

Summary
Keratin intermediate filament (IF) proteins are epithelial cell cytoskeletal components that provide structural stability and protection from cell stress, among other cellular and tissue-specific functions. Numerous human diseases are associated with IF gene mutations, but the function of keratins in the endocrine pancreas and their potential significance for glycaemic control are unknown. The impact of keratins on β-cell organisation and systemic glucose control was assessed using keratin 8 (K8) wild-type (K8+/+) and K8 knockout (K8−/−) mice. Islet β-cell keratins were characterised under basal conditions, in streptozotocin (STZ)-induced diabetes and in non-obese diabetic (NOD) mice. STZ-induced diabetes incidence and islet damage was assessed in K8+/+ and K8−/− mice. K8 and K18 were the predominant keratins in islet β-cells and K8−/− mice expressed only remnant K18 and K7. K8 deletion resulted in lower fasting glucose levels, increased glucose tolerance and insulin sensitivity, reduced glucose-stimulated insulin secretion and decreased pancreatic insulin content. GLUT2 localisation and insulin vesicle morphology were disrupted in K8−/− β-cells. The increased levels of cytoplasmic GLUT2 correlated with resistance to high-dose STZ-induced injury in K8−/− mice. However, K8 deletion conferred no long-term protection from STZ-induced diabetes and prolonged STZ-induced stress caused increased exocrine damage in K8−/− mice. β-cell keratin upregulation occurred 2 weeks after treatments with low-dose STZ in K8+/+ mice and in diabetic NOD mice, suggesting a role for keratins, particularly in non-acute islet stress responses. These results demonstrate previously unrecognised functions for keratins in β-cell intracellular organisation, as well as for systemic blood glucose control under basal conditions and in diabetes-induced stress.
Key words: Keratin, K8, Blood glucose, Diabetes model, Endocrine pancreas
Introduction
The cell cytoskeleton consists of three main structural proteins: actin filaments, microtubules and intermediate filaments (IFs). Keratin (K) proteins form the IFs of epithelial cells and exist as polymeric filaments by pairing of type I (K1-K8, K71-K86) and type II (K9-K28, K31-K40) keratin proteins (Schweizer et al., 2006). IFs are highly regulated by post-translational modifications such as phosphorylation and glycosylation during stress (Eriksson et al., 2009; Omary et al., 2009; Omary et al., 2006; Strnad et al., 2012).
Keratins provide mechanical stability to cells, but are nevertheless highly dynamic structures capable of quickly responding to their cellular environment. They are upregulated and/or modulated during and in recovery from stress (Pekny and Lane, 2007; Toivola et al., 2010). Moreover, keratins and other IF proteins are emerging as important components for regulating cell proliferation and apoptosis (Magin et al., 2007; Marceau et al., 2007), for targeting proteins and organelles in the cell (Styers et al., 2005; Toivola et al., 2005), for regulating translation (Kim and Coulombe, 2010) and in epithelial polarity (Grimm-Günter et al., 2009; Oriolo et al., 2007). Their importance in stress protection is demonstrated by more than 80 human diseases and numerous experimental disease models that are associated with IF mutations, including keratin mutations that cause liver disease and skin-blistering diseases (Omary et al., 2004; Strnad et al., 2012). For example, K8 or K18 mutations are associated with liver disease in ∼12% of patients but remain silent unless the liver is exposed to additional stress (Omary, 2009). This suggests that human keratin mutations could be susceptibility factors for several diseases that are affected by ‘second hits’, such as environmental factors (Omary et al., 2009). Keratin function, however, depends on both the types of keratins that are expressed and on the organ or cells in which they are expressed (Pan et al., 2013). This is exemplified by the remarkable stress tolerance of the exocrine pancreas to K8 or K18 mutation or knockout. Despite K8 and K18 being the major keratins in the liver and exocrine pancreas, keratin absence does not increase susceptibility to exocrine-specific pancreatitis induced by caerulein, choline-deficient diet (Toivola et al., 2000a) or acute coxsackievirus-induced pancreatits (Toivola et al., 2009).
It has been shown by immunofluorescence staining that K8 and K18 are the main keratins in the pancreatic islets (Bouwens, 1998), but relatively little is known about their specific functions in the endocrine pancreas under physiological conditions or during stress. Phenotypical or functional defects in the endocrine pancreas for K8 or K18 mutant or knockout mice under basal conditions have not been reported in the literature. However, transgenic mice that overexpress human K18 mutations that prevent K18 glycosylation (K18 S30A, S31A or S49A), are significantly more sensitive to streptozotocin (STZ)-induced death, probably due to multi-organ failure (Ku et al., 2010). Given that STZ is a β-cell toxin, this indicates that keratin mutations might also predispose the endocrine pancreas to injury in a similar way to that described for other epithelial cells. In addition to stress-related functions in the endocrine islets, keratins could also be involved in the regulation of insulin release from β-cells. K8 is strongly upregulated when β-cells are stimulated with glucose in vitro and ectopic expression of epidermal K1 in islets leads to loss of insulin vesicles and development of diabetes (Ahmed and Bergsten, 2005; Blessing et al., 1993; Schubart and Fields, 1984).
