

Abstract
The early steps of HIV-1 replication involve the entry of HIV-1 into the nucleus, which is characterized by viral interactions with nuclear pore components. HIV-1 developed an evolutionary strategy to usurp the nuclear pore machinery and chromatin in order to integrate and efficiently express viral genes. In the current work, we studied the role of nucleoporins 153 and 98 (Nup153 and Nup98) in infection of human Jurkat lymphocytes by HIV-1. We showed that Nup153-depleted cells exhibited a defect in nuclear import, while depletion of Nup 98 caused a slight defect in HIV integration. To explore the biochemical viral determinants for the requirement of Nup153 and Nup98 during HIV-1 infection, we tested the ability of these nucleoporins to interact with HIV-1 cores. Our findings showed that both nucleoporins bind HIV-1 cores suggesting that this interaction is important for HIV-1 nuclear import and/or integration. Distribution analysis of integration sites in Nup153-depleted cells revealed a reduced tendency of HIV-1 to integrate in intragenic sites, which in part could account for the large infectivity defect observed in Nup153-depleted cells. Our work strongly supports a role for Nup153 in HIV-1 nuclear import and integration.
Keywords: HIV-1, Nuclear import, Nup98, Nup153, Integration, Capsid
Introduction
The influence of the physiological state of cells on retroviral replication was initially studied by Rubin and Temin; they demonstrated that stopping cell division by X-rays or UV light prevents Rous Sarcoma virus replication (Rubin and Temin, 1959). Subsequent research established the relationship between cell cycle stage and retroviral infection, revealing that diverse retro-viruses have different requirements for productive infection (Katz et al., 2003; Suzuki and Craigie, 2007; Yamashita and Emerman, 2006). For example, gamma-retroviruses such as murine leukemia virus (MLV) require the host cell to pass through mitosis for productive infection (Lewis and Emerman, 1994; Roe et al., 1993). By contrast, lentiviruses such as HIV-1 show no difference in productive infection when comparing dividing versus non-dividing cells (Lewis et al., 1992). This evidence suggests that lentiviruses have developed specific mechanisms for the infection of non-dividing cells. The ability of HIV-1 to infect non-dividing cells has been attributed to the capacity of the virus to transport the pre-integration complex (PIC) to the nucleus (Miller et al., 1997; Suzuki and Craigie, 2007). Translocation of the HIV-1 PIC into the nucleus is not a simple process, as its size is similar to that of a ribosome, which is at least 56 nm in diameter (Bowerman et al., 1989; Miller et al., 1997). Because of this large size, it is unlikely that the PIC enters the nucleus by passive diffusion (Mattaj and Englmeier, 1998). On the contrary, HIV-1 PIC translocation into the nucleus must be an active process. Several components of the PIC such as matrix, Vpr, integrase and the central DNA flap, have been proposed to be directly involved in PIC transport into the nucleus (De Rijck et al., 2007; Fassati, 2006). Although barely detectable amounts of capsid can be found on the HIV-1 PIC (Bukrinsky et al., 1993; Fassati and Goff, 2001; Iordanskiy et al., 2006; Miller et al., 1997), recent evidence has shown that capsid plays a role in the ability of HIV-1 to infect non-dividing cells (Yamashita and Emerman, 2004, 2006; Yamashita et al., 2007). In agreement, several studies found the HIV-1 core associated to the nuclear pore (Arhel et al., 2007; Di Nunzio et al., 2012). While the mechanism used by the HIV-1 PIC to enter the nucleus is not completely understood, it is accepted that nuclear import of the complex is active and energy-dependent (Suzuki and Craigie, 2007).
In addition to the viral determinants involved in HIV-1 PIC nuclear import, several host factors have been described to play a role in this process: 1) importin 7 (Ao et al., 2007; Fassati et al., 2003; Zaitseva et al., 2009), 2) Importin a3 (Ao et al., 2010), 3) importin α/importin β heterodimer (Fassati et al., 2003; Gallay et al., 1997; Hearps and Jans, 2006), 4) transportin-SR2/TNPO3 (Brass et al., 2008; Christ et al., 2008; Konig et al., 2008; Krishnan et al., 2010; Levin et al., 2010; Thys et al., 2011; Valle-Casuso et al., 2012; Zhou et al., 2008), 5) RANBP2 (Di Nunzio et al., 2012; Ocwieja et al., 2011) and 6), Nup98 and Nup153 (Di Nunzio et al., 2012; Konig et al., 2008; Lee et al., 2010; Matreyek and Engelman, 2011; Woodward et al., 2009).
Recent work has suggested a role for Nup98 and Nup153 in HIV-1 replication (Di Nunzio et al., 2012; Konig et al., 2008; Matreyek and Engelman, 2011). Nup98, which is symmetrically localized to the cytoplasmic and nuclear sides of the nuclear pore, is involved in HIV-1 integration (Di Nunzio et al., 2012; Ebina et al., 2004; Konig et al., 2008). Nup153 that localizes to the nuclear side of the nuclear pore, was initially described to be involved in HIV-1 replication by three independent genome-wide siRNA screens (Brass et al., 2008; Konig et al., 2008; Zhou et al., 2008). Although the C terminus domain of Nup153 has been shown to bind the HIV-1 integrase protein using bacterially purified proteins (Woodward et al., 2009), the genetic determinant for the requirement of Nup153 during HIV-1 infection has been mapped to the capsid protein (Matreyek and Engelman, 2011). In agreement with the latter, HIV-1 bearing mutations on capsid showed different infection sensitivities to the depletion of Nup153 (Lee et al., 2010; Matreyek and Engelman, 2011; Schaller et al., 2011). However, a direct interaction between capsid and Nup153 has not been shown.
The current work explores the role of Nup98 and Nup153 in HIV-1 infection. We initially confirmed the involvement of Nup98 and Nup153 in the early steps of HIV-1 infection by testing infectivity of Nup98 and Nup153 -depleted Jurkat cells. Nup98-depleted cells modestly block HIV-1 infection during integration. By contrast, Nup-153-depleted cells preferentially block HIV-1 infection during nuclear import. In agreement with findings that showed capsid as the determinant for the requirement of Nup153, we demonstrated the ability of Nup153 to interact with the HIV-1 core. Because depletion of Nup153 affects nuclear import but severely diminishes infectivity, we studied the distribution of HIV-1 integration sites in Nup153-depleted Jurkat cells. Our findings revealed that the intragenic/intergenic ratio of integration sites changes in Nup153-depleted cells.
