

SUMMARY
Huntington disease (HD) is a devastating illness, although its autosomal dominant genetic transmission allows a unique opportunity to study apparently healthy individuals before manifest disease. Attempts to study early disease are not unique in neurology (e.g., Mild Cognitive Impairment, Vascular Cognitive Impairment), but studying otherwise-healthy appearing individuals who will go on with nearly 99% certainty to manifest the symptoms of brain disease does provide distinct but valuable information about the true natural history of the disease. The field has witnessed an explosion of research examining possible early indicators of HD during what is now referred to as the “prodrome” of HD. A NIH study in its ninth year (PREDICT-HD) has offered a glimpse into the transition from an apparently healthy state to an obviously diseased state, and can serve as a model for many other genetic diseases, both neurological and non-neurological.
Keywords: Huntington disease, diagnosis, detection, prevention, biomarkers, clinical endpoints, clinical trials
Publication of the human genome has accentuated the desired paradigm shift in medicine from treatment of disease following diagnosis to the prediction and prevention of disease in healthy persons. Huntington disease (HD), an autosomal dominant progressive neurological disease with an identified gene mutation [1], has proven helpful to expedite preparations for preventive clinical trials in neurodegenerative diseases. This is because individuals with the gene mutation can be identified years before clinical diagnosis through genetic testing. HD is a neurodegenerative disorder with loss of medium GABAergic spiny neurons, sparing of the large cholinergic interneurons [2–3], and specific neuronal loss in layers V and VI of cerebral cortex [4]. These physiological changes lead to an insidious decline in motor, cognitive, and psychiatric functioning, a diminished quality of life, and premature death for individuals carrying the expanded polyglutamine repeat sequence [5]. Age of disease diagnosis is associated with length of the expanded gene mutation so that longer repeat lengths have a younger age of diagnosis [6–7].
This review examines the earliest signs and symptoms observed in individuals carrying the gene expansion for HD who do not meet criteria for clinical diagnosis of disease. These groups of so-called “at-risk” participants have been referred to as “asymptomatic,” “presymptomatic,” “preclinical,” “prediagnosed,” “premanifest,” “pre-HD,” or “prodromal HD.” Regardless of the label, research participants with a known expansion in the HD gene but no clinical diagnoses provide an incomparable opportunity to identify the earliest neurobiological and clinical changes in brain disease. We use the term “prodrome” to describe the phase precursory to the manifestation of full disease; the shorthand “prHD” will be used throughout this review to represent the HD prodrome.
Several clinical trials are investigating means to alleviate or reduce symptoms and slow progression in clinically diagnosed HD (http://www.hdtrials.org) [8]. Consistent with other medical conditions, treatments might be ideally initiated at or before the earliest signs of disease. There are at least two primary challenges to the design of clinical trials for prHD: (1) selection of participants who are most likely to show measurable change over the course of a clinical trial; and (2) development of outcome measures that are sensitive to interventions and can demonstrate variation over the natural history of prHD. To meet these and other challenges to preventive clinical trials, indicators of very early disease are needed. Attempts to detect HD earlier by studying individuals in the HD prodrome will be reviewed.
The PREDICT-HD Study
Neurobiological Predictors of Huntington’s Disease (“PREDICT-HD”; NS40058, PI JS Paulsen) is a multi-national, longitudinal, observational study aimed at identifying biological and refined clinical markers of prHD in humans, and then validating the optimal marker(s) and clinical endpoints for use in preventive clinical trials. Participants recruited from 32 sites across the United States, Canada, United Kingdom, Germany, Spain, and Australia undergo annual study visits consisting of a neurological motor examination, cognitive assessment, brain MRI, psychiatric and functional rating scales, and blood draws for genetic and biochemical analyses [9–10].
Currently in its ninth year of funding by the National Institutes of Health and CHDI Foundation, Inc, PREDICT-HD has enrolled more than 880 gene-expanded but not clinically diagnosed participants, as well as a smaller sample of demographically-matched, non-gene-expanded participants (n = 220) that serve as a comparison group. To date, 127 participants from the PREDICT-HD study have been prospectively clinically diagnosed. Clinical diagnosis refers to the current standard for the diagnosis of HD; when a movement disorder specialist is (a) 99% sure that “unequivocal extrapyramidal signs” are present (b) in a person with a family history of HD [58]. In addition, a proxy measure of “estimated years to HD clinical diagnosis” was developed [11], and validated [12] using gene mutation and current age. Data reported using prHD participants often use measures to estimate or “predict” eventual diagnosis using formulae similar to the one cited above. When such formulas are used they are described as “estimated” diagnosis which is distinct from actual clinical diagnosis. This paper summarizes published literature in prHD as well as findings from PREDICT-HD. The HD field has witnessed exciting research growth; the number of HD publications has increased fourfold over the past nine years. This review provides a summary intended to (1) facilitate greater interest in this promising area of research; (2) ignite other research groups to consider earlier detection of disease in healthy individuals; and (3) document challenges to future progress.
Neuroimaging indicators of prHD
Measures of structural neuroimaging in HD historically emphasized the basal ganglia and have been shown to be related to disease duration, magnitude of dementia, severity of movement disorder, cognitive performance, functional capacity [13–16] and longer CAG repeat lengths [17–18]. More recently, however, two major paradigm changes in HD research have occurred. First, since it is established that over 50% of cell death has already occurred at the time of clinical diagnosis [19] and that cognitive, sensory, and psychiatric abnormalities often precede motor symptoms in HD [9, 20–21], neuroimaging in prHD has become of primary interest. Second, with ongoing and rapid advances in technology coupled with longstanding indications of extrastriatal involvement in HD [19], more recent efforts in HD neuroimaging have involved whole brain investigation. Findings have included cerebral spinal fluid volumes [22], regional cortical degeneration [23–24], abnormal thinning of cortical sulci [25], whole brain atrophy [26–27], and significant grey [27] and white matter loss [9, 28–29], even in very early and prHD. White matter findings have been further explored in HD with the application of diffusion tensor imaging (DTI) [30–33], showing that fractional anisotropy is both increased and decreased significantly, and depending upon specific stage of early HD (See Figure 1). Importantly, DTI indicated tissue changes were apparent in prHD participants in the absence of significant white matter volume loss, suggesting that subtle morphological alterations occur before the overt death of neurons.
Figure 1. Diffusion tensor functional anisotropy group statistical maps.
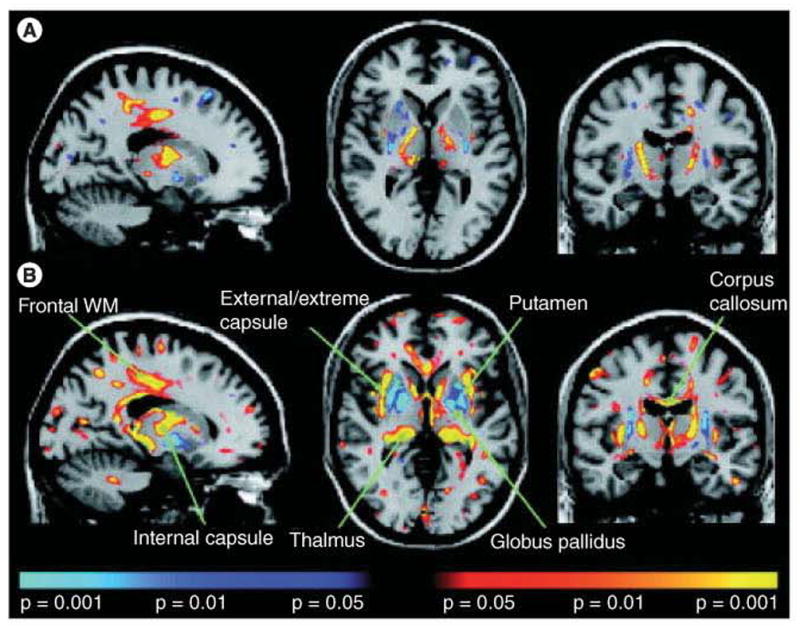
(A) Pre-Huntington’s disease. (B) Huntington’s disease. Exploratory whole-brain analyses demonstrated significant reductions in functional anisotropy in the internal capsule, frontal subcortical WM and portions of the thalamus, and increases in functional anisotropy in the putamen in the group of presymptomatic individuals known to carry the genetic mutation that causes Huntington’s disease (pre-Huntington’s disease), paralleling the results of the region of interest analysis. In the early-stage Huntington’s disease group, significant increases in the putamen and globus pallidus were observed; reductions in functional anisotropy included the internal capsule, corpus callosum, external/extreme capsule, cerebral peduncles, brainstem and WM underlying brain regions, including sensorimotor cortex, frontal, parietal and parieto–occipital areas. Blue areas show areas of statistically significant increases in functional anisotropy; yellow areas show areas of statistically significant reductions in functional anisotropy.
WM: White matter.
Reproduced with permission from [33];.
Despite efforts to consider all brain changes associated with early HD, however, the few reports that have conducted comparisons among imaging indices report the prominence of the basal ganglia. For instance, an examination of percent volume loss for each brain region demonstrated that striatal volume loss was as great as or greater than any other structure examined. An effect size analysis of each brain region validated the percent volume loss and suggested that the most robust differences between matched normal comparison participants were in the basal ganglia regions [34–36] (See Figure 2).
Figure 2. Volume differences and effect sizes based on comparisons of demographicaily matched normals and pre-Huntington’s disease less than 10 years from estimated motor diagnosis.
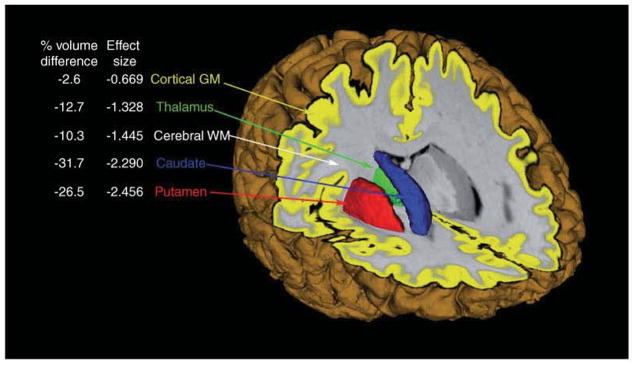
GM: Grey matter; WM: White matter.
Efforts to determine whether brain changes are somewhat independent of one another, however, have revealed that regional cortical thinning as well as white matter atrophy both provide independent contributions to aspects of disease beyond contributions made by the striatum alone. For instance, cortical surface reconstruction maps [23] (See Figure 3) show that regional cortical thinning in prHD remains when adjusting for caudate and putamen volumes. Similarly, a multiple regression on estimated motor diagnosis suggested that white matter volume loss made a significant contribution to predicted years-to-clinical diagnosis above and beyond contributions made by the striatal volumes alone [34–36].
Figure 3. Evidence of regional cortical thinning in pre-Huntington’s disease and its relationship to striatal volume. Cortical surface reconstruction maps.

Significant cortical thinning was present in the pre-Huntington’s disease group compared with controls. When adjusting for caudate volumes, the intergroup variance in thinning was less prominent over portions of superior temporal gyrus; when adjusting for putamen volumes, variance was less prominent over posterior frontal regions. Some areas of thinning appeared to be independent of caudate or putamen volumes. Maps are displayed on an average composite brain, with areas of more thinning transitioning from red (p < 0.05) to yellow (p < 0.001), unadjusted for multiple comparisons.
L: Left; R; Right.
Reproduced with permission from [25].
