

Abstract
A variety of Lewis bases were combined with 9-BBN-NTf2 to establish the requirements for the generation of borenium cations. Five different types of behavior were found, but the most interesting was the combination of Et3N, DABCO, 2,6-lutidine, or Ph3P=S which formed borenium ions exclusively even in sub- or superstoichiometric quantities. The 9-BBN borenium ion complex of 2,6-lutidine was found to rapidly catalyze the hydrosilylation of a variety of ketones in the presence of Et3SiH. Preliminary mechanistic experiments suggest that the reduction involves borenium ion activation of Et3SiH and not the ketone.
Foregoing studies from these laboratories have provided extensive preparative and mechanistic illustration of the counterintuitive concept of Lewis base activation of Lewis acids.1 The majority of the early investigations focused on the activation of silicon tetrachloride with chiral bisphosphoramides to generate a highly reactive, chiral trichlorosiliconium ion complex that catalyzed the diastereo- and enantioselective aldol additions of enoxysilanes derived from aldehydes, ketones, esters, amides, and nitriles.2 In recent years, our attention has turned to the Lewis base activation of Lewis acidic reagents in Group 16 (selenium3 and sulfur4) as well as Group 17 (bromine5 and chlorine6) to effect enantioselective cyclofunctionalizations of unactivated alkenes. Despite the dramatic difference in the chemical transformations involved, the underlying principle of catalysis is the same, namely the electronic redistribution in donor–acceptor complexes first articulated by Gutmann, Figure 1.7 To date, all of our mechanistic studies have demonstrated that the ionization of the dative complexes into highly electrophilic, cationic species is required for catalysis.
Figure 1.
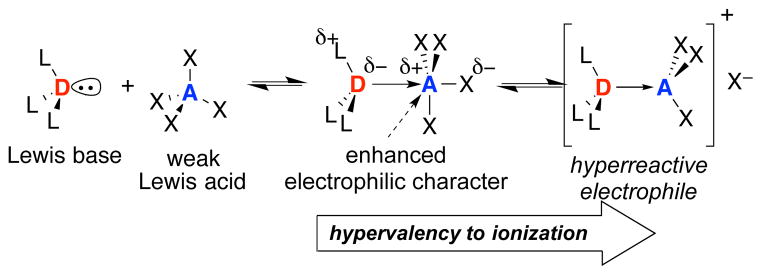
Gutmann formulation of Lewis base activation of Lewis acids.
In continuation of these investigations, we were drawn to the possibility of activating Lewis acids in Group 13 in view of the recent interest in the chemistry of tricoordinate cationic boron species (borenium ions).8 Many different methods for borenium ion generation are on record9 and these highly reactive species have been used as stoichiometric reagents (e.g. in arene and alkane borylation).10 On the other hand, the main focus in borenium cation catalysis has been the development of oxazaborolidine catalysts and other application methods are very rare.11 Very recently, borenium ion catalyzed hydrogenation and hydroboration have been reported. In 2012, Crudden and co-workers reported a novel, metal-free, catalytic method for the reduction of imines using air-stable, nonhazardous boranes. The catalysts for this process are discrete, well-characterized borenium salts derived from pinacolborane.12a In addition, Curran, Vedejs and co-workers reported a borenium ion catalyzed hydroboration of alkenes with N-heterocyclic carbene-boranes,12b Stephan and co-workers described hydrogenation of imines with 9-BBN–NHC borenium ion complexes,12c and Jäckle recently described the preparation of chiral borenium ions that effected asymmetric hydrosilylation of a ketone.12d
To develop a catalytic method for the generation of borenium ions presented far greater challenges than those encountered with Group 14, 16, and 17 elements in previous endeavors such as: (1) the design features for Lewis base activation are entirely different in view of the high affinity of the Lewis acidic elements in this group, (2) the intrinsic Lewis acidity of any precursors presents a major problem for suppression of background reactions, (3) ionization of a labile ligand is essential (at least for boron) to provide the coordinative unsaturation needed for catalysis, and (4) the lack of an established suite of known reactions of borenium ions for which (asymmetric) catalysis would be useful. Thus, we viewed this exercise as an exploratory investigation to learn the rules for the catalytic generation of borenium ions to enable the discovery of new reaction chemistry of these novel species.
