

Summary
Transient disruption of endothelial adherens junctions and cytoskeletal remodeling are responsible for increases in vascular permeability induced by inflammatory stimuli and vascular endothelial growth factor (VEGF). Nitric oxide (NO) produced by endothelial NO synthase (eNOS) is crucial for VEGF-induced changes in permeability in vivo; however, the molecular mechanism by which endogenous NO modulates endothelial permeability is not clear. Here, we show that the lack of eNOS reduces VEGF-induced permeability, an effect mediated by enhanced activation of the Rac GTPase and stabilization of cortical actin. The loss of NO increased the recruitment of the Rac guanine-nucleotide-exchange factor (GEF) TIAM1 to adherens junctions and VE-cadherin (also known as cadherin 5), and reduced Rho activation and stress fiber formation. In addition, NO deficiency reduced VEGF-induced VE-cadherin phosphorylation and impaired the localization, but not the activation, of c-Src to cell junctions. The physiological role of eNOS activation is clear given that VEGF-, histamine- and inflammation-induced vascular permeability is reduced in mice bearing a non-phosphorylatable knock-in mutation of the key eNOS phosphorylation site S1176. Thus, NO is crucial for Rho GTPase-dependent regulation of cytoskeletal architecture leading to reversible changes in vascular permeability.
Key words: Nitric oxide, eNOS, VEGF, Cytoskeleton, VE-cadherin, Cadherin 5, Src
Introduction
Alterations in endothelial barrier function play an important role in the pathogenesis of many disease states, including inflammation, wound healing, edema, acute lung injury, stroke and cancer (Wojciak-Stothard and Ridley, 2002). Endothelial barrier maintenance and the response of the quiescent barrier to locally produced vasoactive agents, such as histamine, prostaglandins, thrombin and VEGF is primarily mediated by dynamic changes in adherens junctions (AJs) and increased centripetal tension created by actomyosin contractility (Dejana, 2004; Feng et al., 1999; Weis and Nelson, 2006).
During an inflammatory process, the microvascular permeability is selectively increased in post-capillary venules, where the AJs are the major type of inter-endothelial junctions. Vascular endothelial cadherin (VE-cadherin, also known as cadherin 5), a cell-type-specific cadherin, is highly expressed on endothelial cells (ECs) and forms a complex with proteins of the armadillo family including p120-catenin, β-catenin and plakoglobin, contributing to the stability of the EC barrier. The disruption of AJs by permeability agents is in part mediated by tightly regulated protein phosphorylation, endocytosis and intramembrane cleavage. In particular, many studies have reported that tyrosine phosphorylation of VE-cadherin and its binding partners induced by histamine (Andriopoulou et al., 1999; Shasby et al., 2002), tumor necrosis factor-α (TNF-α) (Angelini et al., 2006) or VEGF (Esser et al., 1998) correlates with the destabilization of endothelial AJs. Most recently, the crucial importance of AJ regulation during inflammation has been determined in mice expressing a ‘stabilized’ VE-cadherin (Broermann et al., 2011) where the acute responses to inflammatory mediators are markedly attenuated.
VEGF is a potent vascular permeability factor that plays an essential role in both physiological and pathological angiogenesis (Ferrara et al., 2003). VEGF exerts its effects on vascular permeability through the activation of the Src family kinases, in particular c-Src (Eliceiri et al., 1999; Zachary and Gliki, 2001) and phosphoinositide 3 kinase (PI3K)/Akt-dependent phosphorylation and, hence, activation, of endothelial nitric oxide synthase (eNOS) (Ackah et al., 2005; Papapetropoulos et al., 1997). Pharmacological inhibition of Src family protein tyrosine kinases (PTKs) reduces VEGF-induced phosphorylation of AJ components and vascular permeability (Eliceiri et al., 1999; Weis et al., 2004) suggesting that there is a crucial role for c-Src in regulating EC barrier function in response to activation of growth factor receptors.
By contrast, both VEGF-induced vascular leak and increases in blood flow can be blocked pharmacologically by inhibitors of NOS or its downstream targets, soluble guanylate cyclase or protein kinase G (Mayhan, 1999; Murohara et al., 1998; Papapetropoulos et al., 1997; Ziche et al., 1997). Moreover, a cell-permeable peptide derived from the endogenous inhibitor of eNOS function, caveolin, blocks edema formation and hydraulic conductivity in post-capillary venules that is induced by mustard oil, carrageenan, platelet-activating factor and VEGF (Bernatchez et al., 2005; Bucci et al., 2000; Hatakeyama et al., 2006). The inhibitor data are complemented by genetic studies showing that the loss of eNOS, but not other NOS isoforms, abrogates VEGF-induced permeability (Fukumura et al., 2001; Gratton et al., 2003a). The activity of eNOS can be regulated by phosphorylation by several proteins kinases such as AMP-activated protein kinase (AMPK), protein kinase A and Akt. The loss of Akt1 also reduces VEGF-stimulated permeability (Ackah et al., 2005), as well as inflammation and histamine-induced vascular permeability in vitro and in vivo, which, in the latter cases, are mediated by hypo-phosphorylation of VE-cadherin (Di Lorenzo et al., 2009).
Endothelial barrier function is also regulated by the tensile forces within the endothelial monolayer. Rho, Rac and Cdc42, members of the Rho GTPase family, are key regulators of the actin cytoskeleton; in particular Rho governs stress fiber formation, actomyosin bundles and focal adhesions, whereas Rac and Cdc42 have been shown to promote formation of lamellipodia and filopodia, respectively, in cultured cells when stimulated with a vasoactive compound (Ridley, 2001; Ridley and Hall, 1992; Ridley et al., 1992). Rho GTPases can also regulate endothelial cell contractility by controlling the phosphorylation state of myosin light chain (MLC), through the activation of MLC kinases, a Ca2+/CaM-dependent MLC kinase (MLCK) and the Rho-associated kinase (Rho-kinase) (Amano et al., 1996). However, how eNOS-derived NO regulates the mechanisms responsible for cytoskeletal alterations, EC barrier breakdown and vascular permeability remains elusive.
In the present study, we show that eNOS-derived NO regulates the rearrangement of the actin cytoskeleton and the integrity of endothelial barrier by affecting the adhesive properties of AJs. The loss of NO in ECs stabilizes cortical actin structures by increasing the interaction of VE-cadherin with TIAM1 at the cell junctions, leading to an augmentation of Rac activity, thereby promoting cortical actin assembly. Conversely, Rho activation, as well as Src-dependent VE-cadherin phosphorylation, was markedly attenuated in eNOS-deficient cells. The primacy of eNOS during permeability changes in vivo is illustrated by impaired VEGF-, histamine- and inflammation-driven permeability in mice harboring a non-phosphorylatable knock-in mutation of eNOS at S1176, a key residue necessary for eNOS activation. These data show that NO is a key molecule promoting endothelial cell permeability, linking the cytoskeleton to VE-cadherin phosphorylation through regulation of the endothelial-junction-associated Rac guanine-nucleotide-exchange factor (GEF) TIAM1.
Results
eNOS is required for VEGF-induced permeability in endothelial cells
Several studies have demonstrated that eNOS is essential for VEGF-mediated changes in microvascular permeability (Aramoto et al., 2004; Fukumura et al., 1997; Gratton et al., 2003a; Hatakeyama et al., 2006; Murohara et al., 1998). However, the precise mechanism by which eNOS modulates vascular leakage is not known. To address potential mechanisms, VEGF-induced changes in barrier function was assessed in human microvascular endothelial cells (HDEMCs) using a variety of approaches to interfere with eNOS function. Treatment of HDEMCs with eNOS small interfering RNA (siRNA), but not control siRNA resulted in a >80% reduction in eNOS protein levels (Fig. 1A,B). Treatment of ECs with VEGF resulted in the time-dependent activation of VEGFR2, MAPK42 and MAPK44 (hereafter MAPK42/44; also known as MAPK3 and MAPK1, and ERK1 and ERK2, respectively) and phosphorylation of eNOS on S1177 in control cells (Fig. 1A, left panel), and a reduction in the latter in eNOS knockdown cells (Fig. 1A, right panel). Next, VEGF-induced changes in trans-endothelial electrical resistance (TEER) were measured in eNOS-deficient cells. As expected, VEGF induced a drop in TEER followed by a recovery to baseline, consistent with transient changes in barrier function. Silencing of eNOS significantly attenuated VEGF-induced changes in barrier function compared to siRNA control ECs (Fig. 1C). Furthermore, inhibition of eNOS using a conventional NOS inhibitor, L-nitroarginine methyl ester (L-NAME), or the cell-permeable peptide cavtratin, which corresponds to the inhibitory domain of caveolin-1 (Bernatchez et al., 2005; Bucci et al., 2000), also significantly reduced the changes in TEER after VEGF administration (Fig. 1D,E). These data show that eNOS-derived NO promotes physiological changes in EC barrier function in vitro.