Since structural imaging measures appear to be biomarker candidates for prHD clinical trials, consideration of marker purpose is essential. Rapid screening tools for clinical trial inclusion will demand imaging measures that are feasible in terms of time, cost and burden. Recently, some research groups have made efforts to produce and validate imaging protocols that meet these criteria. Progress has included voxel-based morphometry [37], multivariate support vector machines [38], and automated artificial neuronal network segmentations [10,25,28, 34–36,]. Although fewer of these measures have undergone longitudinal study, there is some evidence of adequate and reliable change indices [43–44]. For example, Aylward and colleagues [35] presented data from jnearly 200 prHD individuals and 60 controls, scanned both at baseline and 2-year follow-up. PrHD participants were divided into groups based on proximity to estimated clinical diagnosis: Far (> 15 years from estimated diagnosis); Mid (9–15 years); and Near (< 9 years). All prHD groups showed a faster rate of atrophy than Controls in striatum and cerebral white matter although annual percentage change was greater in striatum than cerebral white matter. However, when normal age-related atrophy (i.e., change observed in the Control group) was taken into account, there appeared to be more disease-related atrophy in white matter than in striatum. The authors concluded that measures of volume change in both the striatum and white matter may serve as good outcome measures for future clinical trials in prHD. These findings emphasize the importance of normal comparison participants in the interpretation of prHD findings and in the design of preventive clinical trials.
In most brain disorders, there is accumulating evidence that the clinical signs of disease do not simply depend on the extent of tissue destruction, but rather represent a complex balance among neuronal dysfunction, tissue repair, and circuitry reorganization. It is generally accepted that neurons endure a period of dysfunction prior to death. Structural imaging characterizes brain volume and cell loss, whereas functional imaging portrays brain performance and cell dysfunction [45]. Based upon this distinction, functional neuroimaging modalities may be more sensitive to the earliest changes in HD than are structural imaging approaches. Functional imaging findings in prHD have proven to be somewhat consistent, demonstrating alterations even when minimal or no volume loss is evident.
Several Positron Emission Tomography (PET) measures in prHD are abnormal prior to motor diagnosis, including D1 and D2 receptor binding, peripheral benzodiazepine binding using PK, as well as glucose metabolism [46–49]. Longitudinal PET studies have suggested annual changes in prHD ranging from 2.3% to 10.9% per year [29, 49–51]. Given the relatively large longitudinal effect sizes suggested by these data, PET measures may prove cost-effective as an outcome for preventive clinical trials. Further research is essential to determine whether PET might offer greater sensitivity, larger effects, and consequential smaller sample sizes for clinical trial design. Additionally, magnetic resonance spectroscopy (MRS) has advantages over PET in availability and cost, but has reported conflicting results [39, 52–53], suggesting that further research elucidating inconsistent findings and assurances of reliability is needed.
Functional magnetic resonance imaging (fMRI) has been used the most frequently as a tool to understand and document early changes in brain function associated with prHD. Although abnormalities are consistently detected, patterns and interpretations are mixed. A more complete review of functional imaging in HD has been produced elsewhere [54]. Briefly, consistent differences in activation patterns and lateralization effects are found between HD, prHD, and NC, which vary dependent on the disease stage. One pattern is characterized by hyperactivation in prHD further from diagnosis followed by hypoactivation in prHD closer to diagnosis [40–42]. Although activation circuits vary depending on the cognitive paradigm used, this general finding has been replicated across studies. Reduced activation in prHD has been described by more studies [55–56], despite variation in tasks. These fMRI activation patterns that emerge across studies are well depicted by Zimbleman and colleagues [57] who show all three patterns: a) equivalent activations between normal comparisons and prHD estimated to be far from diagnosis with diminished activations in prHD closest to motor diagnosis; b) stepwise decreases in activations as diagnosis approaches; and c) hyperactivations in prHD far from diagnosis with hypoactivations in prHD closer to diagnosis (See Figure 4).
Figure 4. Magnetic resonance imaging signal changes.
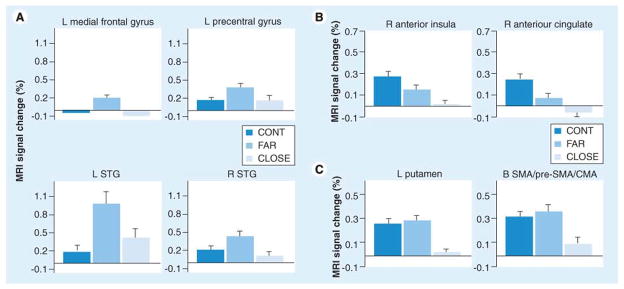
(A) Percentage MRI signal change for each region of interest in which the far group showed significantly greater activation relative to the close and control groups (i.e., far > cont = close). (B) Percentage MRI signal change for each region of interest in which a step-wise reduction in activation was observed between the three groups (i.e. cont > far > close). (C) Percentage MRI signal change for each ROI in which the close group showed significantly lower activation relative to the far and control groups (i.e., cont = far > close).
Error bars = standard error of the mean.
CMA: Cingulate motor area; Cont: Control; L: Left; MRI: Magnetic resonance imaging; R: Right; SMA: Supplementary motor area; STG: Superior temporal gyri.
Reproduced with permission from [58].
Although careful comparison and further study are needed to disentangle fMRI in prHD, the factors to consider are clear and will be mentioned here to encourage careful cross-study comparisons of future research. First, estimations of proximity to motor diagnosis have recently been validated [12] and a consistent utilization of the validated formula would improve cross-study comparisons of samples. The diagnosis estimation formula requires that CAG repeat length and current age are considered in the estimate. Second, cognitive assessment performances are essential to determine whether cognitive impairment is evident, since some studies found that prHD with apparently intact cognitive performances may be using compensatory strategies to achieve normal performances, and may be more likely to demonstrate hyperactivations in other brain areas with or without hypoactivations in the caudate. Third, structural analysis of volume loss is key to ascertain whether hypoactivations reflect decreased volume. Stratifying groups based on striatal volumes assists with staging of prHD, because groups shown to have normal striatum volumes and normal cognitive performances are less likely to show striatal hypoactivation. Finally, consistent use of clinical rating scales [58], will allow all participants to be assessed on the same metric of clinical motor manifestation and diagnosis.
This brief summary provides ample support for the utility of imaging in the detection and tracking of disease in prHD. Future research in this area will be most beneficial when addessing one of three questions. First, does the imaging measure being studied improve upon current available measures? The measure should be compared with regard to reliability, cost, and effect size. That is, a new measure that has greater consistency, less cost and larger effect sizes for prHD change over time might be a better outcome measure than striatal volume. Second, does the imaging measure add non-redundant information? If a new imaging measure were not highly associated with striatal volume and represented an additional component of the disease process (as is suggested by white matter), the measure may suggest new directions for understanding mechanism, treatment development, or further reducing sample size by increasing power. Third, imaging measures have not undergone adequate validity studies to determine whether they remain equally effective outcome measures at varying stages of disease [22]. For example, clinical trials designed to follow participants long past clinical diagnosis are likely to require measures in addition to striatal atrophy since the utility of this measure diminishes with the size of the caudate in diagnosed patients. It is not yet known how late in the HD stages this imaging measure remains valid. The studies described in this summary will make significant contributions to the design of preventive clinical trials for prHD.
Motor signs
The motor signs and symptoms of HD have received the most attention in both clinical care and early research. Many research studies have examined the utility of the neurological examination for the early measurement of HD in at-risk persons. Two studies with sufficient sample sizes report that subtle changes in motor function are present in HD gene carriers who do not exhibit sufficient motor signs to make a clinical diagnosis of HD [59–60]. More specifically, the most sensitive signs included minimal chorea of the extremities and slowing of oculomotor functions. In the largest prHD sample studied to date, Biglan and his colleagues [61] examined 926 participants at-risk for HD (733 cases and 196 controls) and found that elevated total motor scores at baseline were associated with estimated clinical diagnosis (partial R2 0.14, p<0.0001) and smaller striatal volumes (partial R2 0.15, p<0.0001). Nearly all motor domain scores showed greater abnormality with increasing proximity to estimated clinical diagnosis, although bradykinesia and chorea were most highly associated with diagnostic immediacy. Among individual motor items, worse scores on finger tapping, tandem gait, the 3-step Luria, saccade initiation, and chorea show unique association with diagnosis probability.
Although longitudinal studies of motor progression in the prHD epoch are few, findings are mixed. Solomon and her colleagues [62] show accelerating motor declines as diagnosis approaches, whereas Witjes-Ane and her colleagues [63] suggest that no measures show decline over time and that more sensitive assessment tools are needed.
Although the motor exam may be the most widely used method of assessing motor signs in HD, supplementary methods have been explored such as video motion analysis [64] and quantitative eye and motor measures [65]. Using quantitative motor assessments (i.e., force transducer, GAITrite, and saccadometer) Tabrizi [27] found greater antisaccade error rates and greater tongue protrusion variability in diagnosed HD and in prHD with more disease burden [66]. Georgiou-Karistiantis and her colleagues used motor tasks that involve variations in cognitive load and found that difficulty inhibiting automatic responses, movement times, and reaction times for cognitive tasks are associated with estimated diagnosis [67]. Golding and colleagues [68] suggest that saccadic slowing and delayed reflexive saccades are evident only in diagnosed HD, whereas prHD show slowing in voluntary-guided saccades, associated with estimated diagnosis. Blekher and colleagues [69] compared a video-based eye tracking system with clinical ratings and reported that anti-saccades and memory-guided eye movements were more sensitive and may provide a more objective and sensitive method to quantify early HD. Antoniades and colleagues [70] provide a theoretical explanation for saccade measurements in prHD, suggesting that very early eye movements might reflect an impairment of the tonic suppression of the colliculus normally mediated by pathways through the basal ganglia. Finally, Rao and colleagues [71] used a computerized walkway that recorded spatiotemporal variables and reported that prHD volunteers demonstrated reduced gait velocity, decreased stride length and greater variability in step time that were associated with estimated diagnosis. Despite many efforts to utilize improved technology to better assess early motor signs of HD, findings have been mixed and future studies will require careful comparison of published and new findings to better understand how quantified motor abnormalities might better detect prHD.
Since the clinical diagnosis of HD is based upon motor abnormalities, it is no surprise that subtle motor signs are predictive of eventual clinical diagnosis. What is not clear, however, is whether improved motor measures might decrease the variability observed in clinical ratings and diagnoses. Decreased variance is readily translated into increased power for clinical trials. On this basis alone the importance of improved reliability of the motor exam and the development of improved motor measures are paramount for improved clinical trials in all movement disorders. A comprehensive review of early motor signs in prHD is needed with clear guidelines on how to standardize the motor examination for prHD. Future research exploring quantitative methods of motor assessment (such as GAITrite and saccodometry) could then be directly compared with clinical ratings to determine whether the increased sensitivity attained is worth the extra cost required by the inclusion of physiological instrumentation into clinical trials.
Cognitive impairments
It is now well-accepted that prHD participants, in comparison to controls, show poorer performance in measures of attention, processing speed, psychomotor functions, episodic learning and memory, emotion processing, sensory perceptual functions and executive function [20, 62, 72–83]. One report with a very large sample size of prHD participants suggests that cognitive decline can be detected up to 15 years prior to traditional motor diagnosis [9–10]. The papers are too numerous for a comprehensive review, however, since nearly 150 empirical reports of cognitive function in prHD have been published in the past 15 years.