In this paper, islet keratins are characterised using immunofluorescence staining as well as biochemical methods. The analysis confirms that K8 and K18 is the predominant islet keratin pair, and moreover, reveals the presence of minute levels of K7, which has not been reported for mouse endocrine pancreas previously. The results presented here further suggest that keratins have multiple functions in the endocrine pancreas, given that differences are seen in β-cells of K8−/− (Krt8−/−) mice in insulin vesicle ultrastructure and GLUT2 localisation, as well as in the maintenance of glucose homeostasis. Moreover, responses to STZ-induced β-cell injury are modulated in K8−/− mice, and diabetes development in K8+/+ mice as well as in NOD mice is accompanied with marked K8 upregulation, indicating that keratins might have further relevance during conditions of hyperglycaemia and islet stress in diabetes.
Results
K8 and K18 are the major keratins in the K8+/+ endocrine pancreas, with remnant K7 and K18 in K8−/− mice
To determine the expression pattern of keratins in the endocrine pancreas before and after K8 genetic elimination, immunofluorescence staining was performed for K7, K8, K18, K19 and K20. K8 and K18 were the main type II and type I keratins, respectively, expressed in α- (supplementary material Fig. S1 for K8, K18 not shown) and β-cells of normal K8+/+ islets (Fig. 1A). In agreement with a previous study (Blessing et al., 1993), K8 and K18 immunofluorescence was lower in the islets than in the exocrine pancreas (Fig. 1A; Fig. 6A; supplementary material Fig. S1). Western blotting of islets confirmed the presence of K8 and K18 in K8+/+ islet lysates and comparatively very low levels of K18 in K8−/− islets (Fig. 1C). Purification of keratins by high-salt extraction (HSE) verified this keratin pattern, although K18 was barely detectable in the K8−/− HSE owing to the low keratin protein expression levels (Fig. 1C, lane 4). The Hsc70 loading control (Fig. 1C, lanes 1 and 2) verified that the low K18 level in K8−/− islets was not due to lower protein loading onto the gel. By confocal microscopy, small amounts of K18 were observed in the K8−/− endocrine cells (Fig. 1A,B), which were co-expressed with K7 in K8−/− as well as in K8+/+ islet β-cells (Fig. 1B). However, these minor levels of stunted K7 filaments could not be detected in the isolated islets by western blotting (data not shown). In the exocrine pancreas, K18 was found in K8−/− ducts but not in acinar cells (Fig. 1A). K20 was not detected in the islets (data not shown) and K19 was seen in occasional K8+/+ islets, but was not located to α- or β-cells (Fig. 1A), but rather to islet-associated occasional duct-like structures (Fig. 1A, inset) (Bertelli et al., 2001; Stosiek et al., 1990).
Fig. 1.
