

Abstract
Purpose:
Triple-negative breast cancer (TNBC) accounts for 10-20% of newly diagnosed invasive breast cancer. Finding effective targets for chemotherapy-resistant TNBC has proven difficult in part because of TNBC’s molecular heterogeneity. We have previously reported that, likely because of GR’s anti-apoptotic activity in ER-negative breast epithelial and cancer cells, high glucocorticoid receptor (GR) expression/activity in early-stage TNBC significantly correlates with chemotherapy-resistance and increased recurrence. We hypothesized that pre-treatment with mifepristone, a (GR)-antagonist, would potentiate the efficacy of chemotherapy in GR+ TNBC by inhibiting GR’s anti-apoptotic signaling pathways and increasing the cytotoxic efficiency of chemotherapy.
Experimental Design:
TNBC cell apoptosis was examined in the context of physiological glucocorticoid concentrations, chemotherapy, and/or pharmacologic concentrations of mifepristone. We used high-throughput live microscopy with continuous recording to measure apoptotic cells stained with a fluorescent dye, and Western analysis to detect caspase-3 and PARP cleavage. The effect of mifepristone on GR-mediated gene expression was also measured. TNBC xenograft studies were performed in female severe combined immunodeficient (SCID) mice and tumors were measured following treatment with vehicle, paclitaxel or mifepristone/paclitaxel.
Results:
We found that although mifepristone treatment alone had no significant effect on TNBC cell viability or clonogenicity in the absence of chemotherapy, the addition of mifepristone to dexamethasone/paclitaxel treatment significantly increased cytotoxicity and caspase-3/PARP cleavage. Mifepristone also antagonized GR-induced SGK1 and MKP1/DUSP1 gene expression, while significantly augmenting paclitaxel-induced GR+ MDA-MB-231 xenograft tumor shrinkage in vivo.
Conclusions:
These results suggest that mifepristone pre-treatment could be a useful strategy for increasing tumor cell apoptosis in chemotherapy-resistant GR+ TNBC.
Keywords: triple-negative breast cancer, glucocorticoid receptor, mifepristone, breast cancer, xenograft
Introduction
Glucocorticoids (GCs) are secreted from the adrenal gland in response to exposure to emotional and physiological stressors and are responsible for modulating essential metabolic, cardiovascular, immune, and behavioral functions (1-3). The glucocorticoid receptor (GR) belongs to a family of nuclear hormone receptors that are ligand-dependent transcription factors involved in activating and repressing gene expression, thereby changing the complement of proteins regulating key signaling pathways (4, 5). In its ligand-bound state, GR initiates or represses gene expression in a cell type-specific manner (1). For example, GR activation can induce apoptosis in lymphocytes (6, 7), while its activation results in inhibition of apoptosis in breast epithelial cells (8). Furthermore, in a TNBC xenograft model, the activation of tumor GR by dexamethasone (dex), a synthetic GC, diminished chemotherapy effectiveness in vivo (9). Until now, the use of a GR-antagonist in an in vivo model of GR+ triple-negative breast cancer (TNBC) has not been reported.
It was previously shown by our group and others that GR activation initiates potent anti-apoptotic signaling pathways in breast epithelial cells, at least in part, via transcriptional regulation of genes encoding cell survival pathway proteins (5, 10-12). For example, genes encoding the anti-apoptotic proteins serum and glucocorticoid-inducible protein kinase-1 (SGK1) and mitogen-activated protein kinase phosphatase-1 (MKP1/DUSP1) are both upregulated following GR activation (13-15). Moreover, short-hairpin RNAs (shRNAs) expression targeting either SGK1 or MKP1/DUSP1 demonstrated a requirement for the induction of these proteins to induce GR-mediated cell survival (13). Furthermore, mifepristone, a dual GR and PR modulator, significantly antagonizes the induction of SGK1 and MKP1/DUSP1 expression in ER-negative breast cell lines treated with glucocorticoids (13).
In this study, we tested the hypothesis that GR-modulation (using mifepristone) could potentiate chemotherapy-induced cytotoxicity in TNBC models where GR (but neither ER or PR) is expressed. Our data suggest that mifepristone blocks GR-mediated tumor cell survival by antagonizing associated gene expression and inhibiting apoptotic pathways that are usually activated by endogenous glucocorticoids, thereby augmenting chemotherapy-induced cell death and decreasing in vivo TNBC tumor growth.
Experimental Procedures
Materials
Paclitaxel (Sigma Cat. No.T7402) and dexamethasone (Sigma Cat. No. D4902) were purchased from Sigma-Aldrich. Initially, mifepristone was purchased from Enzo Life Sciences (Cat. No. BML-S510-0025) and later experiments were repeated with pharmaceutical-grade mifepristone provided by Corcept Therapeutics (Menlo Park, CA). For in vivo experiments, pharmaceutical-grade paclitaxel liquid suspension was purchased from Bedford Laboratories (Bedford, OH).
Cell culture
MDA-MB-231, BT-20 and MDA-MB-468 cell lines were purchased from American Type Culture Collection. MDA-MB-231 and BT-20 cells were cultured in Dulbecco’s Modified Eagle Medium (Lonza) and MDA-MB-468 cells in RPMI-1640 (Thermo Fischer Scientific), both supplemented with 10% fetal calf serum (FCS)(Gemini Bio-Products) and antibiotics (1% penicillin-streptomycin, Lonza). All cell lines were cultured at 37°C in a humidified atmosphere with 5% CO2. Prior to treatment with glucocorticoids, mifepristone and/or chemotherapy, cells were grown in Dulbecco’s Modified Eagle Medium or RPMI-1640 supplemented with 2.5% charcoal stripped FCS and 1% penicillin-streptomycin.