Although reports cite comprehensive cognitive batteries being administered to detect early cognitive decline in prHD [84–88], the field is well poised to move toward greater specificity in terms of the types of tasks that are sensitive in prHD prior to motor disturbances. We report some specific findings here that are based on over 1,000 participants from the PREDICT-HD study. Findings are based on three stratified groups: a far from clinical diagnosis group, estimated to be more than 15 years from diagnosis; a middle group, estimated to between 9 and 15 years from diagnosis; and a near diagnosis group, estimated to be less than 9 years from diagnosis. We calculated effect sizes for 22 individual cognitive measures in comparison to demographically matched research participants who were at-risk but determined to have normal CAG repeat lengths after undergoing predictive testing. We then chose the most sensitive three tasks in each conceptual cognitive domain and averaged their effect sizes. The orange bars in Figure 5 depict the effect sizes based upon prHD participants near motor diagnosis and demographically matched comparison participants.
Figure 5.
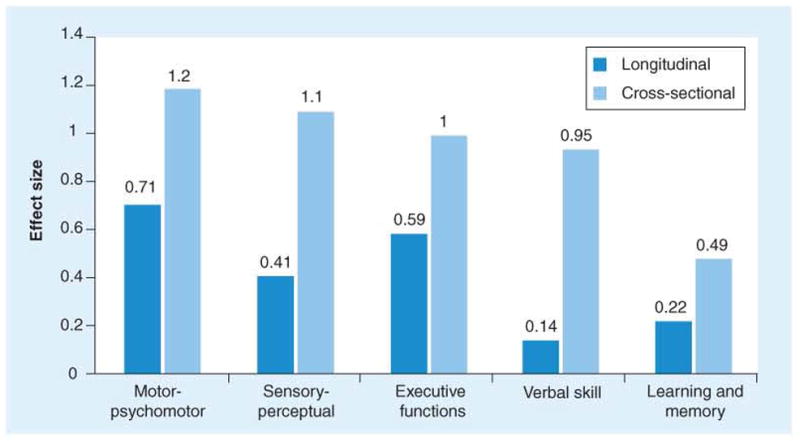
Cognitive domain effect sizes for pre-Huntington’s disease near estimated clinical diagnosis.
Next, we examined the longitudinal data from PREDICT-HD. Although most of the cognitive tasks are only repeated every two years, a few are repeated annually. Again, performance changes in prHD near diagnosis were compared with changes in the normal comparison group and effect sizes for longitudinal data are shown in the blue bars from Figure 5. Figure 6 shows the association between estimated years to motor diagnosis (depicted along the x-axis) and several cognitive measures. For example, finger tapping, timed tap generation, and word list learning all show deterioration in performance at least 15 years before the expected motor diagnosis. As shown in Figure 6, nearly all markers studied appear to follow a similar pattern of decline. Such an acceleration of decline in cognitive and motor measures has been replicated in a separate sample [62]. Rupp and colleagues [89] suggest that only some cognitive measures show acceleration of decline whereas others show a constant rate of decline throughout the prodromal period. Further longitudinal study is critical to the determination of the best cognitive measures for clinical trials in prHD.
Figure 6. Relationship between estimated years to diagnosis in participants with the prodrome for Huntington’s disease and various other measures.
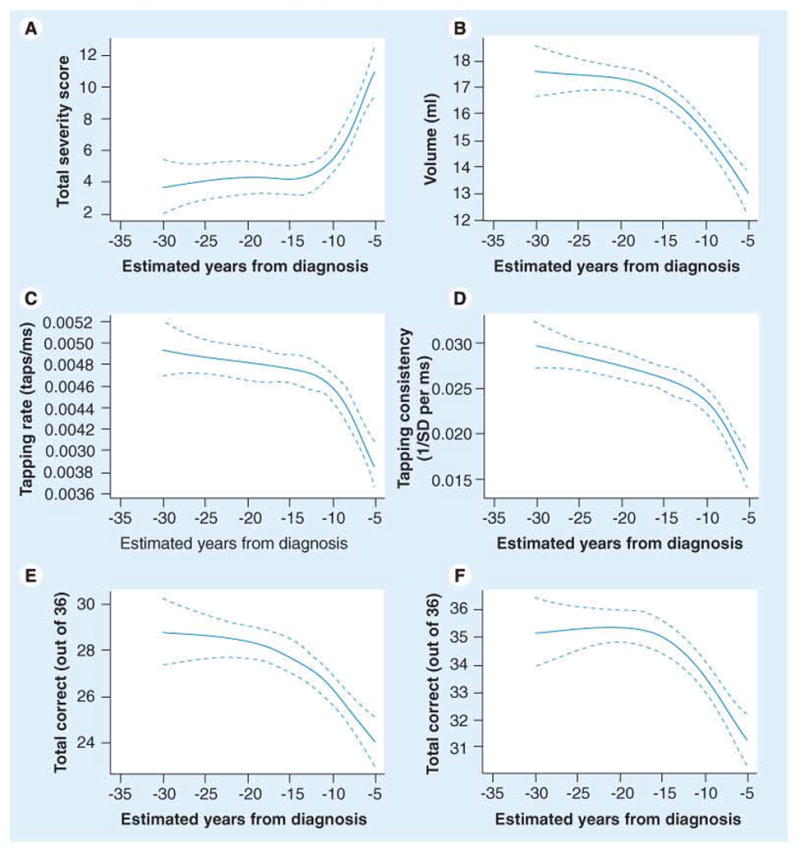
(A) Motor exam score. (B) Striatal volume. (C) Speeded finger tapping. (D) Self-timed finger tapping. (E) Word list learning. (F) Odor identification. Solid line plots the mean; broken lines are 95% confidence limits. All relationships are adjusted for gender, age and education.
SD: Standard deviation.
Reproduced with permission from [10].
Using conventional criteria for mild cognitive impairment (MCI), it has been suggested that at least 38% of prHD show impairment on demographically standardized assessment [90]. Although the utility of MCI has been demonstrated for Alzheimer’s disease [91], the efficacy of the construct for other forms of neurodegenerative diseases is unknown. Recent efforts have begun, however, to determine whether persons with mild cognitive impairment can benefit from clinical care by health care professionals. It is not known whether MCI in persons with eventual HD or other neurodegenerative disorders [92] will show increased probability of more rapid diagnosis of brain disease than their counterparts. Prospective longitudinal study will be critical to determine whether MCI might be a useful concept in the early detection of HD as it has proven utilitarian in Alzheimer’s disease.
These findings reflect the wealth of published studies now showing that cognitive tests are an early and sensitive measure of disease in prHD. In addition, findings suggest that there are several cognitive tests that may be useful for the early detection of disease as well as cognitive tests that may be markers of decline over time. Further research is needed to delineate which specific cognitive domains and which distinct tests will be most useful in selecting participants and providing clinical endpoints for clinical trials in prHD.
Although there is no question that cognitive performance declines during prHD, much remains to be done prior to making a recommended cognitive battery for prHD clinical trials. Much can be learned from our colleagues who have developed collaborative batteries for other brain disorders. For example, the MATRICS Consensus Cognitive Battery (http://www.matricsinc.org/MCCB.htm) [143] was developed with government, industry, and academia collaboration over a period of 4 years and included well-controlled studies of validation. Future research for clinical trials in prHD and HD require that several cognitive studies be conducted and shared among the HD research community. First, cognitive candidates need to undergo rigorous psychometric evaluation to document the reliability, validity and repeatability of tasks considered. For example, it is possible that a task is chosen because it shows a very large effect size although upon more careful consideration the task also shows very large error variance and poor test-retest reliability. Second, cognitive candidates are likely to vary in efficacy dependent upon disease stage. For instance, a task could be chosen due to large change scores over time that disappear when the same task is administered to a prHD participant at an earlier epoch of disease or a diagnosed HD patient. Third, cognitive measures vary greatly in terms of feasibility for clinical trials. The size, portability, cost and stability of computer platforms, the need for peripheral hardware and software, the dependence upon technical stability and lack of variation (not possible in this age of computer advances), and the availability of normative standards (HD has greater normative variability than any other neurodegenerative disorder) all impact choice of cognitive tests. In addition, frustration tolerance, motivational factors, and time constraints will impact the tasks chosen. Finally, in an era where nearly any cognitive measure can show impairment in a well-characterized prHD sample, it is critical that we minimize redundant publication of “just one more test that shows impairment.” We do not need even one more task unless we work closely together to build upon what is known. There are at least two questions that should be addressed in new publications: (a) Does the new cognitive measure being studied improve upon current available measures? The measure should be compared with regard to reliability, cost, and effect size. That is, a new measure that has greater consistency, less cost and larger effect sizes for prHD change over time might be a better outcome measure than Digit Symbol. (b) Does the new cognitive measure add non-redundant information? If a new measure were not highly associated with Digit Symbol and represented an additional component of the disease process it may prove worth pursuing. Further research in the cogntive phenotype of prHD is needed to design clinical trials.
Psychiatric Aspects
Despite variations in prevalence rates for psychiatric disorders in HD [93], there is general agreement that neuropsychiatric symptoms constitute a distressing aspect of HD and often constitute reason for hospitalization [94]. In addition, psychiatric symptoms in diagnosed HD have been reported to have a more robust association with functional outcomes than do motor and cognitive impairments [95–96]. Although reports have suggested that psychiatric symptoms occur as many as 20 years prior to motor diagnosis [97–98], research in prHD has been limited. The few studies in prHD that exist show that irritability [99–100], obsessive checking and pathological impulses [101], disinhibition, apathy and depression [73, 106], and emotional recognition [103] are all significantly impaired in prHD compared with controls. Findings have not been unequivocal, however, as some studies found no difference between gene-positive and gene-negative at-risk participants [104–105]. The lack of consistency in findings could be due to methodological limitations such as small sample sizes, the use of assessment instruments that lack the sensitivity to detect subtle psychiatric changes (categorical versus dimensional measures), measuring psychological functioning in close temporal proximity to genetic testing, unavailable or inadequate information about estimated time to clinical diagnosis of HD, and lacking collateral information about the participants’ psychological functioning. In a study of the largest prHD sample size to date (n = 681), nearly all psychiatric symptoms (e.g., depression, anxiety, obsessive-compulsiveness) were significantly greater in prHD than in normal comparison samples and psychiatric comorbidity increased with proximity to estimated diagnosis [73]. More current is a report of 908 prHD [106], who completed the Frontal Systems Behavioral Scale [107]. Findings showed that prHD had higher scores on all three subscale measures (apathy, disinhibition and executive function) than the non-expanded comparison group and ratings remained significantly higher whether they were assessed via self-report or by companions. Interestingly, ratings between the prHD participant and a companion showed poor agreement and the discrepancy between ratings was associated with time to estimated clinical diagnosis (See Figure 7). The authors have interpreted the discrepancy in behavior ratings as a possible assessment of impaired insight. Although research in clinically diagnosed HD has shown impaired awareness of cognitive, emotional and functional abilities [108], no known study has documented these insight problems in prHD. As prHD participants approach clinical diagnosis, impairments in insight could interfere with accurate reporting of psychiatric symptoms [109], so companion reports and other objective measures might be better indicators of psychiatric distress in prHD.
Figure 7. Difference score between pre-Huntington’s disease participants and companions on ratings of so-called ‘frontal lobe behaviors’.