Fig. 1.

eNOS activity is crucial for VEGF-induced permeability in HDEMCs. (A) HDMECs with control or eNOS siRNA were stimulated with vehicle (control, 0 minutes) or VEGF (100 ng/ml) for 5, 10 and 30 minutes and the cell lysates were analyzed by western blotting for eNOS, P-eNOS1177, P-1175-VEGFR-2, VEGFR-2, P-MAPK42/44, MAPK42/44 (where P indicates the phosphorylated form). (B) Quantification of eNOS levels in HDEMCs by densitometry from four independent experiments and expressed relative to the amount of β-actin. The data shown represent the mean±s.e.m. from five individual experiments. *P<0.05 compared to HDEMCs with siRNA control. (C) TEER measurements of post-confluent HDEMCs transfected with control (black line) or eNOS (red line) siRNA expressed as fractional resistance (%) of the TEER basal values. HDEMCs were stimulated with VEGF (100 ng/ml; addition indicated by the arrow) and TEER measured overtime. *P<0.05 compared to HDEMCs with control siRNA. (D) TEER measurements of HDEMCs treated with L-NAME (1 mM) or vehicle followed by VEGF (100 ng/ml) stimulation. *P<0.05 compared to vehicle-treated HDEMCs. (E) TEER measurements of HDEMCs treated with the eNOS inhibitor cavtratin peptide (10 µM) or the peptide control (antennapedia internalization peptide, AP) for 1 hour, measured after VEGF stimulation for 5 minutes. *P<0.05 compared to AP-treated cells (n = 3). The data shown represent the mean±s.e.m. from three individual experiments.
VEGF-induced stress fiber formation is markedly reduced in the absence of NO
Following VEGF stimulation, changes in TEER are associated with the reorganization of actin filaments into dense stress fibers causing cell contraction and endothelial permeability (Soga et al., 2001; van Nieuw Amerongen et al., 2003). Thus, we examined whether the loss of eNOS would have any effect on VEGF-mediated actin cytoskeletal reorganization. As expected, F-actin (visualized with phalloidin staining) in contact-inhibited monolayers was predominantly localized at the cell periphery (Fig. 2A, panel a), and upon stimulation with VEGF (30 minutes), F-actin reorganized into stress fibers. Interestingly, F-actin reorganization was largely abrogated in HDMECs transfected with siRNA eNOS, which exhibited a distinct pattern of junction-associated belt of cortical actin (Fig. 2A, panel d compared to panel b). The persistence of cortical actin due to the loss of eNOS was confirmed in mouse lung endothelial cells (MLECs) isolated from wild-type (WT) and eNOS−/− mice. As seen in Fig. 2B, WT ECs responded to VEGF with the formation of stress fibers, and this effect was reduced in eNOS−/− ECs. Moreover, the necessity of eNOS for VEGF-induced changes in cytoskeletal reorganization was shown by the rescuing the eNOS knockout phenotype by adenoviral re-expression of eNOS (Ad-eNOS; Gratton et al., 2003b; Scotland et al., 2002) but not a control adenovirus encoding β-galactosidase (Ad-β-Gal; Fig. 2C). Finally, to rule out potential artifacts due to culturing ECs or interactions between ECs and the extracellular matrix, the formation of stress fibers was examined by ‘en face’ imaging of F-actin in endothelial layers of intact blood vessels isolated from WT or eNOS−/− mice. As shown in Fig. 2D, cortical F-actin was clearly delineated between individual ECs from intrapulmonary arteries isolated from WT and eNOS−/− mice. Consistent with cultured EC data, VEGF failed to induce a stress fiber re-organization in ECs of eNOS−/− blood vessels compared to WT endothelium. Collectively, these results demonstrate the importance of eNOS in mediating cytoskeletal rearrangements in ECs upon VEGF stimulation.
Fig. 2.

The loss of eNOS prevents VEGF-induced stress fiber formation in ECs in vitro and ECs of intrapulmonary arteries in situ. (A) HDEMCs with control or eNOS siRNA were stimulated with vehicle (basal) or VEGF (100 ng/ml) for 30 minutes, then fixed and stained with phalloidin to label F-actin (green) and nuclei (blue). (B) WT and eNOS−/− MLECs were stimulated with vehicle or VEGF (100 ng/ml) for 30 minutes and labeled as in A. (C) eNOS−/− MLECs were transduced with adenovirus encoding either control lac Z (Ad β-gal) or eNOS (Ad eNOS) at 100 MOI for 3 days before stimulation with vehicle or VEGF (100 ng/ml) and immunolabeled similarly to in A. These figures are representative of three separate experiments. (D) Intrapulmonary arteries isolated from WT or eNOS−/− mice were stimulated with vehicle or VEGF (100 ng/ml) for 15 or 30 minutes. Blood vessels were then fixed, cut open longitudinally and immunolabeled with phalloidin as described in A. These figures are representative of five animals from each group.
The loss of eNOS affects VEGF-mediated Rac and Rho activation
The Rho family of small GTPases, including RhoA, Rac1 and Cdc42, regulate the dynamics and structures of F-actin and therefore influence cell shape, EC adhesion and endothelial barrier function (Hall, 1998a; Hall, 1998b; Ridley, 2001; Wojciak-Stothard and Ridley, 2002). Because the loss of eNOS increased cortical actin and impaired VEGF-induced stress fiber formation, the activation of Rac was assessed using p21 activated kinase (PAK)-conjugated agarose beads (Benard et al., 1999). Following VEGF stimulation, maximal Rac activation in WT ECs was achieved within 2 minutes and dropped back down to basal levels at 10 minutes, whereas eNOS-deficient ECs exhibited increased basal as well as sustained Rac activation throughout the VEGF timecourse (Fig. 3A). Moreover, treatment of eNOS-depleted HDEMCs with a Rac inhibitor (NSC 23766) was able to disrupt the ring of cortical actin and partially restore stress fiber formation upon VEGF stimulation (Fig. 3C), suggesting that the loss of eNOS results in a hyperactive GTP-bound state of Rac, which cannot be further activated by VEGF.
Fig. 3.

eNOS deficiency enhances Rac activity while reducing Rho activity. (A) WT and eNOS−/− MLECs were stimulated with VEGF (100 ng/ml) for the indicated times and Rac activity was measured as described in the Materials and Methods. Total Rac and eNOS levels were determined in the whole cell lysates before GST–PAK incubation. The graph shows the densitometric ratio of active Rac to total Rac. The data are representative of three independent experiments. (B) WT and eNOS−/− MLEC were stimulated with VEGF (100 ng/ml) for the indicated times and cell lysates processed for Rho activity. Total Rho and eNOS levels were determined in the whole cell lysates before GST–Rhotekin incubation. The graph shows the densitometric of active Rho to total Rho. The data are representative of three independent experiments. (C) HDEMCs with control or eNOS siRNA were pre-incubated with Rac inhibitor (25 µM, 6 hours) and stimulated with VEGF (100 ng/ml) following immunolabeling of F-actin (green) and nuclei (blue). (D) Western blotting for P-T18/19 MLC2 and MLC2 in whole cell lysates from HDEMCs with eNOS or control siRNA stimulated with VEGF (100 ng/ml) for the indicated times. The data represent three independent experiments. (E) HDEMCs with eNOS or control siRNA were stimulated with vehicle or SNAP, an NO donor (100 µM, 1 hour) and immunolabeled for F-actin (green) and nuclei (blue). Images are representative of three experiments.