Results
Depletion of NUP98 and Nup153 expression inhibits HIV-1 infection of human lymphocytes
To test the role of Nup98 and Nup153 in HIV-1 infection, we first silenced the expression of these nucleoporins in human T cell lymphoblast-like cells (Jurkat) by using lentiviral particles expressing specific shRNAs. As shown in Fig. 1A, we used a lentiviral shRNA construct that expresses the green fluorescent protein (GFP) and contains a deletion on the U3 region, which will subsequently facilitate the study of integration sites. These lentiviral vectors carrying shRNA and GFP as internal reporter gene were tittered on HeLa cells by flow cytometry. After transduction of Jurkat cells at the indicated multiplicities of infection (MOI), expression of Nup98 and Nup153 was monitored by western blotting 5 and 3 days after transduction, respectively. As shown in Fig. 1B, the use of lentiviral particles containing the different shRNAs achieved strong knockdown (KD) of Nup98 and Nup153 expression. Next, we challenged nucleoporins-depleted Jurkat cells with a single dose of HIV-1 expressing luciferase (HIV-1-Luc) as a reporter for infectivity (Fig. 1C). Depletion of Nup98 decreases the infectivity of HIV-1 by two-fold when compare to control cells suggesting a small role for Nup98 in HIV-1 infection. Depletion of Nup153 dramatically affected the ability of HIV-1 to infect Jurkat cells by > 10 fold (Fig. 1C), which is in agreement with similar findings in HeLa cells (Di Nunzio et al., 2012; Matreyek and Engelman, 2011). Because Matreyek and colleagues suggested that capsid is the viral determinant for the requirement of Nup153 on HIV-1 infection (Matreyek and Engelman, 2011), we tested the ability of the HIV-1 bearing the capsid mutation N74D, which is a TNPO3-independent mutant (Lee et al., 2010), to infect Nup98 and Nup153 -depleted cells (Fig. 1C). For this purpose, we challenged Nup98 and Nup153 -depleted cells with similar amounts of HIV-1-Luc and HIV-1-N74D-Luc normalized by p24, as described in the Material and methods section. HIV-1-N74D-Luc infection of Nup98-depleted cells was not affected when compared to control cells (Fig. 1C). By contrast, HIV-1–N74D–Luc infection of Nup153-depleted Jurkat cells was inhibited to a similar extent when compared to wild type HIV-1(Fig. 1C). As a control, we infected TNPO3-depleted cells with HIV-1–N74D–Luc (Fig. S1), which is a TNPO3-independent virus. These results suggested that Nup98 and more so Nup153 are required for HIV-1 infection; therefore, we investigated the step of the HIV-1 life cycle that requires Nup98 and Nup153. Similarly, we challenged Nup98- and Nup153-depleted Jurkat cells with p24-normalized HIV-1-Luc and HIV-1-N74D-Luc viruses and measure the production of two long terminal repeat (2LTR) circles by real-time PCR 24 h post-infection (Fig. 1D). The measurement of 2LTR circles is an indirect evidence that the pre-integration complex (PIC) has been transported to the nucleus (Butler et al., 2001). After the viral DNA is imported into the nucleus, it integrates into the genome; however, a small fraction of this viral DNA is ligated to produce circular forms by nuclear DNA ligases (Li et al., 2001); these products are known as 2LTR circles, and they are used as an indirect measure of nuclear import. Although the 2LTR is an indirect measure of PIC nuclear import, this methodology is widely used as a marker of nuclear import (Butler et al., 2001; Di Nunzio et al., 2012; Ebina et al., 2004; Konig et al., 2008; Lee et al., 2010; Matreyek and Engelman, 2011; Valle-Casuso et al., 2012). Depletion of Nup98 did not affect HIV-1–Luc and HIV-1–N74D–Luc production of 2LTR circles during infection, suggesting that Nup98 is not likely to be involved in nuclear import of the PIC. By contrast, HIV-1–Luc production of 2LTR circles during infection of Nup153-depleted cells was reduced indicating that Nup153 might be involved in the nuclear import of HIV-1. In agreement, infection of Nup153-depleted Jurkat cells by HIV-1–N74D–Luc also rendered a reduction in the production of 2LTR circles.
Fig. 1.

Depletion of Nup98 and Nup153 expression affects the early steps of HIV-1 replication. To knockdown the expression of Nup98 and Nup153 in Jurkat cells, we used the pTrip.GFP.H1shRNA vector (A). This vector contains a deletion on the U3 region (ΔU3) and encodes the green fluorescent protein (GFP) under the control of the CMV promoter to monitor transduction. shRNA cassettes are inserted in the 3′ end of the U3 region, and are regulated by the H1 promoter of the RNA polymerase III, which drives the endogenous production of shRNA. Viral particles produced using the pTrip.GFP.H1shRNA vector containing the specific shRNA against Nup98 and Nup153 were used to transduce Jurkat cells at the indicated multiplicity of infection (MOI). Control cells were transduced with the empty pTrip.GFP.H1shRNA vector. The efficiency of the KD was monitored by western blotting using antibodies against the endogenous Nup98 and Nup153 proteins (B). As a loading control, samples were also blotted using antibodies against β-actin. Nup98 and Nup153 -depleted cells were challenged with HIV-1 and HIV-1-N74D containing luciferase as a reporter of infection. Viruses were normalized by p24 as described in the Materials and methods section. Infectivity was determined 48 h post-infection (hpi) by measuring luciferase activity normalized to the amount of protein (C). Infections using the same viruses were performed to measure the production of 2LTR circles. For this purpose, 24 h pi total genomic DNA from infected cells was used to measure 2LTR circles by real-time PCR normalized to actin. The percentage of 2LTR circles respect to the control is shown (D). Infections carried out in the presence of Nevirapine 5 μM led to undetectable levels of both 2-LTR circles. Similarly, 24 h pi total genomic DNA was used to determine proviral integration sites by Alu–PCR, as described in the Materials and methods section. The percentage of proviruses respect to control is shown (E). To correlate the expression levels of Nup98 or Nup153 with HIV-1 infectivity, we challenged cells expressing different amounts of NUp98 or Nup153 with HIV-1 containing luciferase as a reporter of infection (F, G). Infectivity was determined 48 h post-infection by measuring luciferase activity normalized to the amount of protein. Jurkat cells expressing different amounts of Nup98 or Nup153 were prepared by using viral particles containing the specific shRNA against Nup98 or Nup153 at different multiplicities of infection (MOI). The expression levels of Nup98 and Nup153 were measured by Western blotting using antibodies against Nup98 or Nup153. As a loading control, samples were also blotted using antibodies against β-actin.
To assess the integration of HIV-1 viral DNA into the host chromatin, we challenged Nup98 and Nup153 -depleted Jurkat cells with p24-normalized viruses and measure integration by Alu–PCR at 48 h post-infection (Fig. 1E). We observed a reduction of integrated HIV-1 proviruses in Nup98-depleted cells when compared to infection of control cells. However, infection of Nup98-depleted cells by HIV-1-N74D-Luc viruses showed pro-viral integration levels similar to control cells consistent with our infection assays (Fig. 1E). Infection of Nup153-depleted cells by HIV-1–Luc rendered a larger reduction of proviral integration when compared to control cells, which is in agreement with the strong block observed for nuclear import (Fig. 1E). In addition, we observed that the capsid mutant, N74D, showed some dependency for Nup153 (Fig. 1E).
Next we tested whether a correlation exists between expression levels of Nup98 or Nup153 with HIV-1 infectivity (Fig. 1F and G). To obtain cells expressing different amounts of Nup98 or Nup153, we transduced Jurkat cells with viral particles containing the specific shRNA at the indicated multiplicities of infection. After three days of transduction, cells were used to measure expression levels of the Nups and infectivity by an HIV-1-Luc virus (Fig. 1F and G). As shown In Fig. 1F and G, our results correlated the expression levels of the nucleoporins with HIV-1 infectivity. This correlation showed that HIV-1 infection depends upon expression of Nup98 and Nup153.
Overall these studies suggested that the infectivity phenotype observed for Nup98-depleted cells is due to a contribution of Nup98 to HIV-1 integration (Fig. 2). Regarding Nup153, this work suggested that the infectivity phenotype observed in Nup153-depleted cells is due to a major contribution of Nup153 to HIV-1 nuclear import combined to an additional defect in integration (Fig. 2).
Fig. 2.

Correlation between proviral integration and infectivity during HIV-1 infection of Nup98 and Nup153 -depleted cells. Percentages for proviral integration and infectivity were obtained from Fig. 1C and E and used to calculate the linear correlation. The Spearman rank correlation coefficient, rs, is 0.8976 and P value of <0.0001.
Role of integrase in the HIV-1 infectivity phenotype observed for Nup98 and Nup153 -depleted cells
Infection of Nup98-depleted cells suggested a role for Nup98 in HIV-1 infection after nuclear import but prior to integration. This suggested the possibility that Nup98 interacts with HIV-1 integrase. To test the ability of Nup98 to interact with HIV-1 integrase, we performed co-immunoprecipitation experiments with proteins expressed in mammalian cells (Fig. 3A), as previously described (Diaz-Griffero, 2012; Valle-Casuso et al., 2012). In agreement with previous findings, Nup98 did not interact with HIV-1 integrase (Ao et al., 2012). As positive control, we assayed the ability of LEDGF/p75 to interact with HIV-1 integrase using similar salt concentrations (Fig. 3C). These results suggested that Nup98 is not assisting HIV-1 replication through an interaction with integrase.