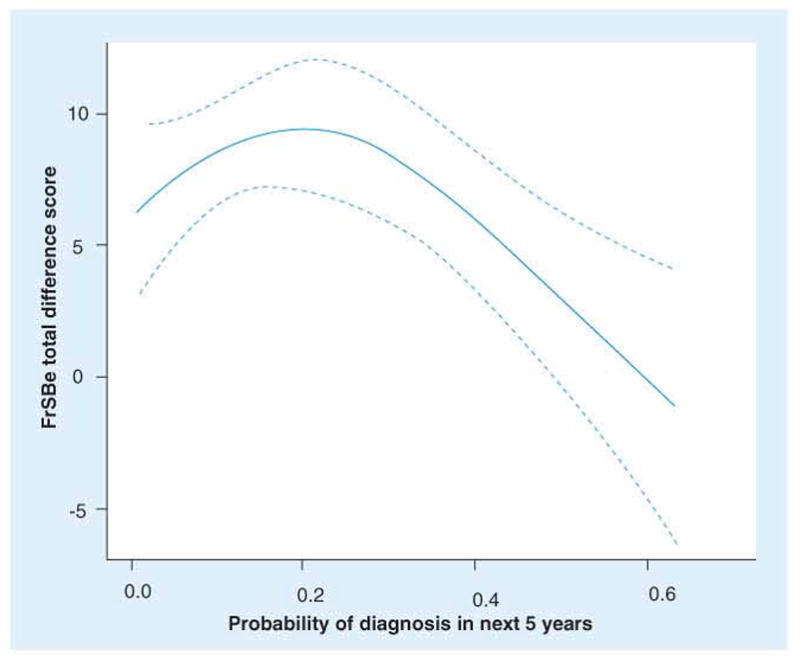
Solid line represents mean and dotted line represents 95% confidence interval. Highly significant association between agreement of manifest frontal behaviors and proximity to manifest motor diagnosis. Adjusted for age (40.65 years), gender (male or female) and education (14 years).
FrSBe: Frontal Systems Behaviour Scale.
Reproduced with permission from [100].
In one of the few longitudinal studies of psychiatric symptoms in prHD, Langbehn and colleagues (2007) showed that psychiatric symptoms were predictive of prospective clinical diagnosis in a cohort of at-risk participants who were not gene tested [110].
Several empirical reports have recently emerged supporting this notion that psychiatric symptoms and even psychiatric disorders precede the onset of motor and cognitive impairments in HD [73, 101, 111]. As suggested by Rosenblatt [112], however, improved quantitative research is needed to refine the psychiatric syndrome of HD. For instance, recent results suggest that obsessive compulsive symptoms in prHD emphasize worrying and checking and rarely reach the levels of severity associated with diagnoses of obsessive compulsive disorders [101] until later in HD following clinical diagnosis [113]. Similarly, an analysis of the depression phenotype in prHD suggests that cognitive difficulties (poor concentration), suicidal thoughts, and low energy characterize this syndrome [21, 102, 115]. Although the range of scores on traditional depression assessment seen in prHD was often as broad as that seen in Major Depressive Disorder (MDD), the profile of these scores was poorly related to overall severity of depressive symptoms (suggesting poor discriminative properties) and appeared distinct from that seen in MDD [117].
Much work remains to be done to characterize the psychiatric phenotype of prHD. Biological psychiatry needs to be embraced to help understand how mood and anxiety manifest in prHD. Reliance on prototypical mental disorders has proven limited to characterize the presentation of psychiatric disturbance in prHD as well as manifest HD. There are at least three research areas requiring clarification. First, imaging and physiology measures would be helpful to better document the pathophysiology of psychiatric disturbance in prHD. Efforts such as those used by Kloppel [116] and Paradiso [118] using functional imaging have offered insight into brain mechanisms of psychiatric symptoms in HD. Second, self-report scales are likely limited by impaired insight, at least in a sizable proportion of prHD. Further study is needed to document insight and/or awareness in both HD and prHD and how clinical rating scales might be developed to allow valid and reliable assessments of psychiatric symptoms without sole reliance on self-report instrumentation. Third, items used from research on other neuropsychiatric disorders may require validation in prHD cohorts. It has been suggested that item performance can vary significantly depnding upon the cohort sampled. Preliminary findings using Item Response Theory (IRT) show that performances of prHD participants are not consisent with those obtained in other psychiatric disorders and will require editing and redeployment [119].
Clinical Endpoints for Clinical Trials
Although often overlooked, the identification of a clinical endpoint is among the most important components of clinical research. According to the FDA, clinical endpoints are the most credible characteristics used to interpret the results of randomized clinical trials and reflect how a patient feels, functions, or survives [120–121]. A major challenge for the neurodegenerative disorders has been the identification of clinical outcome measures that accurately track disease. Over the past few years, research groups have critically reconsidered clinical outcome measures for Multiple Sclerosis [122], Alzheimer disease [91, 123], Amyotrophic Lateral Sclerosis [124], and Parkinson disease [125–126]. Primary outcome measures used in clinical trials of patients with symptomatic HD typically involve a clinical symptom or sign (usually chorea) and a measure of everyday function. One of the most frequently used measures of function in HD is the Total Functional Capacity scale (TFC; [127]. The Total Functional Capacity (TFC; [127] measure is a 5-item clinician rating scale typically completed after a brief interview. The TFC is a broad measure of functional capacity that globally assesses occupation, finances, domestic chores, activities of daily living, and level of care, with scores on each item ranging from 0 to either 2 or 3 (e.g., “Occupation: 0=unable, 1=marginal work only, 2=reduced capacity for usual job, 3=normal). TFC total scores range from 0 to 13 with greater scores indicating higher functioning. Eighty-nine percent of 786 prHD participants from the PREDICT-HD study were rated as having the maximum possible score on the TFC whereas less than eight percent of the participants lost one point, 2% lost 2 points, and 1% lost only 3 points on the TFC. In other words, over 99% of the prHD sample scored over 10/13 on the TFC. Based on these findings, it is likely that outcome measures for an earlier disease epoch may need to be revised to include items of higher functional capacity. For instance, the item typically lost first for participants who later received a clinical diagnosis of HD was the item asking about accustomed employment. It is possible that work functioning measures may provide a better outcome measure for early clinical trials in HD. In addition, more sensitive measures of interpersonal relations may provide appropriate clinical outcomes for early HD. Duff and his colleagues [106] found that personality factors such as apathy, executive control, inhibition, and social cognition were significantly impaired in prHD. To our knowledge, few studies have examined interpersonal relations as early outcome measures for degenerative diseases.
The need for better clinical outcome measures has reached prominent and widespread importance. In 2004, a group in scientists from seven institutions worked in partnership with the National Institutes of Health (NIH) to form a network funded under the NIH Roadmap for Medical Research Initiative to re-engineer the clinical research enterprise. This initiative was called the Patient-Reported Outcomes Measurement Information System (PROMIS; http://www.nihroadmap.nih.gov) and aims to revolutionize the way patient-reported outcome tools are selected and employed in clinical research and practice evaluation. Among the primary objectives of this initiatve is to establish a national resource for accurate and efficient measuresment of health outcomes for clinical practice. PROMIS aims to develop ways to measure patient-reported symptoms (such as pain and fatigue), functional capacity, and aspects of health-related quality of life across a wide variety of chronic diseases and conditions.
Clinical trial considerations in prHD
Clinical diagnosis as endpoint adjusting for baseline risk factors
Given CAG length and a person’s current age, probabilities of clinical diagnosis within an expected time are available [128] and occur throughout the literature in prHD. Given that prospectively diagnosed prHD are available (n=81 clinically diagnosed from n=610 prHD participants evaluated for up to 5 years in PREDICT-HD) it was tested whether biological and/or refined clinical measures would improve the predictability of CAG-age based prognostic models. Using backward stepwise selection procedures, a selection of refined clinial variables were added to CAG-age based prognosis and neurological exam score predictions of clinical diagnosis. Initially, 24 measures were screened and 14 competed for inclusion in the final model. Model results are listed in Table 1. The log-logistic model naturally yields results in terms of odds ratios of diagnosis. These ratios are similar to hazard ratios (more familiar to many readers) in a way analogous to odds ratios versus relative risk in studies with a single follow-up. As expected, results from the CAG-age formula significantly predict the clinical diagnosis of HD in our 81 converters. Ratings from the motor exam and four of the cognitive tests in PREDICT also significantly and independently add to the prediction of HD diagnosis. Although the available sample size from MRIs was smaller, striatal volume (putamen plus caudate) was the strongest MRI predictor, and addition of this variable substantially diminished the independent significance of CAG-age based prognosis, the Stroop and Tower 3 tests, and speeded (but not tone-paced) tapping. Still controlling for motor exam and tone-paced tapping, striatum was a significant predictor (p = .006) with increased odds ratio of clinical diagnosis estimated at 1.55 per cubic cm (2.54 per std. dev. of striatal volume). The total predictive accuracy of this model was nearly identical to the model in Table 1. Provisionally, this suggests that similar prognostic accuracy may be obtainable from volumetric MRI measurement or a combination of genetic information and more detailed clinical examination. However, a firm conclusion should await completion of futher data collection and increased sample sizes.
Table 1.
Multivariate Predictive Model of Diagnosis.
| Predictor | Measurement | Odds Ratio | P |
|---|---|---|---|
| CAG-age | Estimated Clinical Diagnosis | 2.59 | 0.0003 |
| Motor Exam | UHDRS Total Motor Score | 1.93 | 0.0006 |
| Tone-paced tapping | 1/Std. Deviation of tap interval | 1.90 | 0.0062 |
| Speeded tapping rate | Mean tap interval | 1.48 | 0.0116 |
| Tower 3 | Average moves per trial | 1.30 | 0.0512 |
| Stroop interference | Total score | 1.56 | 0.0236 |
Notes: Odds ratios are per standard deviation of each measure among gene-expanded PREDICT-HD subjects. Random frailty effect for raters was significant at p < .0001. Control for age and gender had no substantial effect on the above estimates.
The reduction in variance for cumulative diagnostic probabilities over various lengths of time can be calculated by comparing mean participant-specific (Bernoulli) variances within these nested prognostic models. The variance reductions shown in Table 2 are of substantial practical importance, as they are first-order approximations of the proportional reduction in sample size achievable in a Phase III clinical trial on a similar prHD cohort if the efficacy analysis adjusts for these baseline risk factors. We have confirmed this via computer modeling of 2 and 3-year trials incorporating hypothetical treatments with relative diagnosis risks of 0.6 to 0.8; relative efficacy assumed independent of prognostic markers. Similar prognostic estimations can be calculated using the disease burden formula suggested by Penney and colleagues [66] as used in the TRACK study [27].
Table 2.
Proportion “Variance Explained” in Diagnostic Probability by Prognostic Models
| Yrs Follow Up | CAG and age | Full prognostic model (Table 1) |
|---|---|---|
| 1 | .108 | .245 |
| 2 | .138 | .323 |
| 3 | .210 | .401 |
| 4 | .269 | .441 |
| 5 | .308 | .455 |
Statistics are adjusted R-square, N =522.
Clinical diagnosis as endpoint using enriched samples
Even more dramatic sample size benefits can be shown if participants are limited to only those above median risk in the full prognostic model (See Table 1). Allowing for model-construction bias, we estimate that approximately 95% of all participants diagnosed within 3 years would have higher than median baseline risk and be included in such a study. Computer modeling suggests that sample size is reducible by 50–60% by the use of such screening. If the baseline estimates are further used for statistical adjustment among those participating (similar to Table 2), sample size reductions of 60–70% appear feasible. The above clinical trial scenarios assume that prevention of clinical diagnosis is the efficacy measure and that intervention at this point in HD development can have an impact. Under these narrow assumptions, our results underscore the fact that we can identify those at such low risk for imminent diagnosis that their inclusion in a trial can contribute very little. Balanced against risk, their participation may not only be uneconomical but also unethical.