The first phase of the study required the identification of a suitable boron precursor and its behavior in the presence of a wide range of Lewis bases. The structures of the possible adducts in Scheme 1 could be identified by 11B-, 19F-, and 31P-NMR spectroscopy. From the pioneering work of Vedejs, we chose 9-borabicyclo[3.3.1]nonane bistriflimide (9-BBN-NTf2, 1). Vedejs demonstrated that 1 was effective for the generation of borenium and boronium cations with amine Lewis bases owing to the excellent nucleofugality of the bistriflimide group.13
Scheme 1.
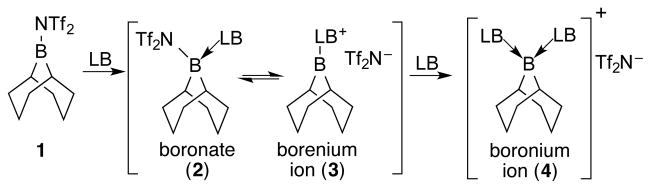
Initially, 11B and 19F NMR spectroscopic analysis of isolated, purified 1 was performed in CH2Cl2 (0.1 M) at room temperature and sharp singlets were observed at 58.9 ppm and −70.0 ppm, respectively. To assay the potential of Lewis bases to generate borenium cations from 1, 0.5, 1.0, and 2.0 equiv of each Lewis base were titrated to the 0.1 M solution of 1 and analyzed by NMR spectroscopy (Table 1). The 19F NMR resonance at ca. −80.0 ppm is characteristic of bistriflimide anion,13 and thus allowed the assignment of the boron-containing fragment as a cation. With this information in hand, DABCO, pyridine, 2,4-lutidine, 2,6-lutidine, HMPA, Ph3P=O, n-Bu3P=O, Ph3P=S, (Me2N)3P=S, pyridine-N-oxide, DMSO, n-Bu3P, and benzophenone were used as Lewis bases and the structures of the 9-BBN-Lewis base complexes were assigned by 11B and 19F NMR spectroscopy titration experiments. Analysis of the results revealed that these Lewis bases could be broadly classified into five different groups: (1) Type I in which weak coordination formed a boronate complex only at all loadings of Lewis base (benzophenone), (2) Type II in which strong coordination formed a boronate complex which spontaneously ionized and bound a second Lewis base to form a boronium ion at all loadings of Lewis base (2,4-lutidine, n-Bu3P), (3) Type III in which strong coordination led to ionization to form borenium ions at all loadings of Lewis base (Et3N, DABCO, 2,6-lutidine, Ph3P=S), (4) Type IV in which strong coordination led to ionization to form borenium ions with 1.0 equiv and then boronium ions with 2.0 equiv of Lewis base (HMPA, Ph3P=O, n-Bu3P=O, (Me2N)3P=S, pyridine-N-oxide, DMSO), and (5) Type V in which strong coordination led to ionization to form only boronium ions at all loadings of Lewis base (pyridine, proton sponge). Notably, for Type III Lewis bases, boronium cations were not formed even with 2.0 equiv of Lewis base. Furthermore, with 1.0 equiv of Type IV Lewis bases, a borenium cation was formed predominantly together with a small amount of the boronium cation.
Table 1.
Titration Experiments with 1 and Various Lewis Bases. 11B-NMR Data.a
| Lewis base | boronate complex (ppm) (2)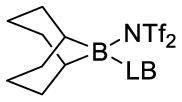
|
borenium ion (ppm) (3)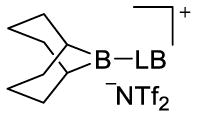
|
boronium ion (ppm) (4)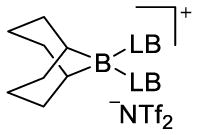
|
|---|---|---|---|
| Et3N (a) | 84.8 (85.1)b | ||
| DABCO (b) | 56.9 | ||
| pyridine (c) | 5.2 | ||
| 2,4-lutidine (d) | 39.4 | 9.7 | |
| 2,6-lutidine (e) | 84.4 | ||
| DMAP (f) | (66.5)b | (3)b | |
| proton sponge (g)c | (16.2)b | ||
| HMPA (h) | 64.9 | 17.8 | |
| Ph3P=O (i) | 67.4 | 17.3 | |
| n-Bu3P=O (j) | 65.1 | 14.5 | |
| Ph3P=S (k) | 80.4 | ||
| (Me2N)3P=S (l) | 80.9 | 46.9 | |
| pyridine-N-oxide (m) | 64.2 | 14.4 | |
| DMSO (n) | 66.1 | 15 | |
| n-Bu3P (o) | 33.1 | −5.4 | |
| benzophenone (p) | 35.6d |
11B NMR spectra were recorded at 128 MHz (CH2Cl2) with proton decoupling. Chemical shifts are reported in ppm from BF3•OEt2 (0.0 ppm) as the external standard.