Fig. 3.

HIV-1 integrase does not interact with Nup98 and Nup153. (A) To test the ability of GFP-Nup98 to interact with HIV-1 integrase, we co-transfected GFP-Nup98 together with FLAG-tagged HIV-1 integrase in human 293T cells (INPUT). Immunoprecipitation of HIV-1 integrase was performed using anti-FLAG antibodies in the presence of 200 mM NaCl, as described in Materials and methods section. Only the Eluted samples were analyzed for the presence of Nup98 and HIV-1 integrase by western blotting using anti-GFP and anti-FLAG antibodies, respectively. (B) Similarly, we tested the ability of Nup153 to interact with HIV-1 integrase at the indicated NaCl concentrations. (C) As a positive control we assayed the known ability of HIV-1 integrase to interact with LEDGF/p75. Experiments were performed in triplicates and a representative experiment is shown. WB, Western blot; IP, Immunoprecipitation.
Next we explored the ability of Nup153 to interact with HIV-1 integrase. Contrary to previous findings (Woodward et al., 2009), we found that GFP-Nup153 and HIV-1 integrase expressed in human 293T cells did not interact with each other at the indicated salt concentrations (Fig. 3B). These results suggested that Nup153 expressed in human cells does not interact with HIV-1 integrase. We believe that the difference between our results and previous observations is based on the system used to analyze the interaction. Contrary to us, Woodward and colleagues used bacterially purified protein to demonstrate the interaction, and they only tested a C-terminal fragment of Nup153 (896–1475) (Woodward et al., 2009).
Nup98 and Nup153 interact with HIV-1 in vitro assembled HIV-1 CA–NC complexes
Genetic experiments suggested that HIV-1 capsid is the viral determinant for the requirement of Nup153 during HIV-1 infection (Matreyek and Engelman, 2011). To test this hypothesis biochemically, we measured the ability of Nup98 and Nup153 to interact with in vitro assembled HIV-1 capsid–nucleocapsid (CA–NC) complexes as previously described (Ganser et al., 1999; Lienlaf et al., 2011; Valle-Casuso et al., 2012). The fusion construct GFP-Nup98 strongly binds in vitro assembled HIV-1 CA–NC complexes (Fig. 4A). As control, we found that GFP alone does not interact with in vitro assembled HIV-1 CA–NC complexes (Fig. 4A). Because HIV-1 bearing the capsid mutation N74D overcomes the requirement for Nup98, we tested the ability of Nup98 to interact with in vitro assembled HIV-1 CA–NC complexes bearing the capsid change N74D. Nup98 bound HIV-1 CA–NC complexes bearing the capsid change N74D with decreased affinity (~2 fold) when compared to wild type CA–NC complexes (Fig. 4B). These experiments demonstrated the ability of Nup98 to interact with in vitro assembled HIV-1 CA–NC complexes. In addition, the decreased binding of Nup98 to CA–NC complexes bearing the capsid change N74D might explain in part the reason that HIV-1–N74D infection is independent of Nup98.
Fig. 4.
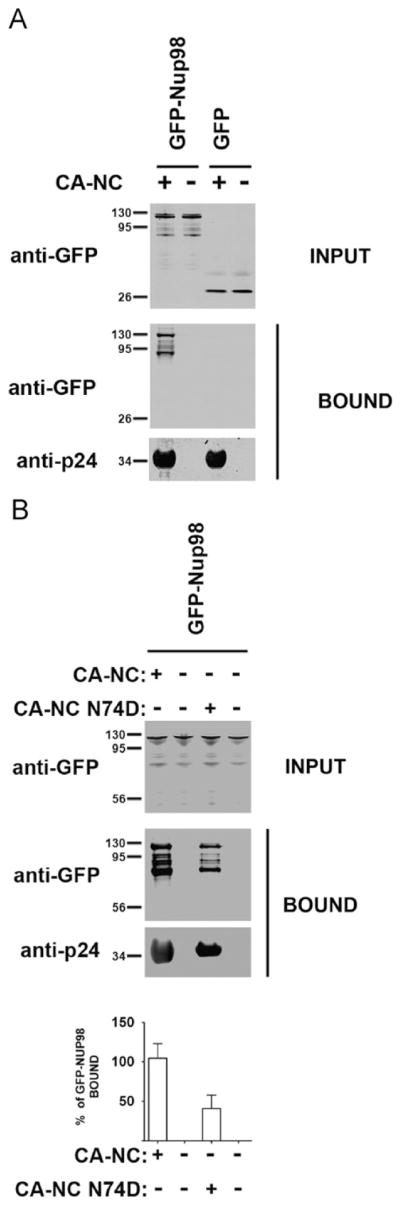
Binding of GFP-Nup98 proteins to in vitro assembled HIV-1 capsid-nucleocapsid (CA–NC) complexes. Human 293T cells were transfected with plasmids expressing GFP-Nup98 or GFP. 48 h after transfection, cells were lysed and incubated at room temperature for 1 h with in vitro assembled wild type (A) or N74D (B) HIV-1 CA–NC complexes. The mixtures were applied to a 70% sucrose cushion and centrifuged. INPUT represents the lysates analyzed by Western blotting before being applied to the 70% cushion. The input mixtures were Western blotted using anti-GFP antibodies. Similarly, the pellets from the 70% cushion (BOUND) were analyzed by Western blotting using anti-GFP and anti-p24 antibodies. The results of three independent experiments were similar; the result of a single experiment is shown. Binding of Nup98 to HIV-1 CA–NC complexes bearing the N74D change was quantified and normalized to the binding ability of Nup98 to the wild type HIV-1 CA–NC complexes (% of GFP-Nup98 BOUND).
Similarly, we tested the ability of Nup153 to interact with in vitro assembled HIV-1 CA–NC complexes. As a positive control, we included the capsid binder GFP–TRIM5αrh (Fig. 5A). Similarly, we found that GFP alone does not interact with in vitro assembled HIV-1 CA–NC complexes (Fig. 5A). In agreement with our infectivity data, we found that GFP-Nup153 binds HIV-1 CA–NC complexes bearing the N74D as strong as the wild type HIV-1 CA–NC complexes (Fig. 5B). These results suggested that Nup153 binds the HIV-1 core during the early stages of infection.
Fig. 5.
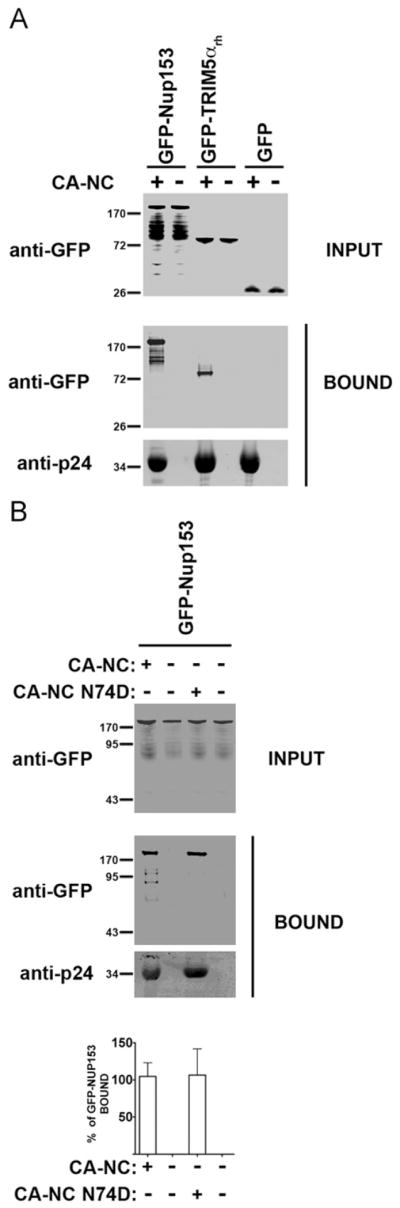
Binding of GFP-Nup153 proteins to in vitro assembled HIV-1 capsid-nucleocapsid (CA–NC) complexes. Human 293T cells were transfected with plasmids expressing GFP-Nup153, GFP–TRIM5αrh or GFP. 48 h after transfection, cells were lysed and incubated at room temperature for 1 h with in vitro assembled wild type (A) or N74D (B) HIV-1 CA–NC complexes. The mixtures were applied to a 70% sucrose cushion and centrifuged. INPUT represents the lysates analyzed by Western blotting before being applied to the 70% cushion. The input mixtures were Western blotted using anti-GFP antibodies. Similarly, the pellets from the 70% cushion (BOUND) were analyzed by Western blotting using anti-GFP and anti-p24 antibodies. The results of three independent experiments were similar; the result of a single experiment is shown. Binding of Nup153 to HIV-1 CA–NC complexes bearing the N74D change was quantified and normalized to the binding ability of Nup153 to the wild type HIV-1 CA–NC complexes (% of GFP-Nup153 BOUND).