Put another way, findings suggest that performance on a simple task such as tapping can significantly improve the prediction of a participant receiving a clinical diagnosis of HD within 5 years. Moreover, these findings demonstrate that our various markers are not redundant. For instance, the tapping task continues to improve prediction even when CAG repeat length, MRI putamen volumes, and the neurologist’s motor rating are already considered. In summary, this preliminary analysis of incident clinical diagnosis of HD suggests several robust markers that can double the predictive power gained from CAG repeat length alone. Sample sizes needed for clinical trials can be reduced up to 70% with various combinations of the PREDICT-HD baseline assessment.
Biological and refined clinical markers as outcomes
Since no endpoints currently exist for a preventive clinical trial in HD, it is encouraging that some of our measures may demonstrate significant longitudinal change over a relatively brief interval. Preventive trials reliant upon eventual clinical diagnosis are costly and time-consuming as larger numbers of participants are needed to enable a comparison of conversions between the treatment and placebo groups. Data from the PREDICT-HD cognitive battery and the MRI have shown that several measures of each show change easily detected within two years [35, 81]. For instance, using a measure of total striatum volume repeated over two years, findings show that participants who are nearer to estimated clinical diagnosis have an effect size of −0.88 when compared with at-risk participants without the CAG expansion. This large effect sizes translates into the sample size needed to use MRI striatal volume as an outcome measure in a two-year clinical trial. To detect a 50% therapeutic effect of a compound, 108 participants per treatment arm would be needed for a two-year clinical trial, assuming a two-sided alpha level of 0.05 and 90% power. One-year longitudinal change indices will be available soon from the multinational TRACK study as well [27].
Outstanding needs prior to preventive trials in prHD
There are several issues that require resolution prior to the launch of a clinical trial for prHD. First, increased sample sizes obtained from PREDICT-HD for prospective diagnoses will be essential to document the association of predictive variables with actual prospective diagnosis. This will test fact, concurrent, and predictive validity for the emergence of refined clinical and new biomakers. Second, the data acquired from the TRACK study for one-year change rates is important to help determine whether change can be detected in one versus two years. It is always a challenge to design a trial long enough to allow robust change to be documented and short enough to expedite clinical trial findings. Third, partnerships between clinical researchers and HD families are improving too slowly for the expeditious implementation of clinical trials. Current HD clinical trials are slow to enroll causing costs to grow at an alarming pace. As treatments are readied for study in humans, it is vital for the participant pool to rapidly volunteer and enroll. Despite efforts by lay and professional organizations, little improvement has occurred in this rate-limiting obstacle for clinical trials.
When does HD begin?
If one were to ask the question, “When does the disease begin?” the correct answer today would be that we don’t know. We haven’t yet examined people with a CAG-expansion early enough in their lives to find a point when they appear to have no indication of HD. To date, we have found clinical and biological indicators of disease even in groups who are estimated to be more than 15 years from estimated clinical diagnosis [10, 25, 27, 40, 49, 81, 129–130].
In light of the extensive findings demonstrating earlier detection of this brain disease years before its traditional diagnosis and the availability of an unequivocal genetic marker in the form of an expanded CAG tract, we must ask ourselves “Why don’t we diagnose HD at an earlier time?” The formal diagnosis of HD is a controversial issue with important political, ethical, legal, social, medical, and research implications. The identification of prHD individuals with varying degrees of risk to receive a clinical diagnosis of HD is appealing from a research perspective, because it could lead to more efficient clinical trials by using enriched samples (i.e., persons with a higher likelihood of clincial diagnosis during a brief time interval). In addition, it is clear that an increased uptake of predictive testing coupled with an augmentation in the propensity of individuals to volunteer for research may have the potential to expedite advances in prHD. Currently these limitations may explain why the study of prHD requires 33 sites in seven countries. Without considerable changes in the research and clinical arena for HD, clinical trials will require such excessive cost that our capacity to find treatments that make a difference for persons with HD will be diminished.
From a clinical standpoint, the early identification of disease could also have advantages, such as advanced planning for medical and legal decision making, initiating treatment plans, and making lifestyle choices (e.g., childcare, work, living arrangements). However, earlier diagnosis must also be weighed with its potential risks, which might include increased stress, changes in social relationships, and discrimination (e.g., loss of insurance coverage [131–132]). From a political perspective, efforts to alter the diagnostic criteria for HD will necessitate the addition of cognitive and psychiatric components, as well as the possibility of imaging criteria. For a disease that has enjoyed mono specialty care for over a century, inclusion of additional criteria may present a challenge to health care professionals.
Earlier identification of disease is universally considered useful when a treatment exists to slow or stop its progression. The argument is raised that until a prevention or treatment is found it is difficult to justify the importance of earlier detection. With regards to HD, however, we need to ask the question “treatment for what?” There may well be treatments for the HD symptoms of depression, irritability, cognitive decline, and so on, that would benefit HD families. Short of a treatment, HD families might benefit from acknowledging the way they feel about what is happening to them as the slowly progressive, insidious changes occur. At present no-one in the prodrome of HD knows to ask about these symptoms or their treatment. Furthermore, possible treating physicians are not yet prepared to offer treatments for HD unless motor signs are evident that make them 99% confident of HD.
Historically, diagnosis in HD seems to have become code language for “when I should tell the patient”. This was likely a combination of personal sensitivity combined with the pre-gene demand to be certain about diagnosis so that genetic studies could determine the locus. Unfortunately, hesitancy to diagnose HD remains. Truth telling in medicine has not always been seen as virtuous. The Hippocratic Oath instructed physicians “what I may see or hear in the course of the treatment or even outside of the treatment in regard to the life of men… I will keep to myself.” As late as the 1960s in America, over 90% of physicians said they would conceal the diagnosis of cancer from patients [133]. In the 1970s truth telling became a requirement for informed consent [134] but patient advocacy groups still had to lobby for greater patient awareness and control over medical decision making. Breaking bad news emerged in the ethics literature as a requirement of professionalism and justice only recently [135]. It has been found that physician attitudes and communication styles can result in behaviors designed to dull the full impact of the news and avoid full statements of a diagnosis [136–137]. Although physicians continue to find it stressful to give a bad diagnosis, it has been reported that patients typically can address the news without increased anxiety and that avoidance of diagnosis can have adverse outcomes [138].
Without adequate information about their condition patients may have an impaired ability to formulate their own goals. According to standard theories of justice and good life, all individuals have the right to make a rational life plan. Such a plan requires knowing one’s aims and facing up to all possible information and circumstances in order to form the best course of action to achieve one’s personal goals [139]. The goal of enabling patients to become full decision making partners with the physician can only be met when the patient has the information necessary to understand the circumstances from which they must formulate a rational life plan. The main problem in withholding information in the case of a debilitating illness “is that the reluctance to make a candid disclosure of the diagnosis … may violate basic moral and legal rights and may deprive patients and caregivers of some of the benefits of early disclosure of diagnosis” [136]. For patients with an early diagnosis of HD the withholding of information may mean the loss of an opportunity to seek early treatment or fulfill lifelong goals on a shortened timeline. To ensure the ability of our patients to make a rational life plan the earliest diagnosis may provide the best opportunity for the pursuit of these important individual aims.
Conclusion
Considerable data are now available regarding early indicators of HD that are detectable years (if not decades) before traditional clinical diagnosis. Usage of any one of the multiple measures reported here results in earlier detection of the pathophysiology of HD. It is important to understand what these findings may mean in the context of clinical care and research. First, the time course for HD has at least doubled. Previous reports had documented that death occurs about 15 years following a clinical diagnosis of HD [140] and current findings suggest that changes in clinical signs are evident 15 years prior to clinical diagnosis [10]. Increased awareness of an over 30-year duration of disease is consistent with patient reports and may assist health care practitioners provide better education and clinical care for HD families.
Second, these efforts to detect earlier disease have resulted in a more comprehensive characterization of HD. What was once a traditional movement disorder is now better described with three key features of basal ganglia disorders: movement, cognitive, and psychiatric disturbances. Although this is not news to the movement disorder specialist who has been working with these patients for decades, it is critical to expand training programs for primary care to better encompass all three components of HD as well as other movement disorders. Similarly, clinical trial efforts are in desperate need of expansion from a narrow emphasis on treatment for motor symptoms to a broader search for treatments that can provide relief for psychiatric and cognitive symptoms [141–142].
Finally, and perhaps most urgently, it is evident that the pathophysiology of HD starts long before the point at which the criteria for traditional diagnosis are satisfied. Moreover, the detectable signs and symptoms of that pathophysiology, especially when coupled with a family history and the ability to genetically confirm (i.e., avoid false positives), provide a clear medical basis for earlier diagnosis. The question is “why not”?
Future perspective
Further examination of the issues raised in this paper is warranted and delay of such discussions is likely to have a direct impact on health care for HD families. Below is a list of general recommendations that may facilitate clinical and research progress in HD.
Integration of the 30-year span of HD into training guidelines for HD health care professionals and families to allow for long-term life and treatment planning as well as an expanded research window.
Design of a multidisciplinary training conference to develop and disseminate the HD information attained over the past decade into education and training programs for all integral specialties, including genetics, genetic counseling, nursing, psychology, psychiatry, neurology, occupational and physical therapy, neuropsychology, speech pathology, radiology, and primary care.
Encouragement for the implementation of a consensus conference to discuss which components of HD might be best used in clinical and research diagnostic criteria for HD. Universal diagnostic criteria that involves all stakeholders across disciplines and countries and has input from the lay organizations and HD families.
Development of more formal involvement of the medical community in policy planning and legal developments such as GINA to protect the HD family from any consequences as they navigate living with a long-term chronic disease.
Executive summary.
Introduction
Huntington disease (HD), an autosomal dominant progressive neurological disease with an identified gene mutation, has demonstrated insidious changes in brain morphology, motor control, cognitive skill, and psychiatric symptoms over a period of 30 years.
The PREDICT-HD Study
A NIH-and CHDI-funded multi-site, longitudinal, observational study in its 9th year aimed at identifying biological and refined clinical markers of early HD in humans who have a gene mutation for future disease, but are currently healthy. Numerous markers have been identified, but require validation before they can be used as outcomes and clinical endpoints for use in preventive clinical trials.
Neuroimaging
Findings from structural as well as functional imaging suggest that disease is present decades prior to traditional clinical diagnosis and several imaging indices appear to be candidates for markers in clinical trials. New technologies are challenging traditional approaches to image analysis and interpretation. Efforts to minimize variability from different scanners and across sites will be critical for all multi-site clinical trials using imaging as an endpoint.
Motor Signs
Subtle motor signs are present long before a clinical diagnosis of HD is given. Findings confront current methods of diagnosis based upon confidence from a movement disorder specialist.
Cognition
Numerous individual tasks show decline and even impairment decades before clinical diagnosis. Additional research is crucial to determine the most robust, but efficient, cognitive assessment of early HD for use in clinical trials. Cognitive domains of speed and dysexecutive functions appear sensitive.
Psychiatric
Nearly all psychiatric symptoms and signs examined are elevated in prHD, yet syndromes do not mirror those seen in prototypical psychiatric disorders. Data is not yet available to determine when psychiatric symptoms begin, since they are already present in research conducted on prHD participants who are very far from estimated diagnosis. Measures of psychiatric ratings are difficult to ascertain possibly due to diminished insight, even in prHD.
Conclusions
Considerable data are now available regarding early indicators of HD that are detectable years (if not decades) before traditional diagnosis. Diagnostic criteria for HD should be considered for revision to include all clinical characteristics (motor, cognitive, psychiatric) as well as biological characteristics (imaging). Earlier diagnosis is likely to advance life planning for individuals as well as research advancements for treatment.