Data in parentheses are from ref. 13.
1,8-bis(dimethylamino)naphthalene.
2.0 equiv of Lewis base.
With a reliable method for the efficient, in situ generation of borenium-Lewis base complexes, the next phase involved the identification of a reaction to evaluate the catalytic potential of these species. The hydrosilylation reaction of ketones was selected as a model reaction inspired by the imine hydrosilylation by Crudden et al.12a By analogy to their proposed catalytic cycle, ketones would be activated by the borenium complex to give the corresponding boronium complex A. Subsequent hydride transfer from triethylsilane would afford borinate complex B that could be exchanged by Et3SiNTf2 to regenerate the borenium-Lewis base complex catalyst (Mechanism I) (Scheme 2).
Scheme 2.

Initial experiments to probe this hypothesis were conducted by treating 4-methylbenzophenone (5a) with Et3SiH (1.5 equiv) in the presence of various boron reagents. It was first established that Et3SiH does not reduce 5a at room temperature after 6 h (Table 2, entry 1). However, in the presence of 9-BBN-NTf2 1 (0.1 equiv) only over-reduced product 6a was obtained in 69% yield in 10 min (entry 2). Whereas no reaction took place in the presence of 0.1 equiv of complex 3b (entry 3), 0.1 equiv of complex 3e afforded the expected reduction product 7a in 95% yield (entry 4). Remarkably, lowering the loading of 2,6-lutidine to 0.05 equiv in the presence of 0.05 equiv of 1 still afforded exclusively 7a albeit much more slowly such that after 20 h only a 69% yield was obtained (entry 5). Clearly, two different mechanisms are operative for the different catalysts, 1 and 3e.
Table 2.
Hydrosilylation of Ketones Catalyzed by Various Boron Reagents.a
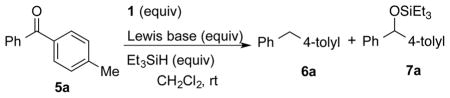
| ||||||
|---|---|---|---|---|---|---|
| entry | 1 (equiv) | Lewis base (equiv) | Et3SiH (equiv) | time | yieldb 6a (%) | yieldb 7a (%) |
| 1 | 0 | – | 1.5 | 6 h | 0 | 0 |
| 2 | 0.1 | – | 1.5 | 10 min | 69 | 0 |
| 3 | 0.1 | DABCO (0.1) | 1.0 | 12 h | 0 | 0 |
| 4 | 0.1 | 2,6-lutidine (0.1) | 1.5 | 30 min | <5 | 95 |
| 5 | 0.05 | 2,6-lutidine (0.05) | 1.5 | 20 h | <5 | 69 |
Reactions were performed at room temperature in an NMR tube (see Supporting Information for details).
Yields determined by integration of product signals versus Cl3CCH2Cl as an internal standard.
The scope of the borenium cation catalyzed hydrosilylation was explored with various ketones 5 and the results are summarized in Table 3. Generally, the use of 0.1 equiv of 3e and 1.5 equivalents of triethylsilane was sufficient for a smooth reaction and, after treatment of the mixture with catalytic amounts of FeCl3 in methanol to remove silyl-protecting group, alcohols 8 were obtained in high yield. Both electron-deficient and electron-rich aryl ketones were compatible with the reduction conditions (entries 1–4). In addition, fused- and heteroaromatic substituents had no negative influence (entries 5 and 7). A secondary alkyl group was also nicely accommodated (entry 8).
Table 3.