Cell death assay
TNBC cell lines (MDA-MB-231 at 2 ×103 cells/well, MDA-MB-468 at 5 × 103 cells/well, and BT-20 at 3.5 × 103 cells/well) were plated in 96-well plates and allowed to adhere overnight in DMEM or RPMI supplemented with 10% FCS. Media was then changed to 2.5% charcoal-stripped serum (CS-FCS) for 48 hours. Cells were treated with vehicle (EtOH 0.1% V/V), dexamethasone (100 nM) or mifepristone (100 nM) alone or dex/mif (100 nM) one hour before paclitaxel (100 nM) treatment for 72-hours. A cyanine dimer nucleic acid dye, YOYO-1 (Life Technologies, Y3601), that causes green fluorescence if the cellular membrane is compromised, was used to detect dead cells. Two images (1.90 × 1.52 mm) in separate regions of each well were captured with a 10x objective at 4-hour intervals using the IncuCyte FLR HD real-time in vitro micro-imaging system (Essen Instruments, Ann Arbor, MI). Cell death (detected as YOYO-1-positive) and total cell counts (phase contrast) were measured computationally by ImageJ Version 1.46r (16) using investigator-coded software for analysis (Supp. Method S1). The “cytotoxic index” represents the number of dead cells/total cells for each image.
Images collected between 12 and 72 hours were used in the analysis. The cytotoxic index was log-transformed to satisfy the normality assumption. Data were analyzed using repeated measures analysis of variance models. A separate model was fitted for each cell line. The fixed effects included were treatment, time, time2, time3, and all corresponding interactions between treatment and time terms. Random effects included random intercept terms for biological and technical replicates, and a random slope for the biological replicate. Correlation between serial measurements was modeled using AR (1) covariance structure. A generalized F-test was used to test the composite hypothesis of no difference between treatment, trt × time, trt ×time2 and trt ×time3, effectively comparing the entire curves over time. Analyses were performed in SAS 9.2.
Clonogenic Assay
MDA-MB-231 cells (10,000 cells per 10 cm dish) were allowed to adhere overnight in DMEM supplemented with 10% FCS. Media was then changed to 2.5% CS-FCS for 48 hours. Cells were treated with vehicle (EtOH 0.1% V/V), dexamethasone (100 nM) or mifepristone (100 nM) alone or dex/mif (100 nM) one hour before paclitaxel (100 nM) treatment for four days. The colonies were fixed with 3.7% paraformaldehyde and stained with 0.05% crystal violet in PBS. Five random 1 cm diameter circles were then drawn with a black marker and the colonies within these five regions were counted under a stereoscope at a magnification of 13X. A minimum of 5 cells per colony were required to meet the criterion for counting as a “colony.”
A mixed effects analysis of variance model was fitted, with colony count as the responsible variable, treatment group as the fixed effect, and biological replicate and technical replicate nested within the biological replicates as the random effects. Dex/pac versus dex/mif/pac groups were compared based on this fitted model, and the reported p-values are unadjusted for multiple comparisons.
Quantitative real-time PCR
MDA-MB-231, MDA-MB-468, and BT-20 cells were seeded at ~50% confluence and allowed to adhere overnight in DMEM with 10% FCS, then cultured in 2.5% CS-FCS for an additional 48 hours. Media was removed and equal volumes of either vehicle (ethanol), dexamethasone (100 nM) or dex/mifepristone (100 nM FC) diluted in DMEM or RPMI supplemented with 2.5% CS-FCS was then added. After four hours of treatment, 100ul of RNA-Solv Reagent (EaZy Nucleic Acid Isolation Kit) supplemented with 2% 2-mercaptoethanol was added to each well to harvest RNA. Total RNA was extracted using the Qiagen All-Prep DNA/RNA Mini kit. cDNA was then reverse-transcribed from 0.5 μg of total RNA with Quanta reverse transcription reagents (Quanta Biosciences) using the GeneAmp PCR System 9700 (Applied BioSystems) per manufacturers instruction. The cDNA was diluted in PerfeCTa SYBR Green FastMix (Quanta Biosciences) and quantitative real-time PCR was carried out in a BioRad PCR System MyIQ (BioRad Life Sciences). The following primers were used: SGK1, 5′-AGGCCCATCCTTCTCTGTTT-3′ (forward) and 5′-TTCACTGCTCCCCTCAGTCT-3′ (reverse); MKP1/DUSP1, 5’-CCTGACAGCGCGGAATCT-3’ (forward) and 5’-GATTTCCACCGGGCCAC-3’ (reverse); NR3C1/GR, 5’-TCTGAACTTCCCTGGTCGAA-3’ (forward) and 5’- GTGGTCCTGTTGTTGCTGTT-3’ (reverse); Actin-B 5’-CAGCGGAACCGCTCATTGCCAATGG-3’ (forward) and 5’-TCACCCCCTGTGCCCATCTACGA-3’ (reverse). The samples were loaded in duplicate. Relative quantification of gene expression was calculated according to the standard curve method, as described by Applied Biosystems User Bulletin 2, October 2001, based on the ΔΔCt approach (15). A ratio of GR target gene expression to Actin-B expression was calculated.
For SGK1 and MKP1/DUSP1 Q-RT-PCR analysis, a mixed-effects ANOVA model was fitted with Ct as the response variable; treatment, gene type (target or reference), and treatment × gene interaction as the fixed effects and replicate as the random effect. A linear contrast was then constructed to estimate ΔΔCt and its confidence interval, and the results were exponentiated to obtain the estimate of 2−ΔΔCt and its confidence intervals; 68% confidence intervals, corresponding to +/− SEM under the normality assumption. Analyses were performed in SAS 9.2.