Bibliography
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers
- 1**.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2*.Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington’s disease. Science. 1985;230:561–3. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 3*.Calabresi P, Centonze D, Pisani A, et al. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington’s disease. Ann Neurol. 1998;43:586–97. doi: 10.1002/ana.410430506. [DOI] [PubMed] [Google Scholar]
- 4*.Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci Lett. 1991;133:257–61. doi: 10.1016/0304-3940(91)90583-f. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen JS, Mikos A. Huntington’s disease. In: Morgan JE, Ricker JH, editors. Textbook of clinical neuropsychology. Taylor & Francis; New York, NY: 2007. pp. 616–635. [Google Scholar]
- 6**.Duyao M, Ambrose C, Myers R, et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. 1993;4:387–92. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- 7*.Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, Ross CA. Correlation between the onset age of Huntington’s disease and length of the trinucleotide repeat in IT-15. Hum Mol Genet. 1993;2:1547–9. doi: 10.1093/hmg/2.10.1547. [DOI] [PubMed] [Google Scholar]
- 8*.Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for disease progression in Huntington’s disease. Cochrane Database Syst Rev. 2009:CD006455. doi: 10.1002/14651858.CD006455.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9**.Paulsen JS, Hayden M, Stout JC, et al. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol. 2006;63:883–90. doi: 10.1001/archneur.63.6.883. [DOI] [PubMed] [Google Scholar]
- 10**.Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–80. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. 2004;65:267–77. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 12**.Langbehn DR, Hayden M, Paulsen JS Predict-HD Investigators of the HSG. CAG-Repeat Length and the Age of Diagnosis in Huntington’s Disease: A Critical Review and Prospective Validation Study of Statistical Approaches. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2009 doi: 10.1002/ajmg.b.30992. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Rosas HD, Salat DH, Lee SY, et al. Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain. 2008;131:1057–68. doi: 10.1093/brain/awn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starkstein S, Brandt J, Folstein SE, et al. Neuropsychological and neuroradiological correlates of Huntington’s disease. Journal of Neurology, Neurosurgery and Psychiatry. 1988;51:1259–1263. doi: 10.1136/jnnp.51.10.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Starkstein SE, Brandt J, Bylsma FW, Peyser CE, Folstein M, Folstein SE. Neuropsychological correlates of brain atrophy in Huntington’s disease: A magnetic resonance study. Neuroradiology. 1992;34:487–489. doi: 10.1007/BF00598956. [DOI] [PubMed] [Google Scholar]
- 16.Sax DS, O’Donnell B, Butters N, Menzer L, Montgomery K, Kayne HL. Computerized tomographic, neurologic, and neuropsychological correlates of Huntington’s disease. International Journal of Neuroscience. 1983;18:21–36. doi: 10.3109/00207458308985874. [DOI] [PubMed] [Google Scholar]
- 17.Aylward EH, Li Q, Stine OC, et al. Longitudinal change in basal ganglia volume in patients with Huntington’s disease. Neurology. 1997;48:394–9. doi: 10.1212/wnl.48.2.394. [DOI] [PubMed] [Google Scholar]
- 18.Rosas HD, Goodman J, Chen YI, et al. Striatal volume loss in HD as measured by MRI and the influence of CAG repeat. Neurology. 2001;57:1025–8. doi: 10.1212/wnl.57.6.1025. [DOI] [PubMed] [Google Scholar]
- 19**.Aylward EH, Sparks BF, Field KM, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63:66–72. doi: 10.1212/01.wnl.0000132965.14653.d1. [DOI] [PubMed] [Google Scholar]
- 20.Solomon AC, Stout JC, Johnson SA, et al. Verbal episodic memory declines prior to diagnosis in Huntington’s disease. Neuropsychologia. 2007;45:1767–76. doi: 10.1016/j.neuropsychologia.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Stout JC. Psychiatric symptoms in Huntington’s disease before diagnosis: the predict-HD study. Biol Psychiatry. 2007;62:1341–6. doi: 10.1016/j.biopsych.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 22*.Squitieri F, Cannella M, Simonelli M, et al. Distinct brain volume changes correlating with clinical stage, disease progression rate, mutation size, and age at onset prediction as early biomarkers of brain atrophy in Huntington’s disease. CNS Neurosci Ther. 2009;15:1–11. doi: 10.1111/j.1755-5949.2008.00068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745–7. doi: 10.1212/01.wnl.0000174432.87383.87. [DOI] [PubMed] [Google Scholar]
- 24.Rosas HD, Liu AK, Hersch S, et al. Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology. 2002;58:695–701. doi: 10.1212/wnl.58.5.695. [DOI] [PubMed] [Google Scholar]
- 25.Nopoulos P, Magnotta VA, Mikos A, Paulson H, Andreasen NC, Paulsen JS. Morphology of the cerebral cortex in preclinical Huntington’s disease. Am J Psychiatry. 2007;164:1428–34. doi: 10.1176/appi.ajp.2007.06081266. [DOI] [PubMed] [Google Scholar]
- 26*.Henley SM, Wild EJ, Hobbs NZ, et al. Whole-brain atrophy as a measure of progression in premanifest and early Huntington’s disease. Mov Disord. 2009;24:932–6. doi: 10.1002/mds.22485. [DOI] [PubMed] [Google Scholar]
- 27**.Tabrizi SJ, Langbehn DR, Leavitt BR, et al. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. doi: 10.1016/S1474-4422(09)70170-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beglinger LJ, Nopoulos PC, Jorge RE, et al. White matter volume and cognitive dysfunction in early Huntington’s disease. Cogn Behav Neurol. 2005;18:102–7. doi: 10.1097/01.wnn.0000152205.79033.73. [DOI] [PubMed] [Google Scholar]
- 29.Ciarmiello A, Cannella M, Lastoria S, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med. 2006;47:215–22. [PubMed] [Google Scholar]
- 30.Magnotta VA, Kim J, Koscik T, et al. Diffusion Tensor Imaging in Preclinical Huntington’s Disease. Brain Imaging and Behavior. 2009:77–84. doi: 10.1007/s11682-008-9051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reading SA, Yassa MA, Bakker A, et al. Regional white matter change in pre-symptomatic Huntington’s disease: a diffusion tensor imaging study. Psychiatry Res. 2005;140:55–62. doi: 10.1016/j.pscychresns.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Rosas HD, Tuch DS, Hevelone ND, et al. Diffusion tensor imaging in presymptomatic and early Huntington’s disease: Selective white matter pathology and its relationship to clinical measures. Mov Disord. 2006;21:1317–25. doi: 10.1002/mds.20979. [DOI] [PubMed] [Google Scholar]
- 33.Seppi K, Schocke MF, Mair KJ, et al. Diffusion-weighted imaging in Huntington’s disease. Mov Disord. 2006;21:1043–7. doi: 10.1002/mds.20868. [DOI] [PubMed] [Google Scholar]
- 34.Nopoulos P, Johnson H, Magnotta V, et al. Global and Regional Brain Morphology in Subjects with Huntington’s Disease Prior to Diagnosis. Neuroimage. 2009;47:S91. [Google Scholar]
- 35.Aylward E, Nopoulos P, Pierson R, Langbehn D, Ross CA, Paulsen J. Longitudinal Change in Striatal Volume in Pre-Clinical Huntington’s Disease. Neuroimage. 2009;47:S93. [Google Scholar]
- 36.Paulsen J, Nopoulos P, Ross CA, et al. Striatum and white matter are the best imaging predictors of estimated diagnosis for Huntington disease. Neuroimage. 2009;47:S95. [Google Scholar]
- 37.Muhlau M, Wohlschlager AM, Gaser C, et al. Voxel-Based Morphometry in Individual Patients: A Pilot Study in Early Huntington Disease. AJNR Am J Neuroradiol. 2008 doi: 10.3174/ajnr.A1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kloppel S, Chu C, Tan GC, et al. Automatic detection of preclinical neurodegeneration: presymptomatic Huntington disease. Neurology. 2009;72:426–31. doi: 10.1212/01.wnl.0000341768.28646.b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Oostrom JC, Sijens PE, Roos RA, Leenders KL. 1H magnetic resonance spectroscopy in preclinical Huntington disease. Brain Res. 2007;1168:67–71. doi: 10.1016/j.brainres.2007.05.082. [DOI] [PubMed] [Google Scholar]
- 40**.Paulsen JS, Zimbelman JL, Hinton SC, et al. fMRI biomarker of early neuronal dysfunction in presymptomatic Huntington’s Disease. AJNR Am J Neuroradiol. 2004;25:1715–21. [PMC free article] [PubMed] [Google Scholar]
- 41*.Saft C, Schutte A, Beste C, Andrich J, Pfleiderer B. Altered auditory sensory processing in premanifest Huntington’s disease: Are there different phases in premanifest Huntington’s disease? Journal of Neurology Neurosurgery and Psychiatry. 2008;79:A11–A11. [Google Scholar]
- 42.Saft C, Schuttke A, Beste C, Andrich J, Heindel W, Pfleiderer B. fMRI reveals altered auditory processing in manifest and premanifest Huntington’s disease. Neuropsychologia. 2008;46:1279–89. doi: 10.1016/j.neuropsychologia.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 43*.Henley SM, Frost C, MacManus DG, Warner TT, Fox NC, Tabrizi SJ. Increased rate of whole-brain atrophy over 6 months in early Huntington disease. Neurology. 2006;67:694–6. doi: 10.1212/01.wnl.0000230149.36635.c8. [DOI] [PubMed] [Google Scholar]
- 44.Kipps CM, Duggins AJ, Mahant N, Gomes L, Ashburner J, McCusker EA. Progression of structural neuropathology in preclinical Huntington’s disease: a tensor based morphometry study. J Neurol Neurosurg Psychiatry. 2005;76:650–5. doi: 10.1136/jnnp.2004.047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frackowiak RSJ. Human brain function. San Diego: Academic Press; 1997. p. xiii.p. 528. [Google Scholar]
- 46*.Tai YF, Pavese N, Gerhard A, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–66. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- 47.van Oostrom JCH, Maguire RP, Verschuuren-Bemelmans CC, et al. Rate of decline of striatal imaging parameters in preclinical carriers of the Huntington’s disease mutation. Journal of Neurology Neurosurgery and Psychiatry. 2005:76. [Google Scholar]
- 48*.van Oostrom JC, Dekker M, Willemsen AT, de Jong BM, Roos RA, Leenders KL. Changes in striatal dopamine D2 receptor binding in pre-clinical Huntington’s disease. Eur J Neurol. 2009;16:226–31. doi: 10.1111/j.1468-1331.2008.02390.x. [DOI] [PubMed] [Google Scholar]
- 49*.Feigin A, Tang C, Ma Y, et al. Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain. 2007;130:2858–67. doi: 10.1093/brain/awm217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andrews TC, Weeks RA, Turjanski N, et al. Huntington’s disease progression. PET and clinical observations. Brain. 1999;122 (Pt 12):2353–63. doi: 10.1093/brain/122.12.2353. [DOI] [PubMed] [Google Scholar]