Role of Nup98 and Nup153 in integration site distribution
Depletion of nucleopore components or karyopherins modifies the integration targeting and reduces the integration efficiency of HIV-1 (Di Nunzio et al., 2012; Konig et al., 2008; Ocwieja et al., 2011); therefore, we hypothesized that depletion of Nup98 and Nup153 may affect HIV-1 integration preferences and efficiency. Having confirmed that knockdown of Nup98 and Nup153 in lymphocytes reduced the efficiency of infection, we examined the role of these factors on integration site selection by preparing integration site libraries using ligation-mediated PCR (LM–PCR), as previously described (Cattoglio et al., 2010). To test this hypothesis, we analyzed integration sites distribution in Nup98 and Nup153 -depleted Jurkat cells. For this purpose, we challenged Nup98 and Nup153 -depleted cells with a single-round HIV-1 expressing the cell surface protein heat-stable antigen (HIV-1-HSA) as a reporter of infection (Fig. 6A). As a control, we challenged Jurkat control cells, which contain the shRNA empty vector, with HIV-1-HSA. To isolate HIV-1HSA integration sites, we digested the genomic DNA isolated from double positive cells for GFP and HSA and ligated the digested products to DNA-linkers (Fig. 6B). We amplified the integration sites by nested PCR using as a template the ligated DNA (Fig. 6B) and sequenced the fragments by 454-pyrosequencing (Cattoglio et al., 2010; Wang et al., 2009). Primers annealing to the HIV-1 U3 region, which is absent in shRNA expressing self-inactivated (ΔU3) lentiviral vectors, were used to selectively amplify HIV-1-HSA integration sites (Fig. 6B and Fig. S2). A total of 798, 1518 and 893 insertion sites were mapped to the human genome (UCSC release hg19) in control, Nup98- and Nup153-depleted Jurkat cells, respectively (Fig. 6C). All UCSC known genes having their transcription start site (TSS) at ±50 kb from the annotated integration target. Integrations were annotated as TSS-proximal when mapping in a ±2.5 kb interval around a TSS of known genes; intragenic when occurring inside the transcribed portion of a known Gene (exons and introns) > 2.5 kb downstream of a TSS; and intergenic in all the other cases (upstream or downstream of known genes) (Cattoglio et al., 2010). Our results showed that Nup98 and Nup153 -depleted cells exhibit a 5% and 8% decrease of integrations occurring in intragenic sites, respectively, when compared to Jurkat control cells (Fig. 6C and Table 1). In parallel, depletion of Nup153 increased HIV-1 integration frequency in intergenic regions (Fig. 6C and Table 1). Nup98-depleted cells also showed a modest increase (2%) in the proportion of intergenic HIV-1 integrations and a 2.7 fold increase in integrations occurring in close proximity to TSSs. Finally, to evaluate the frequency of HIV-1 integration in CpG islands and DNaseI hypersensitive sites in Nup98- and Nup153-depleted cells, we measured the proportion of integrations occurring in regions containing these genomic features, which are hallmarks for open chromatin, by using online available data sets (UCSC track containing annotated CpG islands and ENCODE Jurkat DNase I hypersensitive sites; Fig. S3). Interestingly, Nup153-depleted cells exhibited a reduced fraction of integrations mapping to regions enriched in CpG islands and DNaseI hypersensitive sites (Fig. S3).
Fig. 6.

Contribution of Nup98 and Nup153 to HIV-1 selection of integration sites. (A) To measure HIV-1 infectivity Jurkat Nup98 and Nup153KD depleted cells were infected with HIV-1-HSA and analyzed by Flow cytometry. As a control, we challenged Jurkat control cells, which contain the shRNA empty vector, with HIV-1-HSA. The graphic shows the % of HSA positive cells 3 days post-infection, these cells were sorted by HSA beads before to start LM–PCR. (B) Strategy employed to detect exclusively HIV-1-HSA and not the shRNA lentiviral vector, containing the deletion in the U3 region of LTR, used to achieve knockdown cells. (C) Distribution of HIV-1 integration sites in the genome of control, Nup98KD, Nup153KD Jurkat cells. USCS known genes having their TSS in a ±50 kb window around the integration sites (798, 1518 and 893 for control, Nup98KD, Nup153KD Jurkat cells, respectively) and a dataset of control 40,000 random sites were annotated. Integrations were distributed inside (intragenic), outside (intergenic), or in the promoter region (TSS-proximal) of UCSC known genes. Statistical differences between the integration frequencies in Nup98 KD, Nup153 KD and control Jurkat cells were determined by using a 2-sample test for equality of proportions (*P<0.05; **P<0.005 and ***P<0.001).
Table 1.
Integration site distribution for Nup153 and Nup98 depleted cells.
| Genomic features | Jurkat control | Jurkat Nup98KD | Jurkat Nup153KD | Random |
|---|---|---|---|---|
| n=798 | n=1518 | n=893 | n=40,000 | |
| % Intragenic integrations (±50 kb) | 77.0 | 72.0 | 69.0 | 38.0 |
| % Intergenic integrations (±50 kb) | 21.0 | 23.0 | 28.0 | 59.0 |
| % TSS-proximal integrations (±50 kb) | 1.7 | 4.5 | 3.0 | 3.0 |
| % Integrations on CpG islands (±50 kb) | 67.5 | 69.4 | 63.7 | 38.7 |
| % Integrations in DNaseI hypersensitive sites (±50 kb) | 97.9 | 97.1 | 95.7 | 83.1 |
| Gene density/integration (±50 kb) | 2.0 | 2.2 | 1.9 | 0.9 |
| Average genes in 1 Mb | 46.0 | 47.0 | 44.0 | 30.0 |
The table indicates the percentage of HIV integrations occurring in intragenic, intergenic, transcriptional start site (TSS)-proximal regions, and in close proximity to CpG islands and in DNaseI hypersensitive sites. Computer-generated random control sequences were obtained from Cattoglio et al.(2010).
Overall, these experiments suggested that Nup98 and Nup153 depletion reduces HIV-1 integration preferences for chromosomal regions rich in genes and associated features such as CpG islands, DNAaseI hypersensitive sites, thus suggesting that Nup98 and Nup153 influence HIV-1 integration in transcriptional units and in regions characterized by an open chromatin configuration, as shown for other factors such as TNPO3 and RanBP2 (Ocwieja et al., 2011; Schaller et al., 2011).
We corroborated these studies by doing a second infection and sequencing more than 3000 proviral integration sites (Fig. S4).
Discussion
To study the role of Nup153 and Nup98 in HIV-1-infection, we depleted its expression in human Jurkat cells and study HIV-1 nuclear import and integration. These experiments confirmed in Jurkat cells the observation made in HeLa cells that depletion of Nup153 mainly affects nuclear import while the depletion of Nup98 slightly reduce HIV-1 integration (Fig. 1) (Di Nunzio et al., 2012; Matreyek and Engelman, 2011). Our results suggested that Nup153 play an important role on the nuclear import of HIV-1, which is in agreement with the localization of Nup153 to the nuclear basket region of the nucleopore with the C-terminal domain exposed to the cytoplasmic face of the nucleopore complex, possibly to enhance the nuclear transport activity (Ball and Ullman, 2005; Paulillo et al., 2005, 2006). Similarly, we studied the role of Nup98 in HIV-1 infection. Our findings indicate that the 2-fold decrease on infectivity caused by Nup98 depletion is due to an effect on the integration process, since HIV-1 nuclear import seems not to be affected by depletion of Nup98. Interestingly, we observed that the Nup98 depleted cells showed a an increase of 2.7 fold for integration in transcriptional start sites (TSS) respect to control cells. This observation is in agreement with previous works in which Nup98 has been found to interact with active genes, preferentially far away from the promoter (Capelson et al., 2010; Kalverda and Fornerod, 2010).
Previous studies have suggested that the interaction of HIV-1 integrase with Nup153 is required for the nuclear import of HIV-1; this work showed a direct interaction between the C-terminal region of Nup153 and the integrase of HIV-1 with both proteins produced in bacteria (Woodward et al., 2009). To explore this interaction in proteins expressed in mammalian cells, we tested the ability of full-length Nup153 to interact with HIV-1 integrase using proteins produced in human 293T cells. Our results showed that Nup153 was not able to interact with HIV-1 integrase at any of the tested salt concentrations (Fig. 3). To test that our assay is sensitive to bona fide interactor of HIV-1 integrase, we showed the previously described interaction of HIV-1 integrase with LEDGF/p75 (Cherepanov et al., 2003). We believe that the difference in results resides on the source of the proteins used to test the interaction, bacteria versus mammalian cells. Our results suggested that Nup153 does not interact with HIV-1 integrase in mammalian cells.
Genetic studies have suggested that capsid is the viral determinant for the requirement of Nup153 (Matreyek and Engelman, 2011). To explore this possibility, we tested the ability of Nup153 to bind in vitro assembled HIV-1 CA–NC complexes, which recapitulates the surface of the viral core (Pornillos et al., 2010). This capsid binding assay has been extensively used to characterize the binding of TRIM5αrh variants to capsid (Diaz-Griffero et al., 2009; Lienlaf et al., 2011), among other capsid interactors such as RANBP2, CPSF6 and TNPO3 (Di Nunzio et al., 2012; Lee et al., 2010; Valle-Casuso et al., 2012). In agreement with the genetic studies performed by Matreyek and colleagues, we found that Nup153 binds in vitro assembled HIV-1 CA–NC complexes. This result suggested that the interaction of Nup153 with the HIV-1 core might be important for the ability of HIV-1 to enter the nucleus. Even thought our findings indicate that Nup98 is not required for nuclear import, it binds HIV-1 CA–NC complexes, which is a puzzling observation. Because Nup153 and Nup98 have in common the presence of phenylalanine–glycine (FxG) repeats, one possibility is that the FxG repeats are directly mediating the interaction of nucleoporins with in vitro assembled HIV-1 CA–NC complexes (Matreyek oral communication CSH 2012). In agreement, we have previously shown the binding of HIV cores by Nup358/RanBP2, which is a nucleoporin that contains several FxG repeats (Di Nunzio et al., 2012). Future analysis of the ability of different nucleoporins to bind the HIV-1 core will establish whether FxG repeats are directly involved in the ability of nucleoporins to interact with the HIV-1 core.
Our findings indicate that Nup153 is involved in nuclear import; however, the discrepancy found when comparing infectivity with integration in Nup153-depleted cells suggested that Nup153 might be also involved in integration (Fig. 1). Because the decrease in infectivity observed in Nup153-depleted cells did not directly correlate to a decrease in integration efficiency, we hypothesized that HIV-1 in Nup153-depleted cells is integrating in transcriptionally inactive regions. In agreement with our hypothesis, our findings showed that Nup153-depleted cells exhibit a ~10% decrease in intragenic integrations (regions that are likely to be transcriptionally active), which could in part explain the discrepancy between HIV-1 infectivity and integration in Nup153 depleted cells. In agreement, a recently published report showed that depletion of Nup153 from HeLa cells decreases the ability of HIV-1 to integrate in transcriptionally active regions (Koh et al., 2012). The large phenotype observed for HIV-1 infectivity in Nup153 KD cells suggested that Nup153 could be simultaneously affecting HIV-1 nuclear import and integration. The functional duality of Nup153 could be occurring by a direct or an indirect mechanism. A direct mechanism implies that Nup153 interaction with HIV-1 is modulating nuclear import and integration. An indirect mechanism, more likely, will suggest that depletion of Nup153 interfere with the formation of an intact nucleopore (Hase and Cordes, 2003; Mackay et al., 2009), which could result in the disruption of HIV-1 infection at multiple stages. Future investigations will explain whether the interaction between capsid and Nup153 is functional for the virus.
Although Nup98 and Nup153 are involved in integration, the mechanism by which this process occurs is not known. One possibility is that the Nups target the PIC to specific regions of the chromatin allowing preferential integration. A second possibility will suggest that the Nups are rearranging the chromatin conformation increasing the ability of HIV-1 to integrate in transcriptionally active genes. Future experiments will provide insights on how Nucleoporins facilitate HIV-1 integration.
Overall our work suggests that the defect on infectivity observed for Nup153-depleted cells is due to the contribution of Nup153 to nuclear import and integration, suggesting a link between these two steps. We also demonstrated the ability of Nup153 to bind the HIV-1 core suggesting that this interaction might be required for nuclear import of HIV-1.
Material and methods
Cells, lentiviral vector carrying shRNA and virus production
The Jurkat cells are CD4+ human T cells. The 293T cells are human embryonic kidney cells. The TRIP–GFP vector was used to produce lentiviral particles expressing specific shRNAs against Nup98 and Nup153. TRIP–GFP vector, which is ΔU3, contains the cis-acting sequences required for formation of the central DNA Flap, and encodes the green fluorescent protein (GFP) under the control of the CMV promoter to monitor transduction. shRNA cassettes were inserted in the 3′ U3 region of the vector plasmid to avoid increasing vector size and to increase shRNA expression in transduced cells, since the U3 region is duplicated during reverse transcription (Di Nunzio et al., 2012). To insert the shRNA cassette, a polylinker (MluI, ClaI, MscI, NheI, SalI, XhoI) was inserted at the U3 BamHI restriction site. Complementary oligo-nucleotides coding for shRNAs were annealed and cloned into BglII/HindIII of pSUPER (OligoEngine Invitrogen) downstream of the H1 promoter. The H1–shRNA was then inserted into ClaI/NheI of the vector plasmid polylinker, upstream in respect to the internal pol II marker gene segments. Lentiviral particles expressing specific shRNAs were produced by transient transfection of 293T cells with the vector plasmid, encapsidation plasmid (Δp 8.74), and the VSV-G envelope expression plasmid. After collection of supernatants at 48 h post-transfection, lentiviral particles expressing specific shRNAs were concentrated by ultracentrigation for 1 h at 22,000 rpm at 4 °C and stored at −80 °C. Lentiviral particles expressing specific shRNAs were titered in HeLa P4-CCR5 cells using flow cytometry to assess GFP expression at 4 days post-transduction.
Jurkat cells were transduced with Lentiviral particles expressing specific shRNAs against Nup98 at MOI 50 and 100 and LV-shRNA against Nup153 at MOI 30, 50, 70 and 100 to generate stable knockdown cells.