In addition to Rac, VEGF is known to activate RhoA in ECs, leading to the formation of stress fibers (Burridge and Wennerberg, 2004). Thus, we examined GTP-Rho levels in eNOS-deficient cells using GST–Rhotekin-conjugated agarose beads (Reid et al., 1996). Basal and VEGF-stimulated Rho activation was largely abrogated in eNOS−/− ECs compared to WT cells (Fig. 3B), suggesting that the defective stress fiber formation observed in eNOS−/− cells was due to inactivation of Rho. The infection of eNOS-depleted HDMECs with an adenovirus encoding the constitutively active form of Rho (Ad RhoL63) readily led to the formation of stress fibers in these cells compared to HDMECs treated with eNOS siRNA and infected with a control virus encoding Lac Z (Ad β-Gal) (supplementary material Fig. S1). This suggests that eNOS is upstream of Rho activation (Siddiqui et al., 2011) and that the loss of stress fiber formation due to eNOS deficiency can be restored by a downstream constitutively active Rho. Many studies have indicated that Rho, through its direct downstream effectors, Rho kinases, can phosphorylate myosin light chain (MLC) kinase (Amano et al., 1996) or inhibit MLC phosphatase, which results in increased MLC phosphorylation (Kimura et al., 1996). In addition, Rac-activated PAK can also phosphorylate MLC or myosin II (Kiosses et al., 1999; Zeng et al., 2000). Thus, we investigated whether the loss of eNOS, either through the decreased activation of Rho and/or hyperactivation of Rac, could affect MLC phosphorylation. As shown in the western blots in Fig. 3D, VEGF-induced phosphorylation of MLC was reduced in eNOS-depleted HDMECs compared to the control, suggesting a role of eNOS upstream of Rho and MLC activation in the VEGF-activated signaling cascade. The reduction in MLC phosphorylation in eNOS-deficient cells was confirmed by immunofluorescence labeling of phosphorylated MLC in WT and eNOS−/− cells treated or not with VEGF (supplementary material Fig. S2). The NO dependency of stress fiber formation was tested in eNOS-depleted HDMECs treated with the NO donor, S-nitroso-N-acetylpenicilamine (SNAP). As seen in Fig. 3E, SNAP induced actin filament formation in both control and eNOS-depleted HDMECs (Fig. 3E), suggesting that NO levels per se are able to induce the formation of stress fibers.
eNOS is required for VEGF-driven c-Src-dependent VE-cadherin phosphorylation
To assess the mechanisms underlying eNOS regulation of barrier function, we examined the relationship between eNOS and VE-cadherin, a well-documented pathway that is important for VEGF-induced EC barrier breakdown, (Esser et al., 1998). As shown in Fig. 4A, and quantified in Fig. 4B, treatment of HDMECs with VEGF induced the time-dependent change in VE-cadherin tyrosine phosphorylation, and the co-association of c-Src and eNOS in the VE-cadherin immunocomplex. Interestingly, VE-cadherin tyrosine phosphorylation and recruitment of c-Src was diminished in eNOS-depleted ECs, as well as in cells treated with L-NAME, using either an antibody against VE-cadherin or the anti-phosphorylated-tyrosine antibody PY20 to immunoprecipitate the complex (supplementary material Fig. S3A,B). However, the activation of c-Src (measured as P-418), its downstream target focal adhesion kinase (FAK P-861), VEGFR2 and MAPK phosphorylation in whole cell lysates were not affected by the lack of eNOS (Fig. 4C, with quantitative phosphoprotein:total protein ratios below and supplementary material Fig. S3C). Finally, the localization of c-Src was examined by immunofluorescence microscopy. As seen in Fig. 4D, VEGF-induced c-Src translocation to VE-cadherin-positive junctions was reduced HDMECs treated with eNOS siRNA (Fig. 4D, and quantified in Fig. 4E). Collectively, these results support the importance of eNOS-derived NO in VEGF-induced c-Src recruitment and phosphorylation of VE-cadherin.
Fig. 4.

eNOS is required for VEGF-driven c-Src-translocation to cellular junctions and VE-cadherin phosphorylation. (A) HDEMCs transfected with eNOS or control siRNA were serum starved and stimulated with VEGF for the indicated times. VE-cadherin immunoprecipitates were analyzed by immunoblotting against phosphorylated tyrosine (4G10, PY20 clone), VE-cadherin and also for co-immunoprecipitation of c-Src and eNOS proteins. (B) The amount of tyrosine-phosphorylated VE-cadherin was quantified by densitometry from four independent experiments and expressed as fold increase compared with untreated controls. (C) Western blot analysis on whole cell lysates (WCL) from HDEMCs treated as described above. The activation of additional pathways (P-419-Src, Src, P-861-FAK, FAK, P-p38, p38; where P indicates the phosphorylated form) was examined after VEGF (100 ng/ml) stimulation. Densitometric quantification of phosphorylated:total for each protein is shown below the blot. (D) Immunofluorescent co-staining of VE-cadherin (green) and c-Src (red) in HDEMCs transfected with eNOS or control siRNA, then serum starved and stimulated with VEGF (100 ng/ml) for 15 minutes. Scale bar: 20 µm. The images have been captured at a 0.3 µm slice thickness (z-stack) by using a Zeiss Axiovert epifluorescence microscope and a 63× oil immersion objective, following deconvolution by Openlab software (Improvision, Lexington, MA). Data are representative of at least three experiments. Scale bar: 30 µm. (E) Pearson's correlation analysis of VE-cadherin and Src colocalization to the plasma membrane.
NO regulates VEGF-induced F-actin assembly through localization of TIAM1 to cell junctions
The inter-relationship between Rho GTPases and VE-cadherin plays an important role in the regulation of endothelial barrier integrity. In confluent ECs, the VE-cadherin complex can initiate the activities of many Rho GTPases by engaging cytosolic Rho GEFs including TIAM1 and VAV2 (activators of Rac) as well as Rho GTPase-activating proteins (GAPs) such as p190RhoGAP (an inactivator of RhoA). In confluent ECs, TIAM1-induced Rac activation leads to the formation of F-actin belts at the cell periphery, promoting cadherin-mediated cell-to-cell adhesion (Lampugnani et al., 2002), as well as the partitioning of VE-cadherin molecules into detergent-resistant basolateral domains. Because the loss of eNOS enhanced the levels of Rac-dependent cortical actin assembly, we investigated whether NO influenced cell adhesion strength by determining the partitioning of VE-cadherin into detergent-resistant domains. WT and eNOS−/− ECs were grown to confluence on glass coverslips, and treated with 0.5% Triton X-100 and the detergent-resistant proteins stained with antibodies for VE-cadherin and TIAM1. Immunofluorescent staining showed an increase of VE-cadherin and TIAM1 colocalized in the insoluble junctional domains of eNOS−/− cells compared to that in WT ECs (Fig. 5A). Similar results of enhanced TIAM1 labeling at junctions were obtained in HDMECs treated with L-NAME (Fig. 5B), suggesting that the lack of NO strengthened the cell-to-cell adhesion in a TIAM1 and Rac-dependent fashion. To directly test whether the loss of NO leads to augmented TIAM1 junctional localization, we evaluated the fraction of TIAM1 that co-immunoprecipitated with VE-cadherin in HDMECs treated with L-NAME. The antibody for TIAM1 detects multiple proteins in western blots, therefore siRNA against TIAM1 was used to identify the specificity of the antibody. As shown in supplementary material Fig. S4, siRNA against TIAM1 reduced the levels of TIAM1 (molecular mass band of ∼220 kDa) but did not reduce the other bands. Inhibition of eNOS with L-NAME increased the formation of a VE-cadherin–TIAM1 immunocomplex (Fig. 5C). Similar experiments were performed in WT and eNOS−/− ECs in the absence of VEGF stimulation. As shown in Fig. 5D, the loss of eNOS increased the basal interaction with VE-cadherin, and VEGF-induced increases in VE-cadherin–TIAM1 immunocomplexes were not observed in eNOS−/− cells. Because the fine-tuning of Rho GTPase function regulates the balance of cortical actin versus stress fibers, and enhanced junctional VE-cadherin–TIAM1 could in principle drive Rac activation, we partially knocked down levels of TIAM1 in control and eNOS-depleted HDEMCs. Notably, reducing TIAM1 levels augmented VEGF-driven stress fiber formation in eNOS-depleted cells (Fig. 5E). These findings suggest that eNOS-derived NO regulates TIAM1-dependent activation of Rac.
Fig. 5.