- 51.Antonini A, Leenders KL, Spiegel R, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain. 1996;119 (Pt 6):2085–95. doi: 10.1093/brain/119.6.2085. [DOI] [PubMed] [Google Scholar]
- 52.Reynolds NC, Jr, Prost RW, Mark LP. Heterogeneity in 1H-MRS profiles of presymptomatic and early manifest Huntington’s disease. Brain Res. 2005;1031:82–9. doi: 10.1016/j.brainres.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 53*.Gomez-Anson B, Alegret M, Munoz E, Sainz A, Monte GC, Tolosa E. Decreased frontal choline and neuropsychological performance in preclinical Huntington disease. Neurology. 2007;68:906–10. doi: 10.1212/01.wnl.0000257090.01107.2f. [DOI] [PubMed] [Google Scholar]
- 54.Paulsen JS. Functional imaging in Huntington’s disease. Exp Neurol. 2009;216:272–7. doi: 10.1016/j.expneurol.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hennenlotter A, Schroeder U, Erhard P, et al. Neural correlates associated with impaired disgust processing in pre-symptomatic Huntington’s disease. Brain. 2004;127:1446–53. doi: 10.1093/brain/awh165. [DOI] [PubMed] [Google Scholar]
- 56.Reading SA, Dziorny AC, Peroutka LA, et al. Functional brain changes in presymptomatic Huntington’s disease. Ann Neurol. 2004;55:879–83. doi: 10.1002/ana.20121. [DOI] [PubMed] [Google Scholar]
- 57.Zimbelman JL, Paulsen JS, Mikos A, Reynolds NC, Hoffmann RG, Rao SM. fMRI detection of early neural dysfunction in preclinical Huntington’s disease. J Int Neuropsychol Soc. 2007;13:758–69. doi: 10.1017/S1355617707071214. [DOI] [PubMed] [Google Scholar]
- 58**.Unified Huntington’s Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord. 1996;11:136–42. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- 59*.Kirkwood SC, Siemers E, Hodes ME, Conneally PM, Christian JC, Foroud T. Subtle changes among presymptomatic carriers of the Huntington’s disease gene. J Neurol Neurosurg Psychiatry. 2000;69:773–9. doi: 10.1136/jnnp.69.6.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.McCusker E, Richards F, Sillence D, Wilson M, Trent RJ. Huntington’s disease: neurological assessment of potential gene carriers presenting for predictive DNA testing. J Clin Neurosci. 2000;7:38–41. doi: 10.1054/jocn.1998.0151. [DOI] [PubMed] [Google Scholar]
- 61**.Biglan KM, Ross CA, Langbehn DR, et al. Motor Abnormalities in Pre-Manifest Persons with Huntington’s Disease: The PREDICT-HD Study. Movement Disorders. 2009 doi: 10.1002/mds.22601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Solomon AC, Stout JC, Weaver M, et al. Ten-year rate of longitudinal change in neurocognitive and motor function in prediagnosis Huntington disease. Mov Disord. 2008;23:1830–6. doi: 10.1002/mds.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63*.Witjes-Ane MN, Mertens B, van Vugt JP, Bachoud-Levi AC, van Ommen GJ, Roos RA. Longitudinal evaluation of “presymptomatic” carriers of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2007;19:310–7. doi: 10.1176/jnp.2007.19.3.310. [DOI] [PubMed] [Google Scholar]
- 64.Delval A, Krystkowiak P, Blatt JL, et al. Role of hypokinesia and bradykinesia in gait disturbances in Huntington’s disease: a biomechanical study. J Neurol. 2006;253:73–80. doi: 10.1007/s00415-005-0929-2. [DOI] [PubMed] [Google Scholar]
- 65.Ali FR, Michell AW, Barker RA, Carpenter RH. The use of quantitative oculometry in the assessment of Huntington’s disease. Exp Brain Res. 2006;169:237–45. doi: 10.1007/s00221-005-0143-6. [DOI] [PubMed] [Google Scholar]
- 66*.Penney JB, Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann Neurol. 1997;41:689–92. doi: 10.1002/ana.410410521. [DOI] [PubMed] [Google Scholar]
- 67.Farrow M, Chua P, Churchyard A, Bradshaw JL, Chiu E, Georgiou-Karistianis N. Proximity to clinical onset influences motor and cognitive performance in presymptomatic Huntington disease gene carriers. Cogn Behav Neurol. 2006;19:208–16. doi: 10.1097/01.wnn.0000213914.64772.b6. [DOI] [PubMed] [Google Scholar]
- 68*.Golding CV, Danchaivijitr C, Hodgson TL, Tabrizi SJ, Kennard C. Identification of an oculomotor biomarker of preclinical Huntington disease. Neurology. 2006;67:485–7. doi: 10.1212/01.wnl.0000218215.43328.88. [DOI] [PubMed] [Google Scholar]
- 69.Blekher T, Johnson SA, Marshall J, et al. Saccades in presymptomatic and early stages of Huntington disease. Neurology. 2006;67:394–9. doi: 10.1212/01.wnl.0000227890.87398.c1. [DOI] [PubMed] [Google Scholar]
- 70*.Antoniades CA, Altham PM, Mason SL, Barker RA, Carpenter R. Saccadometry: a new tool for evaluating presymptomatic Huntington patients. Neuroreport. 2007;18:1133–6. doi: 10.1097/WNR.0b013e32821c560d. [DOI] [PubMed] [Google Scholar]
- 71*.Rao AK, Muratori L, Louis ED, Moskowitz CB, Marder KS. Spectrum of gait impairments in presymptomatic and symptomatic Huntington’s disease. Mov Disord. 2008;23:1100–7. doi: 10.1002/mds.21987. [DOI] [PubMed] [Google Scholar]
- 72.Duff K, Schoenberg MR, Patton D, et al. Regression-based formulas for predicting change in RBANS subtests with older adults. Arch Clin Neuropsychol. 2005;20:281–90. doi: 10.1016/j.acn.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 73*.Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Stout JC Predict HDIotHSG. Psychiatric symptoms in Huntington’s disease before diagnosis: the predict-HD study. Biol Psychiatry. 2007;62:1341–6. doi: 10.1016/j.biopsych.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 74.Farrow M, Churchyard A, Chua P, Bradshaw JL, Chiu E, Georgiou-Karistianis N. Attention, inhibition, and proximity to clinical onset in preclinical mutation carriers for Huntington’s disease. J Clin Exp Neuropsychol. 2007;29:235–46. doi: 10.1080/13803390600657693. [DOI] [PubMed] [Google Scholar]
- 75.Hinton SC, Paulsen JS, Hoffmann RG, Reynolds NC, Zimbelman JL, Rao SM. Motor timing variability increases in preclinical Huntington’s disease patients as estimated onset of motor symptoms approaches. J Int Neuropsychol Soc. 2007;13:539–43. doi: 10.1017/S1355617707070671. [DOI] [PubMed] [Google Scholar]
- 76.Ho AK, Sahakian BJ, Robbins TW, Barker RA. Random number generation in patients with symptomatic and presymptomatic Huntington’s disease. Cogn Behav Neurol. 2004;17:208–12. [PubMed] [Google Scholar]
- 77.Larsson M, Lundin A, Wahlin TBR. Olfactory functions in asymptomatic carriers of the Huntington disease mutation. Journal of Clinical and Experimental Neuropsychology. 2006;28:1373–1380. doi: 10.1080/13803390500473746. [DOI] [PubMed] [Google Scholar]
- 78.Larsson MU, Almkvist O, Luszcz MA, Wahlin TB. Phonemic fluency deficits in asymptomatic gene carriers for Huntington’s disease. Neuropsychology. 2008;22:596–605. doi: 10.1037/0894-4105.22.5.596. [DOI] [PubMed] [Google Scholar]
- 79.Peretti CS, Ferreri F, Blanchard F, et al. Normal and pathological aging of attention in presymptomatic Huntington’s, Huntington’s and Alzheimer’s disease, and nondemented elderly subjects. Psychotherapy and Psychosomatics. 2008;77:139–146. doi: 10.1159/000116607. [DOI] [PubMed] [Google Scholar]
- 80.Pirogovsky E, Gilbert PE, Jacobson M, et al. Impairments in source memory for olfactory and visual stimuli in preclinical and clinical stages of Huntington’s disease. J Clin Exp Neuropsychol. 2007;29:395–404. doi: 10.1080/13803390600726829. [DOI] [PubMed] [Google Scholar]
- 81.Rowe KC, Langbehn DR, Duff K, et al. Self-Paced Timing Task Detects Subtle Changes in Pre-diagnosed Huntington’s Disease. Neuropsychology. 2009 doi: 10.1037/a0018905. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnson SA, Stout JC, Solomon AC, et al. Beyond disgust: impaired recognition of negative emotions prior to diagnosis in Huntington’s disease. Brain. 2007;130:1732–44. doi: 10.1093/brain/awm107. [DOI] [PubMed] [Google Scholar]
- 83.Paulsen JS, Zhao H, Stout JC, et al. Clinical markers of early disease in persons near onset of Huntington’s disease. Neurology. 2001;57:658–62. doi: 10.1212/wnl.57.4.658. [DOI] [PubMed] [Google Scholar]
- 84.Lemiere J, Decruyenaere M, Evers-Kiebooms G, Vandenbussche E, Dom R. Cognitive changes in patients with Huntington’s disease (HD) and asymptomatic carriers of the HD mutation--a longitudinal follow-up study. J Neurol. 2004;251:935–42. doi: 10.1007/s00415-004-0461-9. [DOI] [PubMed] [Google Scholar]
- 85.Verny C, Allain P, Prudean A, et al. Cognitive changes in asymptomatic carriers of the Huntington disease mutation gene. Eur J Neurol. 2007;14:1344–50. doi: 10.1111/j.1468-1331.2007.01975.x. [DOI] [PubMed] [Google Scholar]
- 86.Witjes-Ane MN, Vegter-van der Vlis M, van Vugt JP, et al. Cognitive and motor functioning in gene carriers for Huntington’s disease: a baseline study. J Neuropsychiatry Clin Neurosci. 2003;15:7–16. doi: 10.1176/jnp.15.1.7. [DOI] [PubMed] [Google Scholar]
- 87.Wahlin TBR, Larsson M, Luszcz M. Depression and suicidal ideation in preclinical Huntington’s disease in Sweden. Australian and New Zealand Journal of Psychiatry. 2007;41:A74–A74. [Google Scholar]
- 88.Wahlin TBR, Lundin A, Dear K. Early cognitive deficits in Swedish gene carriers of Huntington’s disease. Neuropsychology. 2007;21:31–44. doi: 10.1037/0894-4105.21.1.31. [DOI] [PubMed] [Google Scholar]
- 89.Rupp J, Blekher T, Jackson J, et al. Progression in Prediagnostic Huntington Disease. J Neurol Neurosurg Psychiatry. 2009 doi: 10.1136/jnnp.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duff K, Beglinger LJ, Theriault D, Allison J, Paulsen JS. Cognitive deficits in Huntington’s disease on the Repeatable Battery for the Assessment of Neuropsychological Status. J Clin Exp Neuropsychol. 2009:1–9. doi: 10.1080/13803390902926184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Petersen RC. Conversion. Neurology. 2006;67:S12–3. doi: 10.1212/wnl.67.9_suppl_3.s12. [DOI] [PubMed] [Google Scholar]
- 92.Paulsen JS, Duff K. Extending MCI beyond Alzheimer disease. Neurology. 2009;72:1116–7. doi: 10.1212/01.wnl.0000345369.41620.fc. [DOI] [PubMed] [Google Scholar]
- 93*.van Duijn E, Kingma EM, van der Mast RC. Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. 2007;19:441–8. doi: 10.1176/jnp.2007.19.4.441. [DOI] [PubMed] [Google Scholar]
- 94.Paulsen JS, Nehl C, Guttman M. Movement Disorders of the Basal Ganglia. In: Rizzo M, Eslinger PJ, editors. Principles and practice of behavioral neurology and neuropsychology. W.B. Saunders; Philadelphia, Pa: 2004. pp. 525–550. [Google Scholar]
- 95.Nehl C, Paulsen JS. Cognitive and psychiatric aspects of Huntington disease contribute to functional capacity. J Nerv Ment Dis. 2004;192:72–4. doi: 10.1097/01.nmd.0000106004.67587.57. [DOI] [PubMed] [Google Scholar]