Substrate Scope.a
| |||||
|---|---|---|---|---|---|
| entry | R | ketone | time (min) | yieldb (%) | alcohol |
| 1 | 4-Me–C6H4 | 5a | 30 | 92 | 8a |
| 2 | 3-Me–C6H4 | 5b | 10 | 90 | 8b |
| 3 | 4-MeO–C6H4 | 5c | 180 | 91 | 8c |
| 4 | 4-Br–C6H4 | 5d | 10 | 93 | 8d |
| 5 | 1-naphthyl | 5e | 60 | 86 | 8e |
| 6 | 2-furyl | 5f | 10 | 82 | 8f |
| 7 | 2-thiophen | 5g | 60 | 83 | 8g |
| 8c | isopropyl | 5h | 60 | 77 | 8h |
Unless otherwise noted, reactions were performed on 0.1 mmol scale with 1.5 equiv of Et3SiH in dichloromethane (1.0 mL).
Yield of isolated, purified product.
0.2 equiv of 3e was used as catalyst.
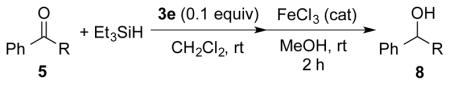
Although 3e functioned as a catalyst for the hydrosilylation of ketones as described above, the reaction mechanism remained unclear. Several control experiments were carried out to provide mechanistic insight. First, the operation of Mechanism I (Scheme 2) was probed by the attempted generation of intermediates A and B and subjecting them to the reaction conditions. However, the combination of 3e and benzophenone in CH2Cl2 revealed no interaction by 11B NMR spectroscopic analysis of the mixture.
The preparation of intermediate B was attempted by treatment of boronate 9a15 with N-(trimethylsilyl)bis(trifluoromethanesulfonyl) imide (TMSNTf2) and 2,6-lutidine. However, 9a was not converted to either silyl ether 7a or over-reduced product 6a instead, it slowly converted to the corresponding lutidinium salt 10a. In addition, 10a was also observed in presence of Et3SiH (Scheme 3). These experiments suggest that the hydrosilylation reaction catalyzed by 3e does not proceed via Mechanism I.16
Scheme 3.

In a different mechanism for the borenium ion catalyzed ketone hydrosilylation reaction, hydride abstraction from Et3SiH by the borenium cation could generate 9-BBN-hydride-2,6-lutidine complex 11 as the active reducing reagent (Scheme 4). To investigate this possibility required the independent preparation of 11. However, 11 did not form from 9-BBN-dimer and 2,6-lutidine.17 Instead, the existence of 11 was probed indirectly by a 11B NMR experiment between 3e and Et3SiH. Although 11 was not detected via 11B NMR spectroscopy, 3e was slowly converted to 9-BBN-H dimer. Furthermore, on mixing 9-BBN-H dimer with TMSNTf2 in the presence of 2,6-lutidine, 9-BBN-H dimer was also slowly converted to 3e. Thus, the equilibrium between 3e and 9-BBN-H dimer, as well as the ability of 3e to activate triethylsilane toward hydrogen transfer were confirmed (Scheme 4).
Scheme 4.

With this information in hand, two plausible catalytic cycles can be constructed; one catalyzed by borenium ion 3e (Mechanism II) and one catalyzed by Et3SiNTf2 (Mechanism III) (Scheme 5). Mechanism II (borenium ion activation of Et3SiH followed by carbonyl activation via silyl transfer (path a) to form silyloxycarbenium ion D and consummated by hydride transfer to regenerate 3e) finds excellent precedent in the Piers mechanism for (C6F5)3B catalyzed hydrosilylation of ketones which has been thoroughly investigated.18 Mechanism III involves the Tf2N− assisted activation of Et3SiH to form Et3SiNTf2 (path b) that in turn forms ion D which is reduced by Et3SiH.
Scheme 5.

To differentiate between these two catalytic cycles, control experiments were conducted. Mechanism III posits that the reduction involves Et3SiNTf2 as the activator and Et3SiH as the reducing agent but does not require 9-BBN-H. Accordingly, 5a, Et3SiH, and 0.1 equiv each of TMSNTf2 and 2,6-lutidine were combined in CH2Cl2 at room temperature. The reaction required 6 h to reach full conversion and afforded 7a in 92% yield. In contrast, an identical reaction containing 0.1 equiv of 9-BBN-H dimer was complete within 10 min to afford 7a in 96% yield (Scheme 6). Thus, although Mechanism III is viable, it does not compete with Mechanism II under the established reaction conditions.
Scheme 6.
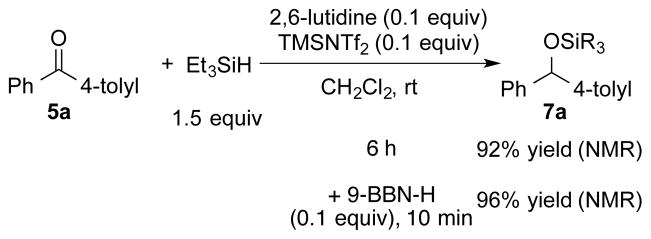
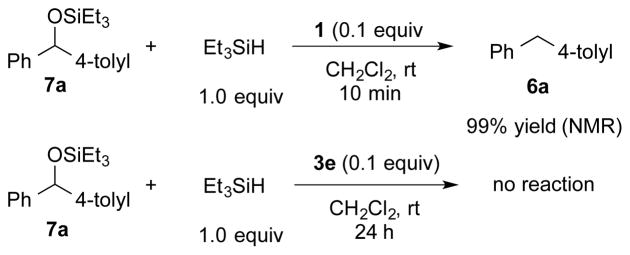
In a final set of experiments, the stability of silyl ether 7a in the presence of Et3SiH and both catalysts was tested. Treatment of 7a with 1 resulted in the formation of 6a in 99% yield after 10 min. However, 7a did not react with borenium cation 3e even after in 24 h. These results further support the operation of Mechanism II and also explain the formation of the reduced product 6a in reaction without 2,6-lutidine (Scheme 7).
Scheme 7.
In conclusion, the interaction of various Lewis bases with 9-BBN-NTf2 has been investigated. The stable borenium cation complex 3e catalyzed the hydrosilylation of ketones. Mechanistic studies have revealed that this reaction takes place via a hydride abstraction pathway similar to the Piers mechanism for (C6F5)3B catalyzed hydrosilylation of ketones. Further studies on this concept, including the development of other activation modes and asymmetric reactions, will be reported in due course.
Supplementary Material
Acknowledgments
Y.U. thanks the Japan Society for the Promotion of Science for a postdoctoral fellowship. We are grateful to the National Institutes of Health for generous financial support (R01 GM85235).
Footnotes
Notes
The authors declare no competing financial interests.
Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 2.(a) Beutner GL, Denmark SE. Angew Chem, Int Ed. 2013 doi: 10.1002/anie.201302084. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beutner GL, Denmark SE. Top Organomet Chem. 2013;44:55–89. [Google Scholar]; (c) Denmark SE, Fujimori S. In: In Modern Aldol Reactions. Mahrwald R, editor. Vol. 2. Vol. 7 Wiley-VCH; Weinheim: 2004. [Google Scholar]; (d) Denmark SE, Stavenger RA. Acc Chem Res. 2000;33:432–440. doi: 10.1021/ar960027g. [DOI] [PubMed] [Google Scholar]
- 3.(a) Denmark SE, Kalyani D, Collins WR. J Am Chem Soc. 2010;132:15752–15765. doi: 10.1021/ja106837b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark SE, Collins WR. Org Lett. 2007;9:3801–3804. doi: 10.1021/ol701617d. [DOI] [PubMed] [Google Scholar]
- 4.(a) Denmark SE, Jaunet A. J Am Chem Soc. 2013;135:6419–6422. doi: 10.1021/ja401867b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark SE, Kornfilt DJP, Vogler T. J Am Chem Soc. 2011;133:15308–15311. doi: 10.1021/ja2064395. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Denmark SE, Collins WR, Cullen MD. J Am Chem Soc. 2009;131:3490–3491. doi: 10.1021/ja900187y. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Vogler T. Chem Eur J. 2009;15:11737–11745. doi: 10.1002/chem.200901377. [DOI] [PubMed] [Google Scholar]
- 5.Denmark SE, Burk MT. Org Lett. 2012;14:256–259. doi: 10.1021/ol203033k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark SE, Burk MT. Proc Nat Acad Sci. 2010;107:20655–20660. doi: 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burk MT. Ph D Thesis. University of Illinois; Urbana-Champaign: 2012. [Google Scholar]
- 7.(a) Gutmann V. The Donor-Acceptor Approach to Molecular Interactions. Plenum Press; New York: 1978. [Google Scholar]; (b) Gutmann V. Coord Chem Rev. 1975;15:207–237. [Google Scholar]
- 8.(a) De Vries TS, Prokofjevs A, Vedejs E. Chem Rev. 2012;112:4246–4282. doi: 10.1021/cr200133c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Piers WE, Bourke SC, Conroy KD. Angew Chem, Int Ed. 2005;44:5016–5036. doi: 10.1002/anie.200500402. [DOI] [PubMed] [Google Scholar]; (c) Kölle P, Nöth H. Chem Rev. 1985;85:399–418. [Google Scholar]
- 9.Interestingly, one of the very early methods described for the generation of borenium ions termed “nucleophilic addition” by Nöth or “nucleophilic addition – heterolysis” by Vedejs is, in fact, Lewis base activation of the boron Lewis acid.
- 10.(a) Del Grosso A, Singleton PJ, Muryn CA, Ingelson MJ. Angew Chem, Int Ed. 2011;50:2102–2106. doi: 10.1002/anie.201006196. [DOI] [PubMed] [Google Scholar]; (b) Genaev AM, Nagy SM, Salnikov GE, Shubin VG. Chem Commun. 2000:1587–1588. [Google Scholar]; (c) De Vries TS, Prokofjevs A, Harvey JN, Vedejs E. J Am Chem Soc. 2009;131:14679–14687. doi: 10.1021/ja905369n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Genaev AM, Nagy SM, Salnikov GE, Shubin VG. Chem Commun. 2000:1587–1588. [Google Scholar]
- 11.Corey EJ. Angew Chem, Int Ed. 2009;48:2100. doi: 10.1002/anie.200805374. [DOI] [PubMed] [Google Scholar]
- 12.(a) Eisenberger P, Bailey AM, Crudden CM. J Am Chem Soc. 2012;134:17384–17387. doi: 10.1021/ja307374j. [DOI] [PubMed] [Google Scholar]; (b) Prokofjevs A, Bourssonnière A, Li L, Bonin H, Lacôte E, Curran DP, Vedejs E. J Am Chem Soc. 2012;134:12281–12288. doi: 10.1021/ja305061c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Farrell JM, Hatnean JA, Stephan DW. J Am Chem Soc. 2012;134:15728–15731. doi: 10.1021/ja307995f. [DOI] [PubMed] [Google Scholar]; (d) Chen J, Lalancette RA, Jäckle F. Chem Commun. 2013:4893–4895. doi: 10.1039/c3cc41556b. [DOI] [PubMed] [Google Scholar]
- 13.Prokofjevs A, Kampf JW, Vedejs E. Angew Chem, Int Ed. 2011;50:2098–2101. doi: 10.1002/anie.201005663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boronate9awas prepared by reaction of 9-BBN-H with authentic carbinol8a; see the Supporting Information.
- 16.Hydrosilylation of imines with neutral boranes proceeds by a differnt mechanism than by borenium ions. Mewald M, Oestreich M. Chem Eur J. 2012;18:14079–14084. doi: 10.1002/chem.201202693.
- 17.9-BBN-H Dimer was treated with 2,6-lutidine in CH2Cl2 at room temperature. After 24 h, 11B NMR analysis showed no change.
- 18.(a) Piers WE, Marwitz AJV, Mercier LG. Inorg Chem. 2011;50:12252–12262. doi: 10.1021/ic2006474. [DOI] [PubMed] [Google Scholar]; (b) Parks DJ, Blackewell JM, Piers WE. J Org Chem. 2000;65:3090–3098. doi: 10.1021/jo991828a. [DOI] [PubMed] [Google Scholar]; (c) Parks DJ, Piers WE. J Am Chem Soc. 1996;118:9440–9441. [Google Scholar]; (d) Rendler S, Oestreich M. Angew Chem, Int Ed. 2008;47:5997–6000. doi: 10.1002/anie.200801675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.