HIV-1 viruses were produced by co-transfection of 293T cells with NL4.3-IRES-HSA ENV− or NLX Luc ENV− and the Vesicular Stomatitis Virus glycoprotein (VSV-G) envelope expression plasmid pHCMV-G (VSV-G). The viruses harvested from 293T cells 48 h post-transfection were treated with 25 U/mL of DnaseI (Roche) and with 100 mM MgCl2 at 37 °C for 30 min. Viruses normalization were performed by p24 ELISA according to the manufacturer’s instructions (Perkin Elmer).
Cell separation and LM–PCR
Twenty million of Jurkat cells were infected with 10 μg of p24 antigen of NL4.3-IRES-HSA ENV−. Three days later, virus-infected cells were sorted using the biotinylated anti-HSA antibody (clone M1/69, BD Biosciences) used at a final concentration of 3 μg/ml for 30 min on ice followed by 15 min with beads anti biotin (BD). After seven washes in PBS/0.5%BSA/2mMEDTA, a small fraction of sorted cells was labelled with antibody PE-conjugated anti-HSA rat (BD 532262) and analysed by flow cytometry. Genomic DNA was extracted by QIAamp DNA micro kit (QIAGEN) and digested with the 4-cutter enzyme BfaI and BglII to prevent the amplification of internal 3′ LTR fragments. A BfaI double-stranded linker was then ligated, and viral 5′ LTR-genome junctions were amplified by LM–PCR using primers specific for the linker and the 5′ LTR of HIV-1 (primer linker: 5′ AGGGCTCCGCTTAAGGGAC 3′; primer HIV-1 5′ LTR: 5′ TGGTAGATCCACAGATCAAGGA 3′). HIV-1 5′ LTR primer anneals to the U3 region of 5′ LTR present only in NL4.3-IRES-HSA ENV- after viral integration and absent in Trip self-inactivated LV-shRNA (Fig. S2). A further PCR step was performed to include sample specific barcodes for parallel sequencing using the 454/Roche pyrosequencing platform as previously described (Cartier et al., 2009; Cattoglio et al., 2007, 2010). Raw sequence reads were processed through an automated bioinformatic pipeline that eliminated small and redundant sequences and were mapped to the UCSC hg19 release of the human genome (Cattoglio et al., 2010). Sequences with 90% or greater identity to the human genome were considered genuine integration sites. As control dataset, we used 40,000 random sites from Cattoglio et al. (2010).
Genomic features (CpG islands and DNaseI hypersensitive sites) were annotated when their genomic coordinates overlapped for at least 1 nucleotide with a 50-kb interval surrounding each integration site. We used UCSC tracks for both cytosine–phosphate–guanosine (CpG) islands and Jurkat DNaseI hypersensitive sites.
Western blotting and co-Immunoprecipitation
Proteins were extracted on ice from control and KD cells using RIPA buffer (20 mM HEPES p H 7.6, 150 mM NaCl, 1% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 2 mM EDTA, complete protease inhibitor [Roche Diagnostics]), and protein concentration was quantified using the Dc Protein Assay (Bio-Rad Laboratories) with bovine serum albumin as standard. 100 μg of total protein lysate was loaded onto SDS–PAGE 6% Tris–glycine gel (Invitrogen) for Nup153, and 20 μg onto 4–12% Bis-Tris gel (Invitrogen) for Nup98. Revelation was carried out using the ECL Plus Western Blotting kit (GE Healthcare). Primary antibodies used for Western blotting (WB) were rat anti-Nup98 (Abcam WB 1:4,000, IF 1:100), anti-mouse Nup153 (SA1, kind gift from B. Burke, WB 1:500, IF 1:10). Secondary or conjugated antibodies used for Western blotting were Beta Actin HRP conjugated antibody (Abcam, 1:2,500), anti-mouse IgG HRP (GE Healthcare, 1:5,000), anti-rabbit IgG HRP (GE Healthcare, 1:5,000),anti-rat IgG HRP (Sigma, 1:80,000), and LEDGF/P75 (Bethyl Laboratories, Inc. cat. A300-847A). Human 293T cells were co-transfected with different HIV-1 integrase containing a FLAG epitope tag on the C-terminus, LEDGF/p75-HA tag, pGFP-Nup98 and pGFP-Nup153 (kindly gift by Dr. Jan Ellenberg). Transfected cells were lysed in extraction buffer [0.5% Triton X-100, 50 mM Tris–HCl, pH=8, 2 mM MgCl2, 5% glycerol and protease inhibitors (Roche)] containing 100 mM, 200 mM or 300 mM NaCl as indicated. Extracts were pre-cleared using protein-A agarose beads (Sigma) at 4 °C for 1 h. Integrase proteins were immunoprecipitated using agarose beads linked to anti-FLAG antibodies (Sigma). Beads were washed 3 times, and immunocomplexes were eluted by boiling the samples in 1 × SDS sample buffer. Samples were then analyzed by Western blotting using antibodies against FLAG (Sigma), antibody anti-HA (Covance MMS-101R) and antibody anti-GFP (Clontech 632459).
HIV-1 CA–NC expression and purification
The HIV-1 CA–NC protein was expressed, purified and assembled as previously described (Ganser et al., 1999). The pET11a expression vector (Novagen) expressing the CA–NC protein of HIV-1 was used to transform BL-21(DE3) E. coli. CA–NC expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) when the culture reached an optical density of 0.6 at 600 nm. After 4 h of induction, the cells were harvested and resuspended in 20 mM Tris–HCl (pH 7.5), 1 μM ZnCl2, 10 mM 2-mercaptoethanol and protease inhibitors (Roche). Lysis was performed by sonication, and debris were pelleted for 30 min at 35,000 × g. Nucleic acids were stripped from the solution by using 0.11 equivalents of 2 M (NH4)2SO4 and the same volume of 10% polyethylenimine. Nucleic acids were removed by stirring and centrifugation at 29,500 × g for 15 min. The protein was recovered by addition of 0.35 equivalents of saturated (NH4)2SO4. The protein was centrifuged at 9820 × g for 15 min and resuspended in 100 mM NaCl, 20 mM Tris–HCl (pH 7.5), 1 μM ZnCl2 and 10 mM 2-mercaptoethanol. Lastly the CA–NC protein was dialyzed against 50 mM NaCl, 20 mM Tris–HCl (pH 7.5), 1 μM ZnCl2 and 10 mM 2-mercaptoethanol, and stored at −80 °C.
In vitro assembly of CA–NC complexes
HIV-1 CA–NC particles were assembled in vitro by diluting the CA–NC protein to a concentration of 0.3 mM in 50 mM Tris–HCl (pH 8.0), 0.5 M NaCl and 2 mg/ml DNA oligo-(TG)50. The mixture was incubated at 4 °C overnight and centrifuged at 8600 × g for 5 min. The pellet was resuspended in assembly buffer (50 mM Tris–HCl (pH 8.0), 0.5 M NaCl) at a final protein concentration of 0.15 mM (Ganser-Pornillos et al., 2004; Ganser et al., 1999), and stored at 4 °C until needed.
Binding of Nup98 and Nup153 to HIV-1 capsid complexes
293T cells were transfected with plasmids expressing GFP-Nup98, GFP-Nup153, GFP-TRIM5αrh, GFP proteins. 48 h after transfection, cell lysates were prepared as follows: washed cells were resuspended in capsid-binding buffer (10 mM Tris, pH 7.4, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT). The cell suspension was frozen and thawed, and incubated on ice for 10 min. Next, the lysate was centrifuged in a refrigerated Eppendorf microcentri-fuge (~14,000 × g) for 5 min. The supernatant was supplemented with 1/10 volume of 10 × PBS and then used in the binding assay. To test binding, 5 μl of CA–NC particles assembled in vitro were incubated with 200 μl of cell lysate at room temperature for 1 h. A portion of this mixture, henceforth referred to as “INPUT” was stored. The mixture was spun through a 70% sucrose cushion (70% sucrose, 1 × PBS and 0.5 mM DTT) at 100,000 × g in an SW55 rotor (Beckman) for 1 h at 4 °C. After centrifugation, the supernatant was carefully removed and the pellet was resuspended in 1 × SDS–PAGE loading buffer and henceforth referred to as “BOUND.” The level of GFP-fusion proteins was determined by Western blotting with an anti-GFP antibody as described above. The level of HIV-1 CA–NC protein in the pellet was assessed by Western blotting with an anti-p24 capsid antibody.