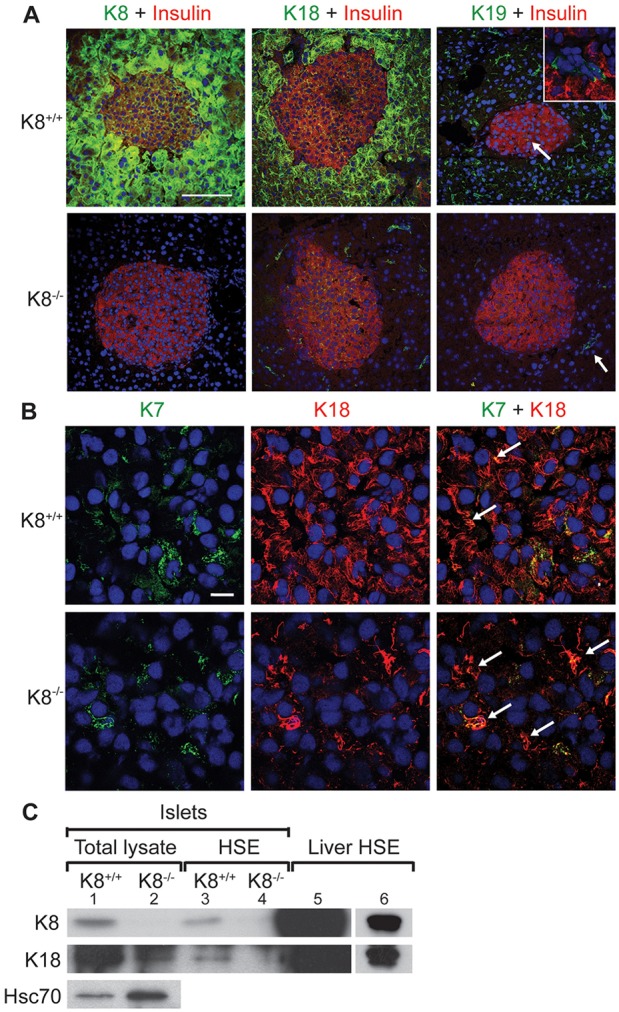
K8 and K18 are main keratins in the endocrine pancreas of K8+/+ mice but low levels of K7 also exist, allowing marginal keratin filament formation in K8−/− mice. (A) Immunostaining of pancreatic sections from K8+/+ and K8−/− mice for insulin (red), K8, K18 or K19 (green) and nuclei (blue). K8 and K18 are the most prominent keratins in the islets and some K19-positive cells are seen in K8+/+ islets. The white arrow in the upper panel points to K19-positive cells and the inset shows a higher magnification of the K19-positive and insulin-negative cells that form occasional duct-like structures within the islet in K8+/+ mice. K8−/− mice lack K8 but express K18 in islets and K19 in exocrine ducts (indicated by the white arrow in the lower panel). Scale bar: 100 µm. (B) Islet cells in pancreatic sections from K8+/+ and K8−/− mice stained for K7 (green), K18 (red) and nuclei (blue). Arrows show colocalisation of K7 and K18. Scale bar: 10 µm. (C) Western blot for K8, K18 and Hsc70 of islet total lysates (lane 1, K8+/+; lane 2, K8−/−), for K8 and K18 of HSE islet lysate (lane 3, K8+/+; lane 4, K8−/−) and liver HSE (lane 5) used as positive control. Lane 6 shows a shorter exposure of the K8 and K18 signals seen in lane 5.
Fig. 6.
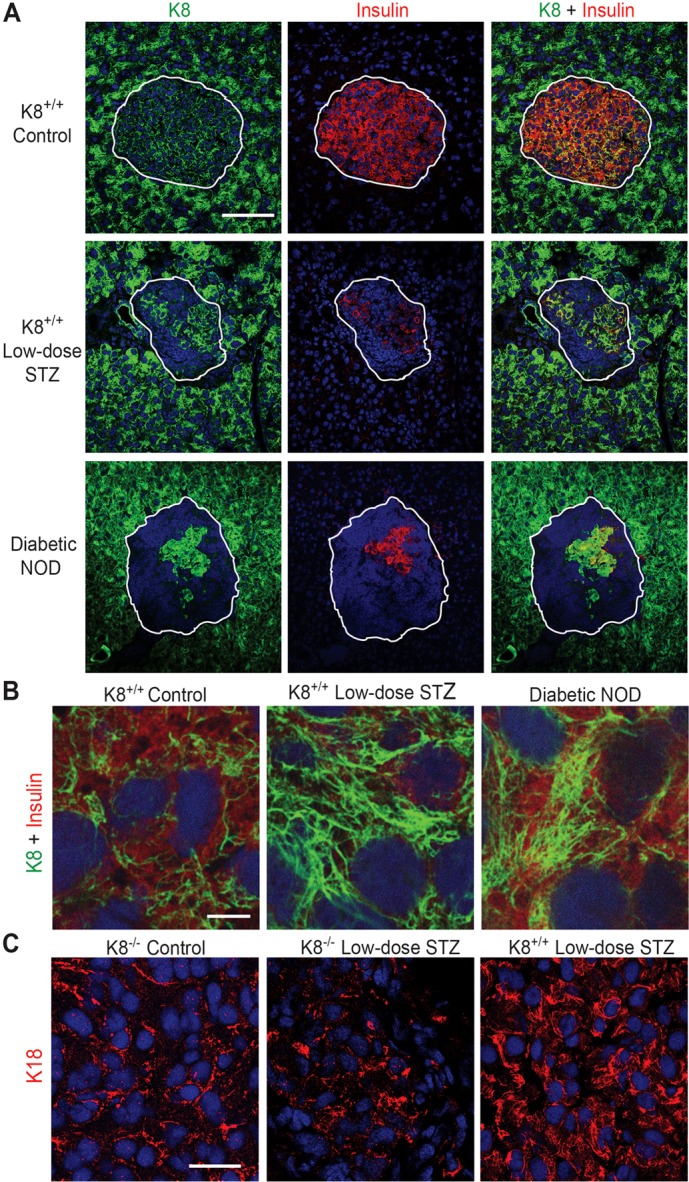
Keratins are upregulated in islets of diabetic NOD mice and low-dose STZ-treated mice. (A) Immunostaining for K8 (green) insulin (red) and nuclei (blue) of pancreatic sections from untreated K8+/+ FVB/n (control) mice, low-dose STZ-induced diabetic K8+/+ mice 2 weeks after STZ and diabetic NOD mice. Cells that coexpress insulin and keratin appear yellow. Islets are indicated with white lines. Note that K8 staining in diabetes models is increased in islet cells compared to untreated control mice, whereas K8 staining in the exocrine pancreas appears unchanged despite diabetes. Scale bar: 100 µm. (B) High magnification of β-cells in untreated K8+/+ mice, low-STZ treated K8+/+ mice and NOD mice stained for K8 (green), insulin (red) and nuclei (blue). Scale bar: 5 µm. (C) Staining for K18 (green) and nuclei (blue) from endocrine pancreas from untreated K8−/− control, low-dose STZ-treated K8−/− and low-dose STZ-treated K8+/+ mice. Scale bar: 20 µm.