NO regulates VEGF-induced F-actin formation through TIAM1 localization to cellular junctions. (A) WT and eNOS−/− MLECs were plated onto glass coverslips, and were treated with 0.5% Triton X-100-containing buffer for 5 minutes on ice. The soluble fraction was removed and the ECs were gently washed with PBS before fixation with paraformaldehyde 4% and immunofluorescent staining of VE-cadherin (red) and TIAM1 (green). (B) Immunofluorescent co-staining of VE-cadherin (green) and TIAM1 (red) in HDEMCs plated on glass-bottomed dishes (MatTek) transfected with eNOS siRNA or control siRNA and treated with 0.5% Triton X-100-containing buffer for 5 minutes on ice. The arrows show TIAM1 at the junctions. (C) HDEMCs were treated with L-NAME or vehicle, and VE-cadherin was immunoprecipitated from the cell lysates. Western blotting for TIAM1 and VE-cadherin was carried out on the whole cell lysates (WCL) and on the immunoprecipitates (IP). (D) Basal and VEGF-stimulated primary WT and eNOS−/− MLEC lysates were subjected to immunoprecipitation of VE-cadherin and immunoprecipitates were analyzed by immunoblotting against TIAM1. The amount of TIAM1 co-immunoprecipitated was quantified and is reported as a ratio to VE-cadherin (VEC). (E) Immunofluorescent staining of F-actin (green) in HDEMCs transfected with eNOS siRNA or control siRNA, or co-transfected with eNOS siRNA and TIAM1 siRNA, serum starved and stimulated with VEGF (100 ng/ml) for 30 minutes. Arrows indicate cortical actin; arrowheads indicate stress fibers formed upon VEGF stimulation. The images have been captured with Zeiss Axiovert epifluorescence microscope and a 63× oil immersion objective, using Openlab software (Improvision, Lexington, MA). Data are representative of three independent experiments.
The eNOS–TIAM1 pathway regulates VE-cadherin phosphorylation
TIAM1-induced Rac activation can promote cadherin-mediated cell-to-cell adhesion in epithelial cells (Hordijk et al., 1997; Sander et al., 1999; Sander et al., 1998), fibroblasts (Sander et al., 1999) and ECs (Lampugnani et al., 2002). The stabilization of mature junctions in ECs also correlates with reduced VE-cadherin phosphorylation (Lampugnani et al., 1997) and the loss of eNOS abrogated VEGF-induced changes in barrier function, stress fiber formation and VE-cadherin phosphorylation. To study whether TIAM1 contributes to NO-regulated VE-cadherin phosphorylation, HDEMCs were treated with control, eNOS siRNA or eNOS and TIAM1 siRNAs. As shown in Fig. 6A, the loss of eNOS reduced basal and VEGF-stimulated VE-cadherin tyrosine phosphorylation after 5 minutes of stimulation (lanes 3 and 4), whereas partial knockdown of TIAM1 in an eNOS-depleted background, enhanced VE-cadherin tyrosine phosphorylation (lane 6), as well as c-Src recruitment to cell junctions (Fig. 6B). Collectively these findings support the concept that eNOS regulates the dynamics of TIAM1–VE-cadherin interactions and the balance of cortical actin versus stress fibers required for reversible changes in EC barrier function. Mechanistically, our data are consistent with the model that NO upregulates Rho activation via p190RhoGEF tyrosine nitration, thereby increasing Rho activity and c-Src recruitment and VE-cadherin phosphorylation. Owing to competition between c-Src and TIAM1 for cadherins, in the absence of NO (and low Rho activity), TIAM1 is recruited to the VE-cadherin complex driving cortical actin localization. An additional possibility is that NO could directly nitrosylate TIAM1 reducing its interaction with VE-cadherin. However, as seen in supplementary material Fig. S4, we could not detect tyrosine nitration of TIAM1 in COS cells transfected with TIAM1 treated with the peroxynitrite-generating compound, SIN-1 (supplementary material Fig. S5).
Fig. 6.
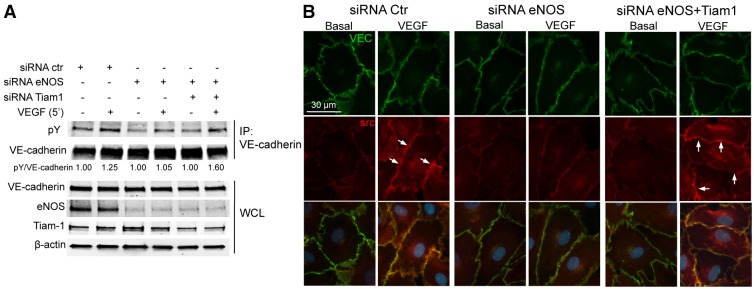
Partial knockdown of TIAM1, in eNOS-deficient cells, restores the phosphorylation of VE-cadherin and the recruitment of c-Src to cellular junctions upon VEGF stimulation. (A) HDEMCs transfected with siRNA control, or siRNA against eNOS or eNOS and TIAM1 were serum starved and stimulated with VEGF (100 ng/ml) for 10 minutes. VE-cadherin was immunoprecipitated and the associated proteins were analyzed by immunoblotting against phosphorylated tyrosine (pY; 4G10, PY20 clone) and VE-cadherin. The whole cell lysates (WCL) were analyzed by western blotting for VE-cadherin, eNOS, TIAM1 and β-actin expression before the incubation with anti-VE-cadherin antibody. (B) HDEMCs transfected with eNOS siRNA or control siRNA, or with eNOS siRNA and TIAM1 siRNA, were co-labeled for VE-cadherin (green) and c-Src (red) after before and incubation with VEGF (100 ng/ml) for 15 minutes. The images have been captured with a Zeiss Axiovert epifluorescence microscope and a 63× oil immersion objective, using Openlab software (Improvision, Lexington, MA). Arrows depict c-Src recruitment to the plasma membrane, which colocalizes with VE-cadherin. Data are representative of three independent experiments.
eNOS phosphorylation at S1176 is essential for agonist- and inflammation-induced changes in vascular permeability
Prior work has demonstrated that eNOS-derived NO is necessary for vascular leakage induced by VEGF and inflammatory irritants (Bucci et al., 2000; Fukumura et al., 2001; Gratton et al., 2003a; Murohara et al., 1998). The molecular mechanisms by which VEGF activates eNOS is through Ca2+-calmodulin and phosphorylation of eNOS on S1177 (equivalent to S1176 in the mouse), both mechanisms increasing the rate of electron transfer from the eNOS reductase to the oxygenase domain resulting in enhanced NO release (Sessa, 2004). The phosphorylation of eNOS at S1176 can be mediated by multiple kinases; however, in the context of angiogenesis, it is known that the serine/threonine kinase Akt1/PKB is the principal kinase because expression of eNOS S1176D, but not eNOS S1176A, knocked into the endogenous eNOS locus in mice, rescues the angiogenic defects in Akt1-deficient mice (Schleicher et al., 2009). To directly test whether this site of eNOS phosphorylation is necessary for vascular leakage, VEGF- and histamine-induced vascular permeability was assessed in vivo using a modified Miles assay. Intradermal injection of VEGF (Fig. 7A and images in Fig. 7B) and histamine (Fig. 7C), promoted the extravasation of Evan's blue (a tracer that binds albumin as index of protein leakage into tissue) in WT and eNOS S1176D mice suggesting that both agonists maximally phosphorylate this residue. In contrast, agonist-induced permeability was reduced in eNOS S1176A mice. Moreover, to assess whether this phosphosite in eNOS is crucial for edema and leukocyte infiltration to sites of inflammation, carrageenan was instilled into air pouches on the backs of mice. Carrageenan-induced edema and leukocyte infiltration was reduced in eNOS S1176A mice compared to WT and S1176D eNOS mice (Fig. 7D,E), demonstrating that eNOS phosphorylation at the S1176 plays a crucial role in vascular permeability and leukocyte infiltration.
Fig. 7.
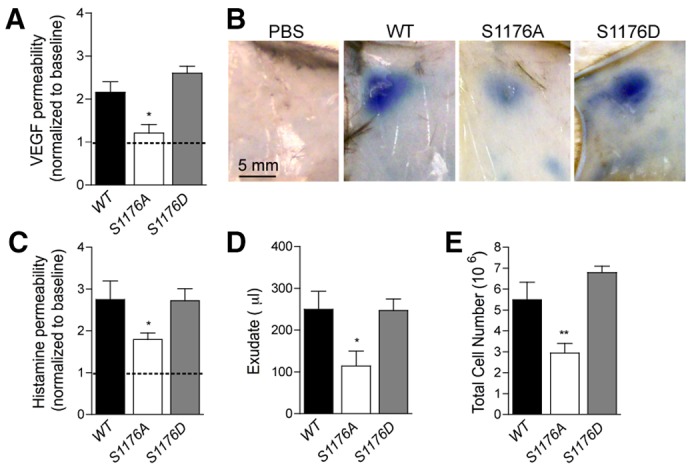
The phosphorylation of eNOS on S1176 is crucial for vascular permeability and inflammation in vivo. (A) VEGF-induced dermal vascular permeability was reduced in S1176A eNOS (loss of function) mice compared to WT and S1176D eNOS (gain of function) mice. The data are plotted as the VEGF:PBS ratio of individual mice. *P<0.05; n = 5 mice per group. (B) Representative images of Evan's blue leakage upon PBS or VEGF injection of the mice described in A. (C) Histamine-induced dermal vascular permeability was reduced in S1176A eNOS mice compared to WT and S1176D eNOS mice. (D,E) S1176A eNOS mice displayed a significant reduction of carrageenan-induced (D) exudate formation and (E) neutrophil recruitment into the air pouches 4 hours post-instillation compared to WT and S1176D eNOS mice (n = 5, per group). The data shown represent the mean±s.e.m. *P<0.05, **P<0.01 compared to WT group.