Antibodies and Western blotting
Cells were allowed to adhere overnight in media containing 10% FCS. The following day, media was changed to 2.5% CS-FCS and cultured for 48 hours, then lysed in buffer containing phosphatase and protease inhibitor cocktails (Roche). Protein concentrations were measured using the BCA Assay Kit (Thermo Scientific) and 2x Laemmli buffer supplemented with 5% 2-mercaptoethanol was added to an equivalent volume of protein lysate. Total proteins (60 μg per lane) were resolved by SDS-PAGE and then transferred to polyvinylidene difluoride membranes (Bio-Rad). After the membranes were washed three times, they were incubated with 5% BSA (Fisher Scientific) in 0.1% Tween20 in TBS (TBS/T) for 1 hour at room temperature, followed by overnight incubation at 4°C with primary antibody against human GR-alpha (GR-XP Cell Signaling), Caspase-3 (Cell Signaling), PARP (Cell signaling) or human β-Actin (Sigma-Aldrich). After additional washing, membranes were incubated for 1 hour at room temperature with either Alexa Fluor 680 goat anti-rabbit (Invitrogen) or 800 goat anti-mouse (LI-COR) secondary antibody, rinsed and scanned using the Odyssey infrared imaging system (LI-COR) at a wavelength of 700 or 800 nm.
Mammary fat pad xenograft studies in female SCID mice
All experiments were performed in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals and approved by the University of Chicago Institutional Animals Care and Use Committee. Suspensions of MDA-MB-231 cells (1 × 107) in 50 μl of PBS were injected subcutaneously into the right pectoral mammary fat pad of a 5-6 week old female SCID mouse (Taconic). Tumors were allowed to reach ~200mm3 and then mice were treated via intraperitoneal (IP) injection with paclitaxel (10 mg/kg) suspended in castor oil (1:10 v/v); vehicle-treated animals received two injections and paclitaxel-treated animals received an additional injection of mifepristone (15 mg/kg/day) or vehicle (ethanol and sesame seed oil 1:10 v/v) one hour prior to the paclitaxel (17). The longest (L) and shortest (S) diameters of the tumors were measured three times a week with electronic calipers and tumor volume was calculated using the formula for an ellipsoid sphere: S2 × L × (0.52) (9). Tumors were dissected and cut lengthwise into mirror image sections. One section was minced in lysis buffer and frozen (for protein analysis), and the other was fixed in 10% neutral buffered formalin (for IHC and IF). Tumor growth data was analyzed using repeated measures analysis of variance models as previously described (9).
Histopathological examination
For anti-GR IF analysis of tumor xenografts, samples were formalin-fixed for 24 hours and embedded in paraffin immediately after necropsy. Sections (5 μm thick) were adhered to positively charged slides, de-waxed in xylene, and hydrated using graded ethanol washes. Heat-induced antigen retrieval was performed using Tris-EDTA Buffer (10mM Tris Base, 1mM EDTA Solution, pH 9.0) incubation in a pressure cooker for 3 min. After 30 min of blocking in 10% normal goat serum in PBS, slides were incubated with either a 1:100 dilution of anti–GR rabbit polyclonal antibody (Santa Cruz, H-300 sc-8992) followed by a secondary Alexa Fluor goat anti-rabbit IgG (Cell Signaling).
Entire scans of each tumor section were captured using the CRi Panoramic Whole Slide Scanner (PerkinElmer Life Sciences). Twenty random individual images of each scan were then analyzed from different locations of the slide. The proportion of GR+ staining cells over the total cell count for each slide was calculated (details in Supp. Method S2) and log-transformed to satisfy the normality assumption. A mixed effects model was fitted, with treatment as the fixed effect, and tumor as the random effect, to account for the correlation between multiple images of the same tumor section.
Results
Mifepristone enhances paclitaxel-induced TNBC cell death
We have previously demonstrated that high GR-expressing ER-negative breast cancer cells exposed to physiological stress dose concentrations of glucocorticoids (1 uM) are relatively resistant to chemotherapy-induced cell death (13). Here we examined whether or not pharmacologically relevant concentrations of mifepristone (100 nM) could reverse the cytoprotective effects of physiological glucocorticoid concentrations (100 uM) in MDA-MB-231, MDA-MB-468, and BT-20 TNBC cell lines (18). We used the IncuCyte system, a real-time microscopic-imaging system (16), to determine the percentage of cell death continuously over several days using software outlined in Supp. Method S1. Cells were treated with vehicle (EtOH 0.1% V/V), dex (100nM), mifepristone (100 nM) or dex/mif (100nM) one hour before paclitaxel (100nM) before being placed in the IncuCyte assay system. Fig. 1A shows the percentage of cell death measured over several days. The addition of mifepristone to MDA-MB-231 (Fig. 1A, top) and BT-20 (Fig. 1A, middle) cells significantly reversed the protective effect of physiological glucocorticoid concentrations (dexamethasone, 100 nM). The addition of mifepristone to MDA-MB-468 cells also increased cell death from dex/paclitaxel, although not significantly (p=0.68 Fig. 1A, bottom). Interestingly, treating MDA-MB-231, BT-20, and MDA-MB-468 cells with mifepristone alone (no chemotherapy) had no significant effect on either apoptosis (Fig. 1) or cell proliferation (Supp. Fig. S1). A representative image of cells prior to counting is shown in Supp. Fig. S2. Fig. 1B shows images from the IncuCyte detection system where green cells reflect apoptotic cells reported in Fig. 1A (19). Trypan blue exclusion assays (Supp. Fig. S3) further supported the conclusion that mifepristone treatment partially reversed GR-mediated protection from chemotherapy-induced apoptosis in TNBC.
Figure 1. In vitro analysis of paclitaxel-induced cell death +/− mifepristone in MBA-MB-231, BT-20, and MDA-MB-468 cells.