Luciferase assays
Luciferase (Promega) activity was measured 48 h p.i according to manufacturer’s instructions, using a microplate fluorimeter (Victor, Perkin Elmer). Protein quantification by Bio-Rad protein assay was carried out on the same lysates to normalize the luciferase data for protein content.
Quantitative PCR
Infected cells and control infected cells cultured in the presence of 5 μM nevirapine were treated for 30 min at 37 °C with 1000 U of DnaseI (Roche). Total cellular DNA was then isolated using the QIAamp DNA micro kit (QIAGEN). Two long terminal repeat (2-LTR) containing circles were detected using primers MH535/536 and probe MH603 (Butler et al., 2001), using as standard curve the pUC2LTR plasmid, which contains the HIV-1 2-LTR junction. Assessment of integration by Alu-PCR was performed as previously described (Brussel and Sonigo, 2003).
To rule out amplification of LV-shRNA, the 5′ primer used for the first amplification step was designed to recognize a unique sequence in HIV-1 U3 not present in LV–shRNA. As in Brussel and Sonigo (2003) this primer was extended with the lambda phage-specific heel sequence shown in bold (5′-atgccacgtaagcgaaactttccgctggggactttccaggg). The U3 modified primer was used in combination with two Alu primers (5′-tcccagctactggggaggctgagg) and (5′-gcctcccaaagtgctgggattacag). DNA generated from WT-infected cells was endpoint diluted in DNA prepared from uninfected cells to generate the integration standard curve and serial dilutions were made starting from 50,000 infected cells. Each sample amplified contained 104 infected cells mixed at 40,000 uninfected cells. The control of the first round PCR was the amplification without Alu primers using rTth DNA polymerase XL as recommended by the manufacturer (Applied Biosystems Inc, Foster City, CA N808-0187). Dilution 1:10 of the first round was amplified using the phage lambda Spa primer (5′-atgccacgtaagcgaaact) and U5-specific primer (5′-ctgactaaaagggtctgagg) with the probe (6FAM-ttaagcctcaataaagcttgccttgagtgc-TAMRA). Both 2-LTR and Alu-PCR reactions were carried out at 24 h p.i and normalized by amplification of the housekeeping gene β-Actin using the following primers and probe (5′-aacaccccagccatgtacgt), (5′-cggtgaggatcttcatgaggtagt), (6FAM-ccagccaggtccagacgcagga-BBQ). Reactions contained 1X FastStart Universal Probe Master Mix (Rox) 2X (Roche), 300 nM forward primer, 300 nM reverse primer, 100 nM probe primer and template DNA in a 20 μl volume. After initial annealing (50 °C for 2 min) and denaturation steps (95 °C for 15 min), 40 cycles of amplification were carried out (95 °C for 15 s, 58 °C for 30sec, and 72 °C for 30 s). As a control, single cycle Luciferase activity assays were systematically carried out to certify the phenotype.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful to Ian Ellenberg for sharing his GFP–Nup153 and GFP–Nup98 plasmids, Michel J. Tremblay for sharing the vector NL4.3-IRES–HSA ENV−. We thank Marco Severgnini and Daniela Sartori for help with 454 pyrosequencing experiments. This work was funded by the ANRS (Agence Nationale de Recherche sur le SIDA), the Sidaction, the Pasteur Institute and the European Union, and an NIH R01 AI087390 award to FDG. ER and MS were supported by FIRB-Futuro in ricerca grant RBFR08U07M. TF, JCV and PP were supported by an NIH R01 AI087390 award to FDG.
Appendix. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/http://dx.doi.org/10.1016/j.virol.2013.02.008.
References
- Ao Z, Danappa Jayappa K, Wang B, Zheng Y, Kung S, Rassart E, Depping R, Kohler M, Cohen EA, Yao X. Importin alpha3 interacts with HIV-1 integrase and contributes to HIV-1 nuclear import and replication. J Virol. 2010;84 (17):8650–8663. doi: 10.1128/JVI.00508-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao Z, Huang G, Yao H, Xu Z, Labine M, Cochrane AW, Yao X. Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication. J Biol Chem. 2007;282 (18):13456–13467. doi: 10.1074/jbc.M610546200. [DOI] [PubMed] [Google Scholar]
- Ao Z, Jayappa KD, Wang B, Zheng Y, Wang X, Peng J, Yao X. Contribution of host nucleoporin 62 in HIV-1 integrase chromatin association and viral DNA integration. J Biol Chem. 2012;287 (13):10544–10555. doi: 10.1074/jbc.M111.317057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prevost MC, Allen TD, Charneau P. HIV-1 DNA flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 2007;26 (12):3025–3037. doi: 10.1038/sj.emboj.7601740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball JR, Ullman KS. Versatility at the nuclear pore complex: lessons learned from the nucleoporin Nup153. Chromosoma. 2005;114 (5):319–330. doi: 10.1007/s00412-005-0019-3. [DOI] [PubMed] [Google Scholar]
- Bowerman B, Brown PO, Bishop JM, Varmus HE. A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 1989;3 (4):469–478. doi: 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319 (5865):921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Brussel A, Sonigo P. Analysis of early human immunodeficiency virus type 1 DNA synthesis by use of a new sensitive assay for quantifying integrated provirus. J Gen Virol. 2003;77 (18):10119–10124. doi: 10.1128/JVI.77.18.10119-10124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky MI, Sharova N, McDonald TL, Pushkarskaya T, Tarpley WG, Stevenson M. Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc Nat Acad Sci USA. 1993;90 (13):6125–6129. doi: 10.1073/pnas.90.13.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7 (5):631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- Capelson M, Doucet C, Hetzer MW. Nuclear pore complexes: guardians of the nuclear genome. Cold Spring Harb Symp Quant Biol. 2010;75:585–597. doi: 10.1101/sqb.2010.75.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrere F, Blanche S, Audit M, Payen E, Leboulch P, l’Homme B, Bougneres P, Von Kalle C, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326 (5954):818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B, Schmidt M, von Kalle C, Howe S, Thrasher AJ, Aiuti A, Ferrari G, Recchia A, Mavilio F. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110 (6):1770–1778. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- Cattoglio C, Pellin D, Rizzi E, Maruggi G, Corti G, Miselli F, Sartori D, Guffanti A, Di Serio C, Ambrosi A, De Bellis G, Mavilio F. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood. 2010;116 (25):5507–5517. doi: 10.1182/blood-2010-05-283523. [DOI] [PubMed] [Google Scholar]
- Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem. 2003;278 (1):372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- Christ F, Thys W, De Rijck J, Gijsbers R, Albanese A, Arosio D, Emiliani S, Rain JC, Benarous R, Cereseto A, Debyser Z. Transportin-SR2 imports HIV into the nucleus. Curr Biol. 2008;18 (16):1192–1202. doi: 10.1016/j.cub.2008.07.079. [DOI] [PubMed] [Google Scholar]
- De Rijck J, Vandekerckhove L, Christ F, Debyser Z. Lentiviral nuclear import: a complex interplay between virus and host. Bioessays. 2007;29 (5):441–451. doi: 10.1002/bies.20561. [DOI] [PubMed] [Google Scholar]