Discussion
The disruption of endothelial barrier function causes a marked increase in vascular permeability, a critical and initial event in the pathophysiology of many inflammatory diseases, including stroke, cancer and atherosclersosis. eNOS-derived NO controls vascular permeability in vivo through multiple actions, including modulation of blood flow in post capillary venules, protein kinase G activation and NO-mediated chemical modifications of junctional proteins. However, the relationship between NO, Rho GTPases and junctional permeability remains elusive. Here, we show that the eNOS-derived NO regulates VEGF-induced stress fiber formation and VE-cadherin phosphorylation by controlling the balance of Rho versus Rac activation. This conclusion is supported by in vitro data in human and murine ECs lacking eNOS and in ex vivo intact pulmonary arteries from WT and eNOS−/− mice, showing that the loss of NO stabilizes the cortical actin belt in a TIAM1–Rac-dependent manner and abrogates Rho-dependent F-actin formation upon VEGF stimulation. Moreover, a central role for NO in regulating VEGF-mediated downstream signaling is supported by experiments showing that the absence of NO reduces VEGF-induced phosphorylation of VE-cadherin by impairing Src recruitment to cell junctions, yet stimulates the recruitment of TIAM1 to the VE-cadherin complex. The partial knockdown of TIAM1 in the absence of NO re-establishes the formation of F-actin-rich stress fibers and Src-dependent VE-cadherin phosphorylation in response to VEGF, supporting the concept that NO controls endothelial barrier function by spatially regulating TIAM1 versus p190RhoGAP (see Fig. 8 for model).
Fig. 8.

Schematic view of VEGF-induced NO regulation of the VE-cadherin complex. In basal conditions, the accumulation of TIAM1 at the VE-cadherin complex in basal conditions is limited by eNOS-derived NO (left panel), while VEGF-induced Rho activation and Src-dependent VE-cadherin phosphorylation are promoted by the NO. The lack of NO increases the amount of VE-cadherin-bound TIAM1, leading to the formation of stable cortical actin in basal and VEGF-stimulated conditions (right panel), reduces stress fiber formation and VE-cadherin phosphorylation, with consequent improvement of endothelial barrier function.
The crucial role of NO in modulating vascular permeability has been extensively corroborated by many studies in vivo (Aramoto et al., 2004; Fukumura et al., 2001; Fukumura et al., 1997; Gratton et al., 2003a; Mayhan, 1999) and in vitro (Di Lorenzo et al., 2009; Lal et al., 2001). Recently, it was demonstrated that eNOS activation is augmented in ECs lacking the caveolae coat protein caveolin-1, and that eNOS hyperactivation results in nitration and impairment of p190RhoGAP-A function and increased Rho activation, leading to increased vascular permeability (Siddiqui et al., 2011). These findings are consistent with our model demonstrating that eNOS deficiency activates Rac, and reduces Rho activation and stress formation, in response to VEGF in vitro and in intact vessels. The eNOS and NO dependency of the response is apparent by experiments showing that reconstitution of eNOS or NO donors both promote cytoskeletal responses.
Of particular interest is the novel observation of cortical actin persistence at the cell periphery of eNOS−/− mouse lung endothelial cells (MLECs) and HDEMCs with siRNA against eNOS. This process is largely determined by Rho GTPases, and our data document that eNOS-derived NO fine-tunes this response in ECs. The persistence of cortical actin is reminiscent of ECs treated with the bioactive lipid sphingosine 1-phosphate (Garcia et al., 2001; Shikata et al., 2003b) or the microtubule-stabilizing drug paclitaxel (Verin et al., 2001), both of which enhance endothelial barrier function. Mechanistically, physiological concentrations of S1P result in preferential activation of Rac over Rho (Shikata et al., 2003a; Shikata et al., 2003b) and enhancement of cortical actin rings. Thus, our data are consistent with the idea that the loss of eNOS enhances endothelial barrier function or impedes VEGF-mediated vascular permeability.
It is widely recognized that there is a complex interaction between Rho family members during cytoskeletal remodeling upon growth factor activation or cell migration. Although some studies suggest a role for Rac upstream of Rho (Lee et al., 1999; Ridley, 2001; Wojciak-Stothard and Ridley, 2002), many others have shown that Rac activation can lead to Rho downregulation (Burridge and Wennerberg, 2004; Sander et al., 1999). Here, we showed that in WT MLECs, the activation of Rac at 2 minutes preceded the activation of Rho at 10 minutes after VEGF stimulation, when Rac activation had already begun to decrease, suggesting that Rac and Rho act in concert in mediating the stress fiber formation observable after 15–30 minutes. The loss of eNOS disrupted this fine balance and probably the lack of stress fiber formation was due in part to an inhibitory action of active Rac on Rho and in part by a direct effect on p190RhoGAP (Siddiqui et al., 2011).
The sustained activation of Rac by stable overexpression of the Rac-GEF TIAM1, results in increased cadherin-mediated cell-to-cell adhesion, cell spreading and downregulation of Rho activity. Interestingly, the expression of active Rho in cells overexpressing TIAM1 was able to restore the cell morphology, strengthening the concept of Rho being downstream of Rac (Sander et al., 1999). Our data demonstrate an increase of TIAM1 at the cell junctions in the absence of eNOS, resulting in stable cortical actin formation and Rho downregulation in response to VEGF. The partial knockdown of TIAM1 was able to rescue Rho-activation in response to VEGF, in agreement with the idea of Rho being downstream of Rac.
Previous studies have shown that the localization of c-Src at the cell membrane is necessary for its biological functions (Catling et al., 1993), and specifically that the translocation of c-Src from the cytoplasm to the plasma membrane upon PDGF stimulation was inhibited by cytochalasin D, an inhibitor of actin polymerization (Fincham et al., 1996). These studies support our data showing that partial knockdown of TIAM1 was able to restore the defective VEGF-induced translocation of c-Src at the AJ and VE-cadherin phosphorylation in the absence of eNOS. These findings suggest that NO influences F-actin formation to regulate VEGF-induced c-Src translocation at the junctional membrane and, therefore, the signaling cascade, leading to disruption of the endothelial barrier. Although it has been reported that NO is able to modulate c-Src activity through cysteine modifications (Rahman et al., 2010), our data showed that the lack of NO did not affect the VEGF activation of c-Src, as confirmed by the phosphorylation of its downstream substrate focal adhesion kinase (FAK) at Y861. This could be explained by the usage of different cell types and/or different agonists. However, future studies on the role of eNOS in the regulation of Rac GEFs might provide further insight into the modulation of VEGF-induced Src translocation to the membrane.
We have recently shown that phosphorylation of eNOS on S1176 appears crucial for the actions of Akt in promoting angiogenesis because eNOS S1176D, but not eNOS S1176A, alleles rescue the defects caused by loss of Akt1 in mice (Schleicher et al., 2009). However, the crucial role of this phosphorylation site in regulating barrier function was unknown. Here, we demonstrate that the increase in permeability induced by VEGF and histamine, as well as the acute inflammatory response to carrageenan, are markedly reduced in knock-in mice expressing the less-active eNOS S1176A compared to mice expressing the active form of the enzyme, eNOS S1176D. This suggests that this phosphorylation site of eNOS is crucial for its function during changes in in vivo permeability and inflammation.
Collectively our data show that endogenous eNOS-derived NO ‘fine tunes’ the levels of Rho GTPases by regulating VE-cadherin-mediated changes in junctional permeability. We show that pharmacological blockade of eNOS or the genetic loss of endogenous eNOS stabilizes cortical actin by sustained recruitment of TIAM1 to the VE-cadherin complex, thereby promoting Rac activation and AJ stabilization. This effect is reversible by NO, TIAM1–Rac inhibition and is physiologically relevant as revealed in eNOS knockin mice. The regulation of AJ dynamics strongly supports the autocrine role of NO in endothelial cells, separate from the canonical, paracrine role of eNOS in controlling blood flow and hemodynamics. The idea that eNOS-derived NO regulates junctional dynamics is supported by recent results showing that NO post-translationally nitrosylates β-catenin and p120-catenin promoting its dissociation from VE-cadherin (Marín et al., 2012; Thibeault et al., 2010), and tyrosine nitrates p190RhoGAP (Siddiqui et al., 2011) effects leading to enhanced permeability. Our data showing enhanced TIAM1–VE-cadherin-mediated stabilization of cortical actin explains why mice deficient in eNOS or mice harboring a S1176A allele exhibit reduced vascular leakage in vivo and sheds light on why anti-VEGF therapy reduces vascular permeability but promotes increased blood pressure in patients.
Materials and Methods
Animals
WT C57BL/6 and eNOS−/− mice were purchased from Jackson's laboratory (∼8–10 weeks of age). Knock-in mice carrying the S1176A and S1176D mutations in the endogenous eNOS locus were generated as previously described (Schleicher et al., 2009). Congenic male, 8- to 14-week-old S1176D eNOS, S1176A eNOS and control littermates were used. All experiments were approved by the Institutional Animal Care and Use Committee of Yale University.
Carrageenan air-pouch
Air-pouches were generated as previously described (Di Lorenzo et al., 2009). On day 6, 0.5 ml of carrageenan suspension (1% w/v) in sterile saline was injected into the pouch. Exudates and leukocytes were recovered from the pouches at 4 hours post-carrageenan. Migrated leukocytes, mainly neutrophils, were counted by use of Trypan Blue, and the total volume of exudate was measured.
Cell culture
Primary human dermal microvascular endothelial cells (HDEMCs) were cultured in EGM2-MV growth medium (Cambrex) as previously described (Di Lorenzo et al., 2009) and were used between passages four and eight for the experiments described. Eco-Pack2-mT packaging cells producing retroviruses containing the Polyoma middle T-antigen (Primo et al., 2000), were cultured in high glucose DMEM with 10% FBS. Mouse lung endothelial cells (MLECs), isolated from WT C57BL/6 or congenic eNOS−/− mice, were immortalized by the Polyoma middle T-antigen and cultured in EBM-2/EGM-2 MV (Cambrex) supplemented with 4 mM L-glutamine, penicillin-streptomycin, 100 mg/ml heparin and 15% heat-inactivated FBS, in 37°C and 5% CO2. Immortalized MLECs were used between passages 3 and 12.
Immunofluorescent staining in cultured cells
HDEMCs treated with eNOS and control siRNA, or WT and eNOS−/− MLECs were grown to confluence, serum starved with 0.15% FBS/EBM-2 overnight and stimulated with 100 ng/ml VEGF (Genentech) for the indicated time. In some experiments, ECs were treated with vehicle, Rac inhibitor (50 µM, 6 h, Calbiochem) or SNP (Sigma). Cells were washed with PBS and fixed in 3.7% paraformaldehyde for 10 minutes, blocked with 5% BSA in PBS for 10 minutes, permeabilized with 0.2% Triton X-100 in PBS for 5 minutes, and incubated overnight with antibodies against VE-cadherin (1∶200, Santa Cruz Biotechnology for HDEMCs; 1∶200, Pharmingen for MLECs) and Src (Cell Signaling). Appropriate secondary antibodies, conjugated to Alexa-Fluor-594 or Alexa-Fluor-488 (1∶200, Molecular Probes) were applied and Alexa-Fluor-488-labeled phalloidin (1∶50, Molecular Probes) was used to visualize the F-actin. DAPI was used to visualize nuclei. Images were captured with a Zeiss Axiovert epifluorescence microscope followed by deconvolution using the Openlab software (Improvision, Lexington, MA). Images in Fig. 2E and Fig. 6B have been captured at a 0.3 µm slice thickness (z-stack) by using a Zeiss Axiovert epifluorescence microscope and a 63× oil immersion objective, following deconvolution by Openlab software (Improvision, Lexington, MA).
In situ phalloidin labeling of F-actin in whole blood vessels
Intrapulmonary arteries from WT or eNOS−/− mice were dissected, cut open longitudinally, pinned with surgical needles and incubated in serum-free DMEM for 16 hours at 37°C to quiesce the blood vessels. Specimens were stimulated with 100 ng/ml VEGF (Genentech) for 15 or 30 minutes and immediately fixed with 4% paraformaldehyde (15 minutes, 4°C). After permeabilization with 0.3% Triton X-100 (30 minutes at room temperature), Alexa-Fluor-488-labeled phalloidin (1∶100, Molecular Probes) was used for ‘en face’ staining of F-actin (15 minutes at room temperature). Fluorescence images of actin in the endothelium were captured using a Zeiss microscope and analyzed using Openlab software.
In vitro permeability assay
The barrier function of HDEMCs was assayed by measuring the resistance of a cell-covered electrode by using an ECIS instrument (Applied BioPhysics). An 8W10E plate was incubated for 15 minutes with L-cysteine (10 mM) solution, followed by 0.1% gelatin for 30 minutes. ECs were plated on the electrode at 4×103 cells/well. The day after, ECs were transfected with small interference RNA (siRNA) for eNOS or control siRNA. After 72 hours, HDMECs were starved overnight in 0.15% FBS/EBM-2 and stimulated with VEGF (100 nM). In another set of experiments, HDEMCs grown on an 8W10E plate were starved overnight in 0.15% FBS/EBM-2 with L-NAME (1 mM) or vehicle, and stimulated with VEGF (100 ng/ml). Resistance was monitored and expressed as the fractional resistance of the basal value. In a further set of experiments, HDEMCs were plated in fibronectin-coated in vitro permeability transwells (0.45 mm, BD Discovery Labware) were treated with 10 µM control peptide (AP) or 10 µM cavtratin (AP-Cav) saline in 0.15% FBS/EBM-2. Transwells were subjected to transmonolayer resistance (TMR) measurement by placing EVOM voltohmmeter (World Precision Instruments) electrodes above and below the cell monolayer. Resistance readings were taken at 0, 5 and 30 minutes after VEGF stimulation for the peptide- or drug-treated wells, corrected for background and total transwell surface area, and expressed as the percentage decrease in resistance from baseline (time = 0).
Western blotting
WT and eNOS−/− MLECs, HDEMCs treated with control or eNOS siRNA, or treated with L-NAME (1 mM) were cultured for 72–96 hours before quiescing in 0.15% FBS/EBM-2 for 6–8 hours before stimulation with 100 ng/ml of VEGF (Genentech) at different time points. Cells were lysed in modified RIPA buffer containing 50 mM Tris-HCl pH 7.4, 0.1 mM EGTA, 0.1 mM EDTA, 1% NP-40, 0.1% SDS, 0.1% DOC, 20 mM NaF, 1 mM Na4P2O7, 1 mM Pefabloc SC, 1 mM sodium orthovanadate and protease inhibitor cocktail (Roche Diagnostics). Soluble proteins were resolved by SDS-PAGE and western blotted for P-1177-eNOS, P-1175-VEGFR2, VEGFR2, P-416-Src, Src, FAK, MLC, P-Thr18/Ser19-MLC2 (Cell Signaling), P-861-FAK (Invitrogen), eNOS, HSP90 (BD Transduction Laboratories), β-actin (Sigma), MAPK42/44, P-MAPK42/44, VEGFR2, phosphotyrosine (4G10, Millipore) or VE-cadherin (Santa Cruz Biotechnology). The secondary antibodies used for all western blots were conjugated directly to infrared fluorescent dyes (Alexa-Fluor-780 from Molecular Probes or IRDye800 from Li-Cor) to allow direct detection on the Odyssey® Infrared Imaging System (Li-Cor). For immunoprecipitations, cells were lysed in a buffer containing 10 mM HEPES-NaOH pH 7.4, 100 mM NaCl, 50 mM β-glycerophosphate, 2 mM MgCl2, 5 mM EGTA, 5 mM EDTA, 1% Igepal (Sigma), 1 mM orthovanadate, 1 mM NaF and protease inhibitors (Roche). Clarified cell extracts were incubated with 2 µg of antibody against VE-cadherin (Santa Cruz Biotechnology) or phosphotyrosine (PY20). Antigen–antibody complexes were collected using protein-G–Sepharose beads (Sigma). SDS-PAGE and immunoblotting was performed as described above, and used the antibody against VE-cadherin (Santa Cruz Biotechnology) or phophotyrosine (PY20, 4G10).
Rac and Rho assay
MLECs were grown to confluence, quiesced in serum-free EBM-2 for 36–48 hours and stimulated with 100 ng/ml VEGF for the indicated amount of time. After washing, cells were lysed with Mg2+ lysis buffer (MLB, Upstate) containing 25 mM HEPES pH 7.5, 250 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1% Igepal CA-630 and 10% glycerol supplemented with 20 mM NaF, 1 µg/ml Leupeptin, 1 µg/ml Aprotinin and 1 mM sodium orthovanadate. After centrifugation (14,000 rpm, 10 minutes, 4°C) of cell lysates, 250 mg of protein was pre-cleared with 50 ml of 50% GST–Sepharose slurry for 10 minutes at 4°C. Subsequently, 10 mg of GST–PAK-1 agarose beads were added to all sample lysates and incubated at 4°C for 1 hour. The samples were resolved on SDS-PAGE and western blotted for Rac protein. Lysates before GST–PAK-1 pulldown were also western blotted for total amount of Rac. In a similar manner, the Rho assay MLECs were grown to confluence, quiesced in serum-free EBM-2 for 36–48 hours and stimulated with 100 ng/ml VEGF for the indicated amount of time and lysed in Rho lysis buffer containing 50 mM Tris-HCl (pH 7.2), 500 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.1% SDS, 0.5% DOC, 20 mM NaF, 1 µg/ml Leupeptin, 1 µg/ml Aprotinin and 1 mM sodium orthovanadate. The lysates (500 mg of protein from each sample) were pre-incubated with 100 nM GTPgS (30 minutes, 25°C) to label the maximal amount of GTP bound Rho prior to GST-Rhotekin agarose beads (30 minutes, 4°C), followed by wash with Rho wash buffer containing 50 mM Tris-HCl (pH 7.2), 150 mM NaCl, 10 mM MgCl2 and 1% Triton X-100 with protease and phosphatase inhibitors mentioned above. Samples were separated by SDS-PAGE and western blotting for Rho protein. Lysates before GST-Rho pulldown were also western blotted for total amount of Rho.
Supplementary Material
Acknowledgments
We thank Mr. Roger Babbitt for skilled technical assistance. The authors have no competing financial interests.
Footnotes
Author contributions
A.D.L., M.I.L., T.M., S.L.E., M.S., M.K., J.Y. performed critical experiments and analyzed data; P.L.H. generated the mice; A.D.L., M.I.L., P.L.H. and W.C.S. conceptualized the study and wrote the manuscript.
Funding
This work was supported by the American Heart Association (grant to A.D.L.); the National Institutes of Health [grant numbers R01 HL64793, R01 HL61371, R01 HL081190, RO1 HL096670, P01 HL1070205 to W.C.S., RO1 DK082600 to Y.I.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.115972/-/DC1
References
- Ackah E., Yu J., Zoellner S., Iwakiri Y., Skurk C., Shibata R., Ouchi N., Easton R. M., Galasso G., Birnbaum M. J. et al. (2005). Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J. Clin. Invest. 115, 2119–2127 10.1172/JCI24726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K. (1996). Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 10.1074/jbc.271.34.20246 [DOI] [PubMed] [Google Scholar]
- Andriopoulou P., Navarro P., Zanetti A., Lampugnani M. G., Dejana E. (1999). Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler. Thromb. Vasc. Biol. 19, 2286–2297 10.1161/01.ATV.19.10.2286 [DOI] [PubMed] [Google Scholar]
- Angelini D. J., Hyun S. W., Grigoryev D. N., Garg P., Gong P., Singh I. S., Passaniti A., Hasday J. D., Goldblum S. E. (2006). TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. 291, L1232–L1245 10.1152/ajplung.00109.2006 [DOI] [PubMed] [Google Scholar]
- Aramoto H., Breslin J. W., Pappas P. J., Hobson R. W., 2nd, Durán W. N. (2004). Vascular endothelial growth factor stimulates differential signaling pathways in in vivo microcirculation. Am. J. Physiol. 287, H1590–H1598 10.1152/ajpheart.00767.2003 [DOI] [PubMed] [Google Scholar]
- Benard V., Bohl B. P., Bokoch G. M. (1999). Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 274, 13198–13204 10.1074/jbc.274.19.13198 [DOI] [PubMed] [Google Scholar]
- Bernatchez P. N., Bauer P. M., Yu J., Prendergast J. S., He P., Sessa W. C. (2005). Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc. Natl. Acad. Sci. USA 102, 761–766 10.1073/pnas.0407224102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broermann A., Winderlich M., Block H., Frye M., Rossaint J., Zarbock A., Cagna G., Linnepe R., Schulte D., Nottebaum A. F. et al. (2011). Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J. Exp. Med. 208, 2393–2401 10.1084/jem.20110525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci M., Gratton J. P., Rudic R. D., Acevedo L., Roviezzo F., Cirino G., Sessa W. C. (2000). In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 6, 1362–1367 10.1038/82176 [DOI] [PubMed] [Google Scholar]
- Burridge K., Wennerberg K. (2004). Rho and Rac take center stage. Cell 116, 167–179 10.1016/S0092-8674(04)00003-0 [DOI] [PubMed] [Google Scholar]
- Catling A. D., Wyke J. A., Frame M. C. (1993). Mitogenesis of quiescent chick fibroblasts by v-Src: dependence on events at the membrane leading to early changes in AP-1. Oncogene 8, 1875–1886 [PubMed] [Google Scholar]
- Dejana E. (2004). Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5, 261–270 10.1038/nrm1357 [DOI] [PubMed] [Google Scholar]
- Di Lorenzo A., Fernández-Hernando C., Cirino G., Sessa W. C. (2009). Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc. Natl. Acad. Sci. USA 106, 14552–14557 10.1073/pnas.0904073106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri B. P., Paul R., Schwartzberg P. L., Hood J. D., Leng J., Cheresh D. A. (1999). Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 4, 915–924 10.1016/S1097-2765(00)80221-X [DOI] [PubMed] [Google Scholar]
- Esser S., Lampugnani M. G., Corada M., Dejana E., Risau W. (1998). Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 111, 1853–1865 [DOI] [PubMed] [Google Scholar]
- Feng D., Nagy J. A., Pyne K., Hammel I., Dvorak H. F., Dvorak A. M. (1999). Pathways of macromolecular extravasation across microvascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation 6, 23–44 [PubMed] [Google Scholar]
- Ferrara N., Gerber H. P., LeCouter J. (2003). The biology of VEGF and its receptors. Nat. Med. 9, 669–676 10.1038/nm0603-669 [DOI] [PubMed] [Google Scholar]
- Fincham V. J., Unlu M., Brunton V. G., Pitts J. D., Wyke J. A., Frame M. C. (1996). Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J. Cell Biol. 135, 1551–1564 10.1083/jcb.135.6.1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D., Yuan F., Endo M., Jain R. K. (1997). Role of nitric oxide in tumor microcirculation. Blood flow, vascular permeability, and leukocyte-endothelial interactions. Am. J. Pathol. 150, 713–725 [PMC free article] [PubMed] [Google Scholar]
- Fukumura D., Gohongi T., Kadambi A., Izumi Y., Ang J., Yun C. O., Buerk D. G., Huang P. L., Jain R. K. (2001). Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 98, 2604–2609 10.1073/pnas.041359198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J. G., Liu F., Verin A. D., Birukova A., Dechert M. A., Gerthoffer W. T., Bamberg J. R., English D. (2001). Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 108, 689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratton J. P., Lin M. I., Yu J., Weiss E. D., Jiang Z. L., Fairchild T. A., Iwakiri Y., Groszmann R., Claffey K. P., Cheng Y. C. et al. (2003a). Selective inhibition of tumor microvascular permeability by cavtratin blocks tumor progression in mice. Cancer Cell 4, 31–39 10.1016/S1535-6108(03)00168-5 [DOI] [PubMed] [Google Scholar]
- Gratton J. P., Yu J., Griffith J. W., Babbitt R. W., Scotland R. S., Hickey R., Giordano F. J., Sessa W. C. (2003b). Cell-permeable peptides improve cellular uptake and therapeutic gene delivery of replication-deficient viruses in cells and in vivo. Nat. Med. 9, 357–362 10.1038/nm835 [DOI] [PubMed] [Google Scholar]
- Hall A. (1998a). G proteins and small GTPases: distant relatives keep in touch. Science 280, 2074–2075 10.1126/science.280.5372.2074 [DOI] [PubMed] [Google Scholar]