K8 absence affects blood glucose regulation and disrupts β-cell ultrastructure
If K8 plays a significant role in islet function or in glycaemic control systemically, it might be reflected in parameters related to blood glucose regulation after K8 inactivation. The average fasting blood glucose level was lower in K8−/− (4.3 mmol/l) than in K8+/+ (5.4 mmol/l) mice (Fig. 2A) and glucose tolerance, when challenged with 2 g/kg of body weight glucose (Fig. 2B), was increased in K8−/− mice. Moreover, insulin administration (0.75 U/kg of body weight) resulted in lower blood glucose and delayed recovery from hypoglycaemia in K8−/− mice compared to K8+/+ (Fig. 2C). Insulin injections frequently caused visible signs of hypoglycaemia such as tremor and fatigue in K8−/− mice, which were not observed in K8+/+ mice. Fasting serum insulin levels did not differ between K8+/+ and K8−/− mice, but the K8−/− mouse insulin response to glucose stimulation was reduced, with no increase in serum insulin levels 10 minutes after glucose stimulation in contrast to K8+/+ mice, which showed a significant increase (Fig. 2D).
Fig. 2.
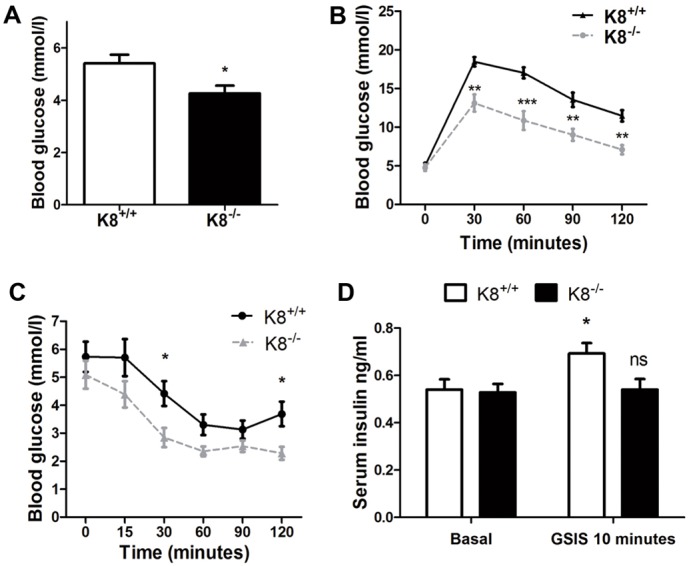
Altered blood glucose regulation in K8−/− mice. (A) Blood glucose levels are shown after overnight fasting in K8−/− mice (black bar) and K8+/+ mice (white bar). Bars represent means±s.e.m., n = 14 K8+/+ and 11 K8−/− mice. (B) Glucose tolerance is shown for K8−/− (grey broken line) and K8+/+ (black line) mice as mean±s.e.m. blood glucose values as a function of time after glucose administration. (C) Insulin tolerance for K8−/− (grey broken line) and K8+/+ (black line) mice is shown as mean±s.e.m. blood glucose values as a function of time after i.p. insulin administration. n = 10 K8+/+ and 8 K8−/− mice for B and C. (D) Basal serum insulin and glucose-stimulated serum insulin levels (GSIS) 10 minutes after i.p. glucose 2 g/kg of body weight in K8+/+ mice (white bar) and K8−/− (black bar) mice shown as mean±s.e.m. serum insulin levels, n = 9 mice/group for fasting insulin, and 3 K8+/+ and 4 K8−/− mice for glucose-stimulated insulin levels. *P<0.05, **P<0.01, ***P<0.001 compared with K8+/+ (A–C) or basal (D).