- 96*.Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74:120–2. doi: 10.1136/jnnp.74.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Folstein SE, Franz ML, Jensen BA, Chase GA, Folstein MF. Conduct disorder and affective disorder among the offspring of patients with Huntington’s disease. Psychol Med. 1983;13:45–52. doi: 10.1017/s0033291700050054. [DOI] [PubMed] [Google Scholar]
- 98.Folstein S, Abbott MH, Chase GA, Jensen BA, Folstein MF. The association of affective disorder with Huntington’s disease in a case series and in families. Psychol Med. 1983;13:537–42. doi: 10.1017/s0033291700047966. [DOI] [PubMed] [Google Scholar]
- 99.Berrios GE, Wagle AC, Markova IS, Wagle SA, Rosser A, Hodges JR. Psychiatric symptoms in neurologically asymptomatic Huntington’s disease gene carriers: a comparison with gene negative at risk subjects. Acta Psychiatr Scand. 2002;105:224–30. doi: 10.1034/j.1600-0447.2002.0o456.x. [DOI] [PubMed] [Google Scholar]
- 100*.Kirkwood SC, Siemers E, Viken R, et al. Longitudinal personality changes among presymptomatic Huntington disease gene carriers. Neuropsychiatry Neuropsychol Behav Neurol. 2002;15:192–7. [PubMed] [Google Scholar]
- 101.Beglinger LJ, Paulsen JS, Watson DB, et al. Obsessive and compulsive symptoms in prediagnosed Huntington’s disease. J Clin Psychiatry. 2008;69:1758–65. doi: 10.4088/jcp.v69n1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duff K, Paulsen JS, Beglinger LJ, et al. Late-breaking research - The “Depression phenotype” in pre-diagnosed Huntington’s disease. Movement Disorders. 2007;22:Iv–Iv. [Google Scholar]
- 103.Johnson SA, Stout JC, Solomon AC, et al. Beyond disgust: impaired recognition of negative emotions prior to diagnosis in Huntington’s disease. Brain. 2007;130:1732–44. doi: 10.1093/brain/awm107. [DOI] [PubMed] [Google Scholar]
- 104.Soliveri P, Monza D, Piacentini S, et al. Cognitive and psychiatric characterization of patients with Huntington’s disease and their at-risk relatives. Neurol Sci. 2002;23 (Suppl 2):S105–6. doi: 10.1007/s100720200091. [DOI] [PubMed] [Google Scholar]
- 105.Shiwach RS, Norbury CG. A controlled psychiatric study of individuals at risk for Huntington’s disease. Br J Psychiatry. 1994;165:500–5. doi: 10.1192/bjp.165.4.500. [DOI] [PubMed] [Google Scholar]
- 106**.Duff K, Paulsen J, Beglinger L, et al. “Frontal” behaviors before the diagnosis Huntington’s disease and its relationship to markers of disease progression: Evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. doi: 10.1176/appi.neuropsych.22.2.196. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grace J, Malloy P Psychological Assessment Resources Inc. FrSBe, frontal systems behavior scale: professional manual. Lutz, FL: Psychological Assessment Resources; 2001. p. 109. [Google Scholar]
- 108.Hoth KF, Paulsen JS, Moser DJ, Tranel D, Clark LA, Bechara A. Patients with Huntington’s disease have impaired awareness of cognitive, emotional, and functional abilities. J Clin Exp Neuropsychol. 2007;29:365–76. doi: 10.1080/13803390600718958. [DOI] [PubMed] [Google Scholar]
- 109.Ho AK, Robbins AO, Barker RA. Huntington’s disease patients have selective problems with insight. Mov Disord. 2006;21:385–9. doi: 10.1002/mds.20739. [DOI] [PubMed] [Google Scholar]
- 110*.Langbehn DR, Paulsen JS Huntington Study G. Predictors of diagnosis in Huntington disease. Neurology. 2007;68:1710–7. doi: 10.1212/01.wnl.0000261918.90053.96. [DOI] [PubMed] [Google Scholar]
- 111.Julien CL, Thompson JC, Wild S, et al. Psychiatric disorders in preclinical Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78:939–43. doi: 10.1136/jnnp.2006.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosenblatt A. Understanding the psychiatric prodrome of Huntington disease. Journal of Neurology Neurosurgery and Psychiatry. 2007;78:913–913. doi: 10.1136/jnnp.2006.112565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Beglinger LJ, Langbehn DR, Duff K, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61:415–8. doi: 10.1016/j.biopsych.2006.04.034. [DOI] [PubMed] [Google Scholar]
- 114.Duff K, Beglinger LJ, O’Rourke ME, Nopoulos P, Paulson HL, Paulsen JS. Risperidone and the treatment of psychiatric, motor, and cognitive symptoms in Huntington’s disease. Ann Clin Psychiatry. 2008;20:1–3. doi: 10.1080/10401230701844802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duff K, Beglinger LJ, Paulsen JS. “Pre-symptomatic” Huntington’s Disease. Handb Clin Neurol. 2008;89:589–98. doi: 10.1016/S0072-9752(07)01255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kloppel S, Stonnington CM, Predrag P, Mobbs D, Tuscher O, Craufurd D, Tabrizi SJ, Frackowiak SJ. Irritability in preclinical HD. Neuropsychologia. 2009 doi: 10.1016/j.neuropsychologia.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Evans K, Vaccarino A, Sills T, et al. Assessing Depression in Huntington Disease: Relationship Between Disease Progression, Depression and Manifestation of Motor Symptoms. World Congress on HD; 2009; Vancouver, BC. [Google Scholar]
- 118.Paradiso S, Turner BM, Paulsen JS, Jorge R, Ponto LL, Robinson RG. Neural bases of dysphoria in early Huntington’s disease. Psychiatry Res. 2008;162:73–87. doi: 10.1016/j.pscychresns.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Evans K, Vaccarino A, Craufurd D, et al. The short version of the problem behaviours assessment for HD (PBA-S): An item response analysis using data from the TRACK-HD study. Clin Genet. 2009;76:1. [Google Scholar]
- 120**.Bren L. The Importance of Patient-Reported Outcomes… It’s All About the Patients. FDA Consumer magazine. 2006 Nov-Dec; 2006. [PubMed] [Google Scholar]
- 121**.Downing GJ, National Institutes of Health (U.S.), United States. Food and Drug Administration. Biomarkers and surrogate endpoints: clinical research and applications: proceedings of the NIH-FDA conference; 15–16 April 1999; Bethesda, Maryland, USA. Amsterdam; New York: Elsevier; 2000. p. xxvii.p. 318. International congress series 1205. [Google Scholar]
- 122.Khan O. Can clinical outcomes be used to detect neuroprotection in multiple sclerosis? Neurology. 2007;68:S64–71. doi: 10.1212/01.wnl.0000275235.43506.d2. discussion S91–6. [DOI] [PubMed] [Google Scholar]
- 123.Oksengard A-R, Winblad B. Dementia Diagnostics Made Evidence-Based: A Critical Evaluation of Cognitive Assessment Tools in Clinical Dementia Diagnostics. Curr Opin Psychiatry. 2005;17:439–442. [Google Scholar]
- 124.Cudkowicz M, Qureshi M, Shefner J. Measures and markers in amyotrophic lateral sclerosis. NeuroRx. 2004;1:273–83. doi: 10.1602/neurorx.1.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kieburtz K. Issues in neuroprotection clinical trials in Parkinson’s disease. Neurology. 2006;66:S50–7. doi: 10.1212/wnl.66.10_suppl_4.s50. [DOI] [PubMed] [Google Scholar]
- 126.Olanow CW, Schapira AH, Agid Y. Neuroprotection for Parkinson’s disease: prospects and promises. Annals of Neurology. 2003;53 (Suppl 3):S1–2. doi: 10.1002/ana.10566. [DOI] [PubMed] [Google Scholar]
- 127.Shoulson I, Fahn S. Huntington disease: clinical care and evaluation. Neurology. 1979;29:1–3. doi: 10.1212/wnl.29.1.1. [DOI] [PubMed] [Google Scholar]
- 128.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR International Huntington’s Disease Collaborative G. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. 2004;65:267–77. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 129.Feigin A, Ghilardi MF, Huang C, et al. Preclinical Huntington’s disease: compensatory brain responses during learning. Ann Neurol. 2006;59:53–9. doi: 10.1002/ana.20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Paulsen JS, Magnotta VA, Mikos AE, et al. Brain structure in preclinical Huntington’s disease. Biol Psychiatry. 2006;59:57–63. doi: 10.1016/j.biopsych.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 131.Bombard Y, Penziner E, Suchowersky O, et al. Engagement with genetic discrimination: concerns and experiences in the context of Huntington disease. Eur J Hum Genet. 2008;16:279–89. doi: 10.1038/sj.ejhg.5201937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Penziner E, Williams JK, Erwin C, et al. Perceptions of discrimination among persons who have undergone predictive testing for Huntington’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147:320–5. doi: 10.1002/ajmg.b.30600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lerner BH. Breast cancer activism: past lessons, future directions. Nat Rev Cancer. 2002;2:225–30. doi: 10.1038/nrc744. [DOI] [PubMed] [Google Scholar]
- 134.Canterbury v Spence. DC Cir. 1972, 464 F2d 772
- 135.Cleary M, Hunt GE, Horsfall J. Delivering difficult news in psychiatric settings. Harv Rev Psychiatry. 2009;17:315–21. doi: 10.3109/10673220903271780. [DOI] [PubMed] [Google Scholar]
- 136**.Karnieli-Miller O, Werner P, Aharon-Peretz J, Eidelman S. Dilemmas in the (un)veiling of the diagnosis of Alzheimer’s disease: Walking an ethical and professional tight rope. Patient Education and Counseling. 2007;67:307–314. doi: 10.1016/j.pec.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 137.Friedrichsen M, Milberg A. Concerns about losing control when breaking bad news to terminally ill patients with cancer: Physicians’ perspective. Journal of Palliative Medicine. 2006;9:673–682. doi: 10.1089/jpm.2006.9.673. [DOI] [PubMed] [Google Scholar]
- 138.Hancock K, Clayton JM, Parker SM, et al. Truth-telling in discussing prognosis in advanced life-limiting illnesses: a systematic review. Palliative Medicine. 2007;21:507–517. doi: 10.1177/0269216307080823. [DOI] [PubMed] [Google Scholar]
- 139.Rawls J. A theory of justice. Cambridge, Mass: Belknap Press of Harvard University Press; 1971. p. xv.p. 607. [Google Scholar]
- 140.Gusella JF, McNeil S, Persichetti F, et al. Huntington’s disease. Cold Spring Harb Symp Quant Biol. 1996;61:615–26. [PubMed] [Google Scholar]
- 141.Beglinger L, Adams W, Paulson H, et al. Randomized Controlled Trial of Atomoxetine for Cognitive Dysfunction in Early Huntington Disease. J Clin Psychopharm. doi: 10.1097/JCP.0b013e3181b2ac0a. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beglinger L, Adams W, Paulsen J, et al. Citalopram and atomoxetine to enhance cognition in early Huntington’s disease: Two pilot clinical trials. World Congress on Huntington’s Disease; 2007; Dresden, Germany. [Google Scholar]
- 143.Nuechterlein KH, Green MF, Kern RS, et al. The MATRICS Consensus Cognitive Battery, Part 1: Test Selection, Reliability, and Validity. Am J Psychiatry. 2008;165:203–213. doi: 10.1176/appi.ajp.2007.07010042. [DOI] [PubMed] [Google Scholar]