- Hall A. (1998b). Rho GTPases and the actin cytoskeleton. Science 279, 509–514 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- Hatakeyama T., Pappas P. J., Hobson R. W. I. I., Boric M. P., Sessa W. C., Durán W. N. (2006). Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J. Physiol. 574, 275–281 10.1113/jphysiol.2006.108175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk P. L., ten Klooster J. P., van der Kammen R. A., Michiels F., Oomen L. C., Collard J. G. (1997). Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science 278, 1464–1466 10.1126/science.278.5342.1464 [DOI] [PubMed] [Google Scholar]
- Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K. et al. (1996). Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273, 245–248 10.1126/science.273.5272.245 [DOI] [PubMed] [Google Scholar]
- Kiosses W. B., Daniels R. H., Otey C., Bokoch G. M., Schwartz M. A. (1999). A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147, 831–844 10.1083/jcb.147.4.831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal B. K., Varma S., Pappas P. J., Hobson R. W., II and Durán W. N. (2001). VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc. Res. 62, 252–262 10.1006/mvre.2001.2338 [DOI] [PubMed] [Google Scholar]
- Lampugnani M. G., Corada M., Andriopoulou P., Esser S., Risau W., Dejana E. (1997). Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J. Cell Sci. 110, 2065–2077 [DOI] [PubMed] [Google Scholar]
- Lampugnani M. G., Zanetti A., Breviario F., Balconi G., Orsenigo F., Corada M., Spagnuolo R., Betson M., Braga V., Dejana E. (2002). VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol. Biol. Cell 13, 1175–1189 10.1091/mbc.01-07-0368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. H., Lee S. H., Kim D., Rhee S., Kim C., Chung C. H., Kwon H., Kang M. S. (1999). Promotion of skeletal muscle differentiation by K252a with tyrosine phosphorylation of focal adhesion: a possible involvement of small GTPase Rho. Exp. Cell Res. 252, 401–415 10.1006/excr.1999.4648 [DOI] [PubMed] [Google Scholar]
- Marín N., Zamorano P., Carrasco R., Mujica P., González F. G., Quezada C., Meininger C. J., Boric M. P., Durán W. N., Sánchez F. A. (2012). S-Nitrosation of β-catenin and p120 catenin: a novel regulatory mechanism in endothelial hyperpermeability. Circ. Res. 111, 553–563 10.1161/CIRCRESAHA.112.274548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhan W. G. (1999). VEGF increases permeability of the blood-brain barrier via a nitric oxide synthase/cGMP-dependent pathway. Am. J. Physiol. 276, C1148–C1153 [DOI] [PubMed] [Google Scholar]
- Murohara T., Asahara T., Silver M., Bauters C., Masuda H., Kalka C., Kearney M., Chen D., Symes J. F., Fishman M. C. et al. (1998). Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest. 101, 2567–2578 10.1172/JCI1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A., García-Cardeña G., Madri J. A., Sessa W. C. (1997). Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 100, 3131–3139 10.1172/JCI119868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primo L., Roca C., Ferrandi C., Lanfrancone L., Bussolino F. (2000). Human endothelial cells expressing polyoma middle T induce tumors. Oncogene 19, 3632–3641 10.1038/sj.onc.1203708 [DOI] [PubMed] [Google Scholar]
- Rahman M. A., Senga T., Ito S., Hyodo T., Hasegawa H., Hamaguchi M. (2010). S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J. Biol. Chem. 285, 3806–3814 10.1074/jbc.M109.059782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid T., Furuyashiki T., Ishizaki T., Watanabe G., Watanabe N., Fujisawa K., Morii N., Madaule P., Narumiya S. (1996). Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J. Biol. Chem. 271, 13556–13560 10.1074/jbc.271.23.13556 [DOI] [PubMed] [Google Scholar]
- Ridley A. J. (2001). Rho family proteins: coordinating cell responses. Trends Cell Biol. 11, 471–477 10.1016/S0962-8924(01)02153-5 [DOI] [PubMed] [Google Scholar]
- Ridley A. J., Hall A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399 10.1016/0092-8674(92)90163-7 [DOI] [PubMed] [Google Scholar]
- Ridley A. J., Paterson H. F., Johnston C. L., Diekmann D., Hall A. (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401–410 10.1016/0092-8674(92)90164-8 [DOI] [PubMed] [Google Scholar]
- Sander E. E., van Delft S., ten Klooster J. P., Reid T., van der Kammen R. A., Michiels F., Collard J. G. (1998). Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 143, 1385–1398 10.1083/jcb.143.5.1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander E. E., ten Klooster J. P., van Delft S., van der Kammen R. A., Collard J. G. (1999). Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 147, 1009–1022 10.1083/jcb.147.5.1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher M., Yu J., Murata T., Derakhshan B., Atochin D., Qian L., Kashiwagi S., Di Lorenzo A., Harrison K. D., Huang P. L. et al. (2009). The Akt1-eNOS axis illustrates the specificity of kinase-substrate relationships in vivo. Sci. Signal. 2, ra41 10.1126/scisignal.2000343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotland R. S., Morales-Ruiz M., Chen Y., Yu J., Rudic R. D., Fulton D., Gratton J. P., Sessa W. C. (2002). Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ. Res. 90, 904–910 10.1161/01.RES.0000016506.04193.96 [DOI] [PubMed] [Google Scholar]
- Sessa W. C. (2004). eNOS at a glance. J. Cell Sci. 117, 2427–2429 10.1242/jcs.01165 [DOI] [PubMed] [Google Scholar]
- Shasby D. M., Ries D. R., Shasby S. S., Winter M. C. (2002). Histamine stimulates phosphorylation of adherens junction proteins and alters their link to vimentin. Am. J. Physiol. 282, L1330–L1338 [DOI] [PubMed] [Google Scholar]
- Shikata Y., Birukov K. G., Birukova A. A., Verin A., Garcia J. G. (2003a). Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J. 17, 2240–2249 10.1096/fj.03-0198com [DOI] [PubMed] [Google Scholar]
- Shikata Y., Birukov K. G., Garcia J. G. (2003b). S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J. Appl. Physiol. 94, 1193–1203 [DOI] [PubMed] [Google Scholar]
- Siddiqui M. R., Komarova Y. A., Vogel S. M., Gao X., Bonini M. G., Rajasingh J., Zhao Y. Y., Brovkovych V., Malik A. B. (2011). Caveolin-1-eNOS signaling promotes p190RhoGAP-A nitration and endothelial permeability. J. Cell Biol. 193, 841–850 10.1083/jcb.201012129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga N., Namba N., McAllister S., Cornelius L., Teitelbaum S. L., Dowdy S. F., Kawamura J., Hruska K. A. (2001). Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp. Cell Res. 269, 73–87 10.1006/excr.2001.5295 [DOI] [PubMed] [Google Scholar]
- Thibeault S., Rautureau Y., Oubaha M., Faubert D., Wilkes B. C., Delisle C., Gratton J. P. (2010). S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol. Cell 39, 468–476 10.1016/j.molcel.2010.07.013 [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen G. P., Koolwijk P., Versteilen A., van Hinsbergh V. W. (2003). Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler. Thromb. Vasc. Biol. 23, 211–217 10.1161/01.ATV.0000054198.68894.88 [DOI] [PubMed] [Google Scholar]
- Verin A. D., Birukova A., Wang P., Liu F., Becker P., Birukov K., Garcia J. G. (2001). Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. Am. J. Physiol. 281, L565–L574 [DOI] [PubMed] [Google Scholar]
- Weis W. I., Nelson W. J. (2006). Re-solving the cadherin-catenin-actin conundrum. J. Biol. Chem. 281, 35593–35597 10.1074/jbc.R600027200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis S., Cui J., Barnes L., Cheresh D. (2004). Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J. Cell Biol. 167, 223–229 10.1083/jcb.200408130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak-Stothard B., Ridley A. J. (2002). Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 39, 187–199 10.1016/S1537-1891(03)00008-9 [DOI] [PubMed] [Google Scholar]
- Zachary I., Gliki G. (2001). Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc. Res. 49, 568–581 10.1016/S0008-6363(00)00268-6 [DOI] [PubMed] [Google Scholar]
- Zeng Q., Lagunoff D., Masaracchia R., Goeckeler Z., Côté G., Wysolmerski R. (2000). Endothelial cell retraction is induced by PAK2 monophosphorylation of myosin II. J. Cell Sci. 113, 471–482 [DOI] [PubMed] [Google Scholar]
- Ziche M., Morbidelli L., Choudhuri R., Zhang H. T., Donnini S., Granger H. J., Bicknell R. (1997). Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J. Clin. Invest. 99, 2625–2634 10.1172/JCI119451 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.