Islet number, α-cell to β-cell ratios (supplementary material Fig. S2) and general islet morphology in untreated mice were similar in the K8+/+ and K8−/− groups (Fig. 3A; Fig. 1A; supplementary material Fig. S2B). Intact islets were also readily isolated from K8−/− pancreata, similar to K8+/+ after collagenase digestion (not shown), indicating that the K8−/− islets are not excessively fragile, despite the lack of K8. Analysis of the K8−/− β-cell ultrastructure demonstrated asymmetries in the morphology of insulin vesicles. Insulin vesicles were frequently clustered together and the insulin cores of the vesicles were reduced in size and deformed (Fig. 3A–C). The area and aspect ratios of the insulin cores were quantified from electron microscopy images of the β-cells to ascertain the size and shape differences between K8+/+ and K8−/− insulin cores (Fig. 3B,C). K8−/− islet insulin cores were on average approximately one-third smaller than the K8+/+ cores (average core size was 12,000 nm2 for K8−/− and 20,000 nm2 for K8+/+ mice; Fig. 3B). In addition, the percentages of insulin cores with aspect ratios above unity (i.e. elongated core shape) were considerably higher in the K8−/− mouse β-cells compared to K8+/+ cells (Fig. 3C). These abnormalities in β-cell insulin vesicles correlated with clearly reduced insulin content in the pancreas of K8−/− compared to K8+/+ mice (Fig. 3D). K8 absence thus alters systemic insulin and glucose control as well as intracellular β-cell organisation and insulin content.
Fig. 3.
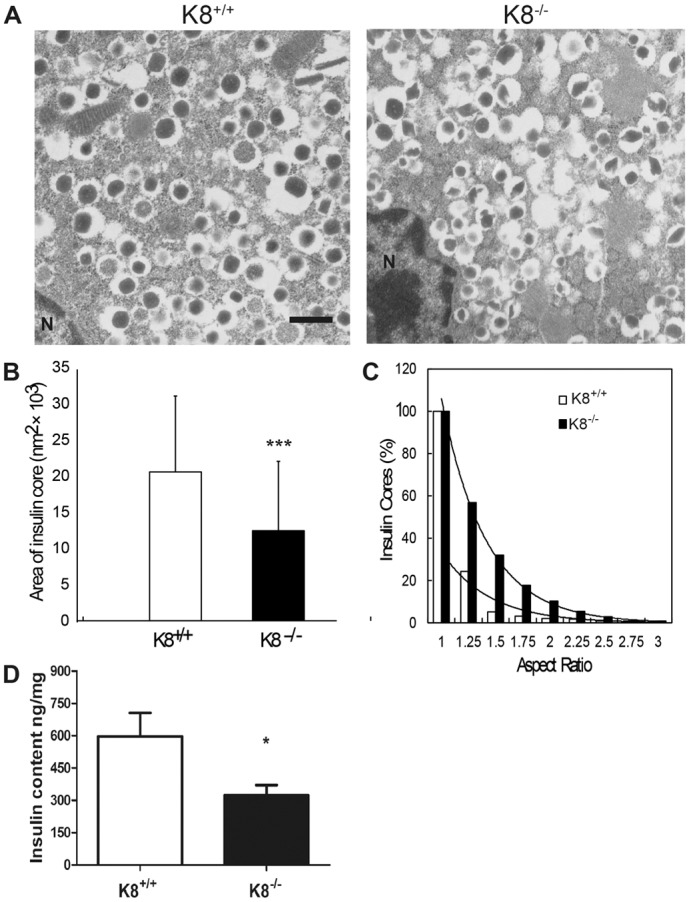
Abnormal β-cell ultrastructure and reduced pancreatic insulin content in K8−/− mice. (A) Electron microscopy images of islet β-cells from K8+/+ and K8−/− mice show that insulin cores (electron-dense vesicle core content) in the vesicles are smaller and more irregular in the K8−/− β-cells. The pictures are representative of islets from two mice per group. N, nucleus. Scale bar: 500 nm. (B) Mean area ± s.d. of individual insulin cores for K8+/+ (white bar) and K8−/− (black bar). (C) Total percentage of insulin vesicle cores in K8+/+ (white bars) and K8−/− mice (black bars) with aspect ratios above the given values (vertical axis) were plotted against the corresponