011;278:587–597. doi: 10.1111/j.1742-4658.2010.07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeakley JM, Tronchere H, Olesen J, Dyck JA, Wang HY, Fu XD. Phosphorylation regulates in vivo interaction and molecular targeting of serine/arginine-rich pre-mRNA splicing factors. J Cell Biol. 1999;145:447–455. doi: 10.1083/jcb.145.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai MC, Lin RI, Huang SY, Tsai CW, Tarn WY. A human importin-β family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J Biol Chem. 2000;275:7950–7957. doi: 10.1074/jbc.275.11.7950. [DOI] [PubMed] [Google Scholar]
- 5.Mathew R, Hartmuth K, Mohlmann S, Urlaub H, Ficner R, Luhrmann R. Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat Struct Mol Biol. 2008;15:435–443. doi: 10.1038/nsmb.1415. [DOI] [PubMed] [Google Scholar]
- 6.Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- 7.Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper and hypophosphorylation inhibit splicing. Mol Cell Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sumanasekera C, Kelemen O, Beullens M, Aubol BE, Adams JA, Sunkara M, et al. C6 pyridinium ceramide influences alternative pre-mRNA splicing by inhibiting protein phosphatase-1. Nucleic Acids Res. 2012;40:4025–4039. doi: 10.1093/nar/gkr1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell. 2012;47:422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, Hannun YA. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem. 2002;277:12587–12595. doi: 10.1074/jbc.M112010200. [DOI] [PubMed] [Google Scholar]
- 11.Kohtz JD, Jamison SF, Will CL, Zuo P, Luhrmann R, Garcia-Blanco MA, Manley JL. Protein–protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 12.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 13.Valcarcel J, Gaur RK, Singh R, Green MR. Interaction of U2AF65 RS region with pre-mRNA branch point and promotion of base pairing with U2 snRNA [corrected] Science. 1996;273:1706–1709. doi: 10.1126/science.273.5282.1706. [DOI] [PubMed] [Google Scholar]
- 14.Velazquez-Dones A, Hagopian JC, Ma CT, Zhong XY, Zhou H, Ghosh G, et al. Mass spectrometric and kinetic analysis of ASF/SF2 phosphorylation by SRPK1 and Clk/Sty. J Biol Chem. 2005;280:41761–41768. doi: 10.1074/jbc.M504156200. [DOI] [PubMed] [Google Scholar]
- 15.Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, et al. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 16.Lai MC, Lin RI, Tarn WY. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc Natl Acad Sci USA. 2001;98:10154–10159. doi: 10.1073/pnas.181354098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yomoda J, Muraki M, Kataoka N, Hosoya T, Suzuki M, Hagiwara M, Kimura H. Combination of Clk family kinase and SRp75 modulates alternative splicing of adenovirus E1A. Genes Cells. 2008;13:233–244. doi: 10.1111/j.1365-2443.2008.01163.x. [DOI] [PubMed] [Google Scholar]
- 18.Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- 19.Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, Fu XD. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J Biol Chem. 1996;271:24569–24575. doi: 10.1074/jbc.271.40.24569. [DOI] [PubMed] [Google Scholar]
- 20.Hagopian JC, Ma CT, Meade BR, Albuquerque CP, Ngo JC, Ghosh G, et al. Adaptable molecular interactions guide phosphorylation of the SR protein ASF/SF2 by SRPK1. J Mol Biol. 2008;382:894–909. doi: 10.1016/j.jmb.2008.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma CT, Velazquez-Dones A, Hagopian JC, Ghosh G, Fu XD, Adams JA. Ordered multi-site phosphorylation of the splicing factor ASF/SF2 by SRPK1. J Mol Biol. 2008;376:55–68. doi: 10.1016/j.jmb.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 22.Ma CT, Hagopian JC, Ghosh G, Fu XD, Adams JA. Regiospecific phosphorylation control of the SR protein ASF/SF2 by SRPK1. J Mol Biol. 2009;390:618–634. doi: 10.1016/j.jmb.2009.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngo JC, Giang K, Chakrabarti S, Ma CT, Huynh N, Hagopian JC, et al. A sliding docking interaction is essential for sequential and processive phosphorylation of an SR protein by SRPK1. Mol Cell. 2008;9:563–576. doi: 10.1016/j.molcel.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams JA. Activation loop phosphorylation and catalysis in protein kinases: is there functional evidence for the autoinhibitor model? Biochemistry. 2003;42:601–607. doi: 10.1021/bi020617o. [DOI] [PubMed] [Google Scholar]
- 25.Aubol BE, Adams JA. Applying the brakes to multisite SR protein phosphorylation: substrate-induced effects on the splicing kinase SRPK1. Biochemistry. 2011;50:6888–6900. doi: 10.1021/bi2007993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aubol BE, Plocinik RM, McGlone ML, Adams JA. Nucleotide release sequences in the protein kinase SRPK1 accelerate substrate phosphorylation. Biochemistry. 2012;51:6584–6594. doi: 10.1021/bi300876h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma CT, Ghosh G, Fu XD, Adams JA. Mechanism of dephosphorylation of the SR protein ASF/SF2 by protein phosphatase 1. J Mol Biol. 2010;403:386–404. doi: 10.1016/j.jmb.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bullock AN, Das S, Debreczeni JE, Rellos P, Fedorov O, Niesen FH, et al. Kinase domain insertions define distinct roles of CLK kinases in SR protein phosphorylation. Structure. 2009;17:352–362. doi: 10.1016/j.str.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams JA, Taylor SS. Energetic limits of phosphotransfer in the catalytic subunit of cAMP dependent protein kinase as measured by viscosity experiments. Biochemistry. 1992;31:8516–8522. doi: 10.1021/bi00151a019. [DOI] [PubMed] [Google Scholar]
- 30.Huynh N, Ma CT, Giang N, Hagopian J, Ngo J, Adams J, Ghosh G. Allosteric interactions direct binding and phosphorylation of ASF/SF2 by SRPK1. Biochemistry. 2009;48:11432–11440. doi: 10.1021/bi901107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haynes C, Iakoucheva LM. Serine/arginine-rich splicing factors belong to a class of intrinsically disordered proteins. Nucleic Acids Res. 2006;34:305–312. doi: 10.1093/nar/gkj424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho S, Hoang A, Sinha R, Zhong XY, Fu XD, Krainer AR, Ghosh G. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci USA. 2011;108:8233–8238. doi: 10.1073/pnas.1017700108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cazalla D, Zhu J, Manche L, Huber E, Krainer AR, Caceres JF. Nuclear export and retention signals in the RS domain of SR proteins. Mol Cell Biol. 2002;22:6871–6882. doi: 10.1128/MCB.22.19.6871-6882.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- 35.Aubol BE, Chakrabarti S, Ngo J, Shaffer J, Nolen B, Fu XD, et al. Processive phosphorylation of alternative splicing factor/splicing factor 2. Proc Natl Acad Sci USA. 2003;100:12601–12606. doi: 10.1073/pnas.1635129100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grant BD, Adams JA. Pre-steady-state kinetic analysis of cAMP-dependent protein kinase using rapid quench flow techniques. Biochemistry. 1996;35:2022–2029. doi: 10.1021/bi952144+. [DOI] [PubMed] [Google Scholar]