- Di Nunzio F, Danckaert A, Fricke T, Perez P, Fernandez J, Perret E, Roux P, Shorte S, Charneau P, Diaz-Griffero F, Arhel NJ. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS One. 2012;7 (9):e46037. doi: 10.1371/journal.pone.0046037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F. The Role of TNPO3 in HIV-1 Replication. Mol Biol Int. 2012;2012:868597. doi: 10.1155/2012/868597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, Lienlaf M, Yokoyama S, Sodroski J. A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J Virol. 2009;83 (20):10737–10751. doi: 10.1128/JVI.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebina H, Aoki J, Hatta S, Yoshida T, Koyanagi Y. Role of Nup98 in nuclear entry of human immunodeficiency virus type 1 cDNA. Microbes Infect. 2004;6 (8):715–724. doi: 10.1016/j.micinf.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Fassati A. HIV infection of non-dividing cells: a divisive problem. Retro-virology. 2006;3:74. doi: 10.1186/1742-4690-3-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassati A, Goff SP. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J Virol. 2001;75 (8):3626–3635. doi: 10.1128/JVI.75.8.3626-3635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassati A, Gorlich D, Harrison I, Zaytseva L, Mingot JM. Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. Embo J. 2003;22 (14):3675–3685. doi: 10.1093/emboj/cdg357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallay P, Hope T, Chin D, Trono D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc Nat Acad Sci USA. 1997;94 (18):9825–9830. doi: 10.1073/pnas.94.18.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol. 2004;78 (5):2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283 (5398):80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- Hase ME, Cordes VC. Direct interaction with nup153 mediates binding of Tpr to the periphery of the nuclear pore complex. Mol Biol Cell. 2003;14 (5):1923–1940. doi: 10.1091/mbc.E02-09-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearps AC, Jans DA. HIV-1 integrase is capable of targeting DNA to the nucleus via an importin alpha/beta-dependent mechanism. Biochem J. 2006;398 (3):475–484. doi: 10.1042/BJ20060466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanskiy S, Berro R, Altieri M, Kashanchi F, Bukrinsky M. Intracytoplasmic maturation of the human immunodeficiency virus type 1 reverse transcription complexes determines their capacity to integrate into chromatin. Retrovirology. 2006;3:4. doi: 10.1186/1742-4690-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalverda B, Fornerod M. Characterization of genome-nucleoporin interactions in Drosophila links chromatin insulators to the nuclear pore complex. Cell Cycle. 2010;9 (24):4812–4817. doi: 10.4161/cc.9.24.14328. [DOI] [PubMed] [Google Scholar]
- Katz RA, Greger JG, Boimel P, Skalka AM. Human immunodeficiency virus type 1 DNA nuclear import and integration are mitosis independent in cycling cells. J Virol. 2003;77 (24):13412–13417. doi: 10.1128/JVI.77.24.13412-13417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh Y, Wu X, Ferris AL, Matreyek KA, Smith SJ, Lee K, Kewalramani VN, Hughes SH, Engelman A. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J Virol. 2012 doi: 10.1128/JVI.01148-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135 (1):49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan L, Matreyek KA, Oztop I, Lee K, Tipper CH, Li X, Dar MJ, Kewalramani VN, Engelman A. The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J Virol. 2010;84 (1):397–406. doi: 10.1128/JVI.01899-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010;7 (3):221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A, Hayouka Z, Friedler A, Loyter A. Transportin 3 and importin alpha are required for effective nuclear import of HIV-1 integrase in virus-infected cells. Nucleus. 2010;1 (5):422–431. doi: 10.4161/nucl.1.5.12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P, Hensel M, Emerman M. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J. 1992;11 (8):3053–3058. doi: 10.1002/j.1460-2075.1992.tb05376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PF, Emerman M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J Virol. 1994;68 (1):510–516. doi: 10.1128/jvi.68.1.510-516.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, Bushman FD. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 2001;20 (12):3272–3281. doi: 10.1093/emboj/20.12.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienlaf M, Hayashi F, Di Nunzio F, Tochio N, Kigawa T, Yokoyama S, Diaz-Griffero F. Contribution of E3-ubiquitin ligase activity to HIV-1 restriction by TRIM5alpha(rh): structure of the RING domain of TRIM5alpha. J Virol. 2011;85 (17):8725–8737. doi: 10.1128/JVI.00497-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DR, Elgort SW, Ullman KS. The nucleoporin Nup153 has separable roles in both early mitotic progression and the resolution of mitosis. Mol Biol Cell. 2009;20 (6):1652–1660. doi: 10.1091/mbc.E08-08-0883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Engelman A. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J Virol. 2011;85 (15):7818–7827. doi: 10.1128/JVI.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaj IW, Englmeier L. Nucleocytoplasmic transport: the soluble phase. Annu Rev Biochem. 1998;67:265–306. doi: 10.1146/annurev.biochem.67.1.265. [DOI] [PubMed] [Google Scholar]
- Miller MD, Farnet CM, Bushman FD. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71 (7):5382–5390. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JA, Chanda SK, Konig R, Malani N, Berry CC, Bushman FD. HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 2011;7 (3):e1001313. doi: 10.1371/journal.ppat.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulillo SM, Phillips EM, Koser J, Sauder U, Ullman KS, Powers MA, Fahrenkrog B. Nucleoporin domain topology is linked to the transport status of the nuclear pore complex. J Mol Biol. 2005;351 (4):784–798. doi: 10.1016/j.jmb.2005.06.034. [DOI] [PubMed] [Google Scholar]
- Paulillo SM, Powers MA, Ullman KS, Fahrenkrog B. Changes in nucleoporin domain topology in response to chemical effectors. J Mol Biol. 2006;363 (1):39–50. doi: 10.1016/j.jmb.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, Yeager M. Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J Mol Biol. 2010;401 (5):985–995. doi: 10.1016/j.jmb.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe T, Reynolds TC, Yu G, Brown PO. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993;12 (5):2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin H, Temin HM. A radiological study of cell-virus interaction in the Rous sarcoma. Virology. 1959;7 (1):75–91. doi: 10.1016/0042-6822(59)90178-3. [DOI] [PubMed] [Google Scholar]
- Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011;7 (12):e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Craigie R. The road to chromatin—nuclear entry of retroviruses. Nat Rev Microbiol. 2007;5 (3):187–196. doi: 10.1038/nrmicro1579. [DOI] [PubMed] [Google Scholar]
- Thys W, De Houwer S, Demeulemeester J, Taltynov O, Vancraenenbroeck R, Gerard M, De Rijck J, Gijsbers R, Christ F, Debyser Z. Interplay between HIV entry and transportin-SR2 dependency. Retrovirology. 2011;8:7. doi: 10.1186/1742-4690-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle-Casuso JC, Di Nunzio F, Yang Y, Reszka N, Lienlaf M, Arhel N, Perez P, Brass AL, Diaz-Griffero F. TNPO3 is required for HIV-1 replication after nuclear import but prior to integration and binds the HIV-1 core. J Virol. 2012;86 (10):5931–5936. doi: 10.1128/JVI.00451-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GP, Levine BL, Binder GK, Berry CC, Malani N, McGarrity G, Tebas P, June CH, Bushman FD. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther. 2009;17 (5):844–850. doi: 10.1038/mt.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward CL, Prakobwanakit S, Mosessian S, Chow SA. Integrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1. J Virol. 2009;83 (13):6522–6533. doi: 10.1128/JVI.02061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Emerman M. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J Virol. 2004;78 (11):5670–5678. doi: 10.1128/JVI.78.11.5670-5678.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Emerman M. Retroviral infection of non-dividing cells: old and new perspectives. Virology. 2006;344 (1):88–93. doi: 10.1016/j.virol.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Perez O, Hope TJ, Emerman M. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog. 2007;3 (10):1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva L, Cherepanov P, Leyens L, Wilson SJ, Rasaiyaah J, Fassati A. HIV-1 exploits importin 7 to maximize nuclear import of its DNA genome. Retrovirology. 2009;6:11. doi: 10.1186/1742-4690-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4 (5):495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.