A, TNBC cell lines were treated with vehicle (EtOH 0.01%), mif (100 nM), paclitaxel (100 nM), dex (100 nM)/paclitaxel (100 nM), or dex (100 nM)/mif (100 nM)/paclitaxel (100 nM). Error bars represent +/−SEM. *Denotes the two treatments compared to derive the p-value shown. B, Representative phase-contrast and cytotoxic fluorescent dye images of three TNBC cell lines using automated IncuCyte imaging. C, Total GR (NR3C1) mRNA expression was first normalized to β-Actin mRNA expression and then compared (fold difference) to one technical replicate associated with MDA-MB-231 NR3C1 levels (error bars reflect +/− SEM of three independent experiments). D, Western blot of glucocorticoid receptor (GR) and β-Actin. GRα translational isoforms are labeled.
We next measured GR (NR3C1) mRNA transcript expression in MDA-MB-231, MDA-MB-468, and BT-20 cells by Q-RT-PCR (Fig. 1C) and total GR protein expression by Western analysis (Fig. 1D). Q-RT-PCR confirmed expression of GR (NR3C1) mRNA levels in all three cells lines. The highest total GR mRNA and protein levels were found in MDA-MB-231 cells; interestingly several translational isoforms (GR-A, GR-B, GR-C, GR-D) were also observed in all three cell lines (20, 21).
Mifepristone induces caspase-3-associated cell death
The molecular mechanisms underlying GR-mediated cell survival in the context of chemotherapy-induced apoptosis are not well understood (12). We therefore characterized caspase-3 and PARP cleavage in association with paclitaxel-induced apoptosis. We found that chemotherapy-induced apoptosis and accompanying caspase-3 and PARP cleavage increased following the addition of mifepristone (Fig. 2). The increased cleavage of caspase-3 and PARP is seen after 24 hours of treatment, but enhanced after 48 hours of treatment. A second biological replicate of caspase-3 cleavage at 24 and 48 hours is reported in Supp. Fig. S4. These data suggest that mifepristone reverses GR-mediated cell survival, at least in part, through blocking an apoptotic mechanism involving increased cleavage of caspase-3 and PARP.
Figure 2. Caspase-3 and PARP cleavage following treatment with mifepristone.

MDA-MB-231 cells were treated with vehicle (EtOH 0.01%), dex (100 nM), mif (100 nM), paclitaxel (100 nM), dex/paclitaxel, or dex/mif/paclitaxel for 24 and 48 hours. The addition of mifepristone resulted in increased cleaved caspase-3 and PARP compared to dex/pac treatment alone.
Mifepristone antagonizes GR-mediated gene expression in TNBC cell lines
SGK1 and MKP1/DUSP1 genes are directly upregulated by GR transactivation in mammary epithelial cells (2). To examine whether mifepristone antagonizes this GR-mediated gene expression, we treated MDA-MB-231, BT-20, and MDA-MB-468 cells with dex (100nM) +/− mifepristone (100nM) for 4 hours. In MDA-MB-231 cells, Q-RT-PCR showed an average dex-associated 2.13-fold increase in SGK1 and 6.62-fold increase in MKP1/DUSP1 mRNA levels over vehicle alone; the dex-mediated increase in both GR-target genes was significantly reversed with the addition of mifepristone (Fig. 3A). In BT-20 cells, SGK1 (2.10-fold increase) and MKP1/DUSP1 (1.70-fold increase) mRNA levels were both inhibited by mifepristone (Fig. 3B). In MDA-MB-468 cells, the dex-associated increase in SGK1 (2.85-fold increase) was significantly inhibited by mifepristone, while MKP1/DUSP1 (1.52-fold increase) expression was reversed although it did not meet statistical significance (Fig. 3C). These data suggest the degree of GR inhibition with mifepristone is variable depending on cell type and the particular gene studied.
Figure 3. SGK1 and MKP1/DUSP1 steady-state mRNA analysis following 4 hrs of dex vs. dex/mif treatment.
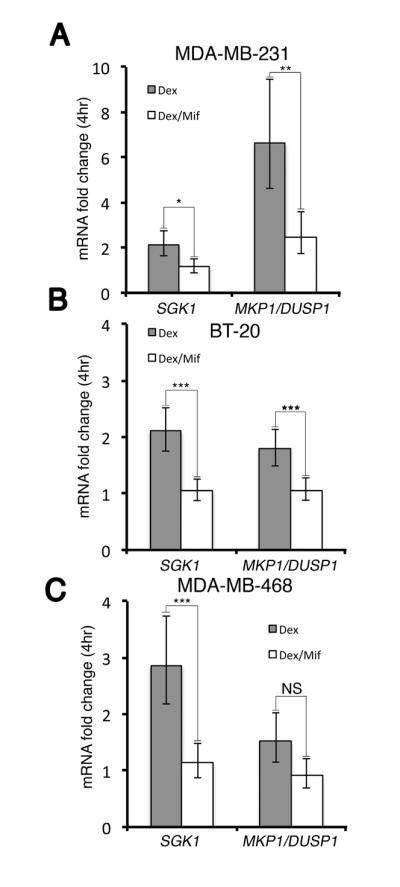
MDA-MD-231, MDA-MB-468, or BT-20 cells were treated with vehicle (EtOH 0.01%), dex (100 nM) or dex/mif (100 nM) for 4 hours. SGK1 and MKP1/DUSP1 mRNA expression was normalized to β-Actin mRNA levels. Normalized SGK1 and MKP1/DUSP1 fold-change relative to vehicle treatment is shown (n=3 experiments +/− SEM). A, In MDA-MB-231 cells, dex-induced SGK1 (P=0.02) and MKP1/DUSP1 (P=0.008) mRNA expression was significantly reversed following concomitant mifepristone treatment. B, BT-20 cells showed significantly less induction of both SGK1 (P=0.004) and MKP1/DUSP1 (P=0.005) mRNA expression with the addition of mifepristone. C, In MDA-MB-468 cells, dex-induced SGK1 (P=0.001) steady-state mRNA levels were significantly higher compared with dex/mif treated cells, although the difference in MKP1/DUSP1 mRNA levels was not significant (NS).
(* P<0.05, ** P<0.01, *** P<0.001).
Mifepristone treatment potentiates paclitaxel effectiveness in an MDA-MB-231 TNBC xenograft model
In cell lines with robust mifepristone-mediated antagonism of GR-target gene expression, we hypothesized that mifepristone administered in vivo might reverse the endogenous GR activity of TNBC and increase chemotherapy sensitivity. Therefore, SCID mice bearing MDA-MB-231 GR+ TNBC mammary fat pad tumor xenografts (~200 mm3) were treated with either vehicle, paclitaxel (10 mg/kg/day), or mifepristone (15 mg/kg/day) one hour prior to paclitaxel (10 mg/kg/day) for five consecutive days (17). Tumors were then measured three times per week for 35 days (Fig. 4A) except for vehicle-treated animals, which were sacrificed once the tumor had reached approximately 3000 mm3. Analysis of the mean tumor volumes +/− SEMs for each treatment group are shown in Fig. 4A. Repeated measures analysis of variance model found a significant treatment × day2 interaction (p<0.001) indicating significant differences in the pattern of tumor growth over time. Differences at each time point in paclitaxel versus mifepristone/paclitaxel treatment groups were evaluated using post-hoc testing. Significant differences between the paclitaxel and mifepristone/paclitaxel treatment groups appeared at day 18 (P=0.02) and remained significantly different throughout the rest of the experiment. The average tumor volumes for the paclitaxel cohort were 500 mm3 (SEM = 14) on day 18 and 2200 mm3 (SEM = 13) on day 35, while the average tumor volumes for the mifepristone/paclitaxel cohort were 300 mm3 (SEM=12) on day 18 and 800 mm3 (SEM = 12) on day 35. Finally, the generalized F-test demonstrated a significant difference between mifepristone/paclitaxel- and vehicle/paclitaxel-treated tumors over time (P=0.0417). Thus, the addition of mifepristone treatment one hour prior to chemotherapy significantly improved the efficacy of paclitaxel in MDA-MB-231 xenografts.
Figure 4. Paclitaxel +/− mifepristone treatment of MDA-MB-231 tumor xenografts.

A, Mice bearing MDA-MB-231 human TNBC xenograft tumors in the thoracic mammary fat pad were treated (arrows) with vehicle (closed squares, n=5), paclitaxel (open circles, n=7) mifepristone (closed triangles, n=6) via IP injection for five consecutive days. Tumors were measured three times a week for 35 days. Daily pre-treatment with mifepristone significantly improved tumor response compared to treatment with paclitaxel alone (P=0.04). To determine the significance of treatment effects on tumor growth, repeated measures ANOVA was performed. Error bars represent +/− SEM. B, Tumors were removed at the endpoint of the experiment, fixed, paraffin-embedded and sectioned followed by fluorescent immunohistochemistry. A higher percentage of GR+ cells remained in tumors following treatment with paclitaxel alone (n=6 animals) compared to tumors from SCID mice treated with mif/paclitaxel (n=5 mice) (P=0.11). C, MDA-MB-231 cells were treated with vehicle (EtOH 0.01%), mif (100 nM), paclitaxel (100 nM), dex (100 nM)/paclitaxel (100 nM), or dex (100 nM)/mif (100 nM)/paclitaxel (100 nM) and colonies were counted after 96 hours of treatment. Error bars represent +/−SEM. *Denotes the two treatments compared to derive the p-value shown.
To explore how the addition of mifepristone treatment increases chemotherapy-induced TNBC tumor shrinkage, paraffin-embedded tumor xenograft sections were examined with a green fluorescense-labeled anti-GR antibody and stained with DAPI (representative image Supp. Fig. S5). As shown in Fig. 4B, tumors from mice (n=6) treated with paclitaxel alone had a trend toward a higher percentage of strongly GR+ residual tumor cells compared to tumors from mice (n=5) treated with mifepristone and paclitaxel (19% versus 8%, P=0.11). Software code described in Supp. Method S2. This difference in GR expression was further supported by Western blot analysis of total GR protein expression in the residual tumors (Supp. Fig. S6). These data suggest that the addition of mifepristone treatment results in improved cytotoxicity of highly GR-expressing MDA-MB-231 cells, ultimately resulting in a smaller population of high GR+ tumor cells and greater tumor shrinkage.
The effect of mifepristone in combination with glucocorticoids and paclitaxel on colony formation in vitro was further evaluated with a clonogenic assay (Fig. 4C). MDA-MB-231 cells were treated with vehicle (EtOH 0.1% V/V), dex (100nM), mifepristone (100 nM) or dex/mif (100nM) one hour before paclitaxel (100nM) in 2.5% CS-FCS. Colonies were counted 96 hours after treatment. The addition of mifepristone to dex/pac significantly decreased the number of colonies formed in comparison to dex/pac alone (P=0.0047). A representative image of each treatment at three different magnifications (15x, 24x and 38x) is reported in Supp. Fig. S7. These data suggest that mifepristone significantly reduces tumor viability in the setting of chemotherapy independently of an effect on cell proliferation.
Discussion
The limited success of targeted treatments for TNBC patients highlights the complex heterogeneity of ER-/PR-/HER2- breast cancer (22, 23). Next-generation sequencing of TNBC samples also strongly suggests that this type of breast cancer can be further classified into several molecular subtypes (24). Defining the major driver pathways dividing TNBC into various phenotypic subtypes is a critical question to be answered to improve TNBC outcome. Over a decade ago, we discovered that GR activation by physiological concentrations of glucocorticoids mediates potent anti-apoptotic signaling in the context of either growth factor deprivation- (8, 14) or chemotherapy-induced apoptosis (13) in GR+, ER-negative pre-malignant breast epithelial and TNBC cell lines. Furthermore, mifepristone, a GR antagonist was observed to reverse the cell survival effects of GR activation in c-Myc overexpressing ER-negative MCF10A cells, an in vitro model for pre-invasive TNBC (8). Here we hypothesized that increased sensitivity to chemotherapy-induced apoptosis would result from concomitant GR antagonism with mifepristone of TNBC. Indeed, we found that co-treatment with the GR-antagonist mifepristone both reverses GR-mediated gene expression in three TNBC cell lines and augments chemotherapy-induced apoptosis.
The mechanism by which GR activation inhibits apoptosis appears to require GR-mediated transcriptional induction of both SGK1 and MKP1/DUSP1 (13). SGK1 overexpression was previously found to inhibit PARP-dependent apoptosis in a variety of cell lines (8), in part through phosphorylation and inactivation of the Forkhead transcription factors (8). Here we find that GR-activation blunts paclitaxel (and growth factor deprivation)-induced cleavage of caspase-3 and PARP. The addition of mifepristone to dexamethasone reversed the reduced caspase-3 and PARP cleavage resulting from growth factor deprivation as well as paclitaxel therapy, suggesting that GR activity is an important mediator of caspase-3- and PARP-dependent apoptosis, regardless of initiating cellular insult (8). We are currently characterizing additional anti-apoptotic GR-target genes and networks modified by GR activity to better understand the transcriptional regulatory mechanisms through which glucocorticoids and mifepristone modify chemotherapy–induced cytotoxicity.
Although most cell lines used in TNBC research express GR-alpha, levels vary (14). In a cohort of over 300 patients with early stage ER-negative cancer, we reported that the highest quartile of GR-expressing primary tumors (by Affymetrix gene arrays) had a significantly worse long-term prognosis compared to patients with tumors expressing the bottom quartile of GR expression (2). In these studies, increased SGK1 and MKP1/DUSP1 tumor expression also correlated with high GR expression, suggesting that these GR-target genes as well as others are likely reflecting increased GR activity (2). Based on the results reported here, we anticipate that GR antagonism will be a particularly useful treatment for patients with GR+ tumors that are likely chemotherapy-resistant TNBC.
Recently, next-generation sequencing of over 100 primary TNBCs identified the NR3C1 (GR-encoding) gene as among the top genes undergoing somatic mutation in association with significant changes in expression of shared GR network genes (24, 25). These findings support the hypothesis that GR activity is a significant driver in the biology of a subset of TNBC.
A previous study by Pietenpol and colleagues (26) characterized somatic mutations in a panel of TNBC cell lines including the MDA-MB-231, BT-20, and MDA-MB-468 lines studied here. MDA-MB-468 cells, the least sensitive to the addition of mifepristone in our assay, were classified as “heavily enriched for mutations relating to cell cycle and cell division pathways” including mutations in both p53 and RB (26) (see Table 1). Interestingly, this cell line was most sensitive to paclitaxel monotherapy (approximately twice the percentage cell death was observed in MDA-MB-468 cells compared to either MDA-MB-231 or BT-20 cells). In contrast, MDA-MB-231 and BT-20 cells predominately harbor somatic mutations in genes encoding proteins related to cell survival and growth factor signaling (respectively PDGFRA and PIK3CA) and were the least sensitive to paclitaxel monotherapy [(25, 26) Table 1)]. This suggests that TNBCs with mutations in survival signaling pathways (rather than cell cycle pathways) could benefit the most from GR antagonism because GR antagonism blocks cell survival but has little effect on proliferation in ER- cells (Supp. Fig S1). Further clinical/translational studies could test the hypothesis that the “basal” type TNBC harboring p53 and RB mutations (both affecting cell cycle) will benefit less from GR antagonism; these tumors also tend to be more sensitive to initial chemotherapy treatment than the “mesenchymal” TNBC subtype that includes MDA-MB-231 (26).
Table 1.
Subtypes and associated somatic mutations in TNBC cell lines (26). Mutations associated with MDA-MB-231, BT-20 and MDA-MB-468 breast cancer cell lines as previously reported (26). TP53 is mutated in all three-cell lines, while mutant CDKN2A (underlined) is present in MDA-MB-231 and BT-20 cells. Only the “basal-like” MDA-MB-468 cell line had both TP53 and RB1 mutations.
| TNBC Subtype | Cell Line | Mutations |
|---|---|---|
| MDA-MB-231 | Mesenchymal-like | TP53, CDKN2A, PDGFRA, BRAF, KRAS, NF2 |
| BT-20 | Unclassified | TP53, CDKN2A, PIK3CA |
| MDA-MB-468 | Basal-like | TP53, RB1, PTEN, SMAD4 |
The residual MDA-MB-231 xenograft tumors we excised and examined approximately one month following treatment with paclitaxel +/− mifepristone suggest that residual tumor GR expression is relatively low following dual paclitaxel and mifepristone treatment compared to tumor remaining after paclitaxel monotherapy. Interestingly, not all individual MDA-MB-231 cells had the same intensity of immunofluorescent GR expression (Fig. 4B). This implies that mifepristone may preferentially target high GR-expressing MDA-MB-231 cells for chemotherapy-induced cell death. Another possibility is that mifepristone results in increased non-metabolized paclitaxel concentrations based on the ability of GR ligands (such as mifepristone and dexamethasone) to inhibit the CYP3A4 liver enzyme, thereby increasing active paclitaxel levels (27). However, we have previously measured plasma paclitaxel levels from SCID mice treated with either vehicle/paclitaxel or dex/paclitaxel and found no significant differences in active paclitaxel concentrations (9). Therefore, the lower percentage of GR-expressing cells following mifepristone treatment is more likely due to greater sensitivity of high GR-expressing cells to chemotherapy-induced apoptosis with the addition of mifepristone.
The steroid binding affinity of mouse corticosterone to human GR is 1.5-3 times lower that cortisol (28). While we observed a statistically significant difference between the pac and mif/pac treatment in vivo, the decreased binding affinity of mouse corticosterone to the human MDA-MB-231 xenografted cells could dampen the antagonistic effect of mifepristone. However, we also performed in vitro cell viability assays with dexamethasone at 100 nM, which is similar to plasma cortisol concentrations in patients (29). In these experiments, we also observed an increase in tumor cell death with the addition of mifepristone to dex/pac. Furthermore, clonogenicity of MDA-MB-231 cells was significantly decreased with dex/mif/pac compared to dex/pac. Taken together, these data support the hypothesis that mifepristone co-treatment targets high GR expressing tumor cells that would otherwise be resistant to chemotherapy alone.
Mifepristone has been studied extensively since the 1980’s for its ability to antagonize progesterone and glucocorticoid receptors in various human tissues (30). However, previous studies using mifepristone in breast cancer have only considered the use of mifepristone to target the PR in ER+/PR+ tumors, never to target the GR in TNBC. For example, several ER+/PR+ breast tumor xenograft studies have suggested a decrease in volume following treatment with mifepristone alone (subcutaneous administration, doses ranging from 25-50 mg/kg) or in combination with anti-estrogen therapy. Additionally, a Phase II clinical trial with 28 patients found that daily single agent oral mifepristone treatment (200 mg) for recurrent ER+/PR+ breast cancer resulted in three partial responses for an overall response rate of 10.7% and only mild to moderate side effects [nausea, lethargy, anorexia, and hot flashes were noted, (31)].
In our studies, an in vitro mifepristone concentration of 100 nM was used to target the GR in PR non-expressing breast tumors based on previous human pharmacokinetic studies where subjects received between 100-800 mg/day of drug. The concentration of mifepristone in the serum 24 hours after administration was found to be approximately 2.0 μM irrespective of dose (32). Additionally, chorionic villi tissue levels in women receiving a single dose of 200 mg of mifepristone found cytosolic concentrations on average of 238 nM (18). These human data suggest that 100 nM is a physiologically achievable mifepristone concentration in tumor tissue.
An ongoing Phase 1 clinical trial is evaluating the safety of mifepristone (300 mg/day) for two days followed immediately on day 2 by a weekly dose of nab-paclitaxel. In this study, the safety and tolerability of mifepristone in combination with chemotherapy for advanced ER-negative, PR-negative, HER2-negative but GR-positive breast cancer will be determined.
Supplementary Material
Acknowledgments
We thank Robert Roe MD (Corcept Therapeutics) for supplying pharmaceutical-grade mifepristone and for helpful discussions on pharmacology. We also thank Christine Labno PhD of the University of Chicago’s Integrated Microscopy Core Facility and Russell Szmulewitz MD for expert help with immunofluorescence. Finally, we thank Paul Volden and other members of the Conzen and Matthew Brady laboratories for valuable feedback and expertise.
Footnotes
Financial Support: NIH R01 CA089208, The University of Chicago Comprehensive Cancer Center NIH P30 CA014599 and Susan G. Komen for the Cure IIR12223772. Corcept Therapeutics provided pharmacological-grade mifepristone only.
Potential Conflicts of Interest: The University of Chicago and Dr. Conzen have a patent application pending that proposes to identify estrogen receptor-negative, glucocorticoid receptor-positive breast cancers as suitable for mifepristone treatment.
Translational Relevance:
Triple-negative breast cancer (TNBC) lacks effective targeted therapies. Approximately 25% of invasive TNBCs are glucocorticoid receptor (GR)-positive (>10% of tumor cells strongly GR-positive by IHC and/or significantly increased tumor NR3C1 (GR) mRNA levels compared with median TNBC NR3C1 expression). High tumor GR expression significantly correlates with earlier relapse in early-stage TNBC. Mifepristone is a potent GR and progesterone receptor (PR) modulator. Here we report that in GR+ TNBC (which lacks expression of the PR), pretreatment with mifepristone potentiates paclitaxel-induced cytotoxicity, presumably by antagonizing GR-activated TNBC cell survival pathways that otherwise contribute to chemotherapy resistance.
Reference
- 1.Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–61. doi: 10.1210/edrv-17-3-245. [DOI] [PubMed] [Google Scholar]
- 2.Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011;71:6360–70. doi: 10.1158/0008-5472.CAN-11-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armaiz-Pena GN, Lutgendorf SK, Cole SW, Sood AK. Neuroendocrine modulation of cancer progression. Brain Behav Immun. 2009;23:10–5. doi: 10.1016/j.bbi.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–74. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 5.Lewis-Tuffin LJ, Cidlowski JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1–9. doi: 10.1196/annals.1351.001. [DOI] [PubMed] [Google Scholar]
- 6.Gibson S, Tu S, Oyer R, Anderson SM, Johnson GL. Epidermal growth factor protects epithelial cells against Fas-induced apoptosis. Requirement for Akt activation. J Biol Chem. 1999;274:17612–8. doi: 10.1074/jbc.274.25.17612. [DOI] [PubMed] [Google Scholar]
- 7.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–6. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 8.Moran TJ, Gray S, Mikosz CA, Cozen SD. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Research. 2000;60:867–72. [PubMed] [Google Scholar]
- 9.Pang D, Kocherginsky M, Krausz T, Kim SY, Conzen SD. Dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumor cell apoptosis. Cancer Biol Ther. 2006;5:933–40. doi: 10.4161/cbt.5.8.2875. [DOI] [PubMed] [Google Scholar]
- 10.Brickley DR, Mikosz CA, Hagan CR, Conzen SD. Ubiquitin modification of serum and glucocorticoid-induced protein kinase-1 (SGK-1) J Biol Chem. 2002;277:43064–70. doi: 10.1074/jbc.M207604200. [DOI] [PubMed] [Google Scholar]
- 11.Huang Y, Johnson KR, Norris JS, Fan W. Nuclear factor-kappaB/IkappaB signaling pathway may contribute to the mediation of paclitaxel-induced apoptosis in solid tumor cells. Cancer Res. 2000;60:4426–32. [PubMed] [Google Scholar]
- 12.Zhang C, Wenger T, Mattern J, Ilea S, Frey C, Gutwein P, et al. Clinical and mechanistic aspects of glucocorticoid-induced chemotherapy resistance in the majority of solid tumors. Cancer Biol Ther. 2007;6:278–87. doi: 10.4161/cbt.6.2.3652. [DOI] [PubMed] [Google Scholar]
- 13.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757–64. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 14.Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276:16649–54. doi: 10.1074/jbc.M010842200. [DOI] [PubMed] [Google Scholar]
- 15.Melhem A, Yamada SD, Fleming GF, Delgado B, Brickley DR, Wu W, et al. Administration of Glucocorticoids to Ovarian Cancer Patients Is Associated with Expression of the Anti-apoptotic Genes SGK1 and MKP1/DUSP1 in Ovarian Tissues. Clin Cancer Res. 2009;15:3196–204. doi: 10.1158/1078-0432.CCR-08-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thon JN, Devine MT, Begonja AJ, Tibbitts J, Italiano JE., Jr High-content live-cell imaging assay used to establish mechanism of trastuzumab emtansine (T-DM1)-mediated inhibition of platelet production. Blood. 2012;120:1975–84. doi: 10.1182/blood-2012-04-420968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masson MJ, Collins LA, Carpenter LD, Graf ML, Ryan PM, Bourdi M, et al. Pathologic role of stressed-induced glucocorticoids in drug-induced liver injury in mice. Biochemical and Biophysical Research Communications. 2010;397:453–8. doi: 10.1016/j.bbrc.2010.05.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang JD, Shi WL, Zhang GQ, Bai XM. Tissue and serum levels of steroid-hormones and RU-486 after administration of mifepristone. Contraception. 1994;49:245–53. doi: 10.1016/0010-7824(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 19.Abraham VC, Towne DL, Waring JE, Warrior U, Burns DJ. Application of a high-content multiparameter cytotoxicity assay to prioritize compounds based on toxicity potential in humans. Journal of Biomolecular Screening. 2008;13:527–37. doi: 10.1177/1087057108318428. [DOI] [PubMed] [Google Scholar]
- 20.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–42. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Gross KL, Oakley RH, Scoltock AB, Jewell CM, Cidlowski JA. Glucocorticoid Receptor alpha Isoform-Selective Regulation of Antiapoptotic Genes in Osteosarcoma Cells: A New Mechanism for Glucocorticoid Resistance. Molecular Endocrinology. 2011;25:1087–99. doi: 10.1210/me.2010-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, et al. Iniparib plus Chemotherapy in Metastatic Triple-Negative Breast Cancer. New England Journal of Medicine. 2011;364:205–14. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 23.Arnedos M, Bihan C, Delaloge S, Andre F. Triple-negative breast cancer: are we making headway at least? Therapeutic advances in medical oncology. 2012;4:195–210. doi: 10.1177/1758834012444711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. Journal of Clinical Investigation. 2011;121:2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He K, Woolf TF, Hollenberg PF. Mechanism-based inactivation of cytochrome P-450-3A4 by mifepristone (RU486) Journal of Pharmacology and Experimental Therapeutics. 1999;288:791–7. [PubMed] [Google Scholar]
- 28.Giannopoulos G, Keichline D. Species-Related Differences in Steroid-Binding Specificity of Glucocorticoid Receptors in Lung. Endocrinology. 1981;108:1414–9. doi: 10.1210/endo-108-4-1414. [DOI] [PubMed] [Google Scholar]
- 29.Weitzman ED, Fukushim D, Nogeire C, Roffwarg H, Gallaghe Tf, Hellman L. 24 hour Pattern of Episodic Secretion of Cortisol in Normal Subjects. Journal of Clinical Endocrinology & Metabolism. 1971;40:850–5. doi: 10.1210/jcem-33-1-14. [DOI] [PubMed] [Google Scholar]
- 30.Spitz IM, Bardin CW. Drug-Therapy – Mifepristone (RU-486) – A Modulator of Progestin and Glucocorticoid Action. New England Journal of Medicine. 1993;329:404–12. doi: 10.1056/NEJM199308053290607. [DOI] [PubMed] [Google Scholar]
- 31.Perrault D, Eisenhauer EA, Pritchard KI, Panasci L, Norris B, Vandenberg T, et al. Phase II study of the progesterone antagonist mifepristone in patients with untreated metastatic breast carcinoma: a National Cancer Institute of Canada Clinical Trials Group study. J Clin Oncol. 1996;14:2709–12. doi: 10.1200/JCO.1996.14.10.2709. [DOI] [PubMed] [Google Scholar]
- 32.Heikinheimo O, Kekkonen R, Lahteenmaki P. The pharmacokinetics of mifepristone in humans reveal insights into differential mechanisms of antiprogestin action. Contraception. 2003;68:421–6. doi: 10.1016/s0010-7824(03)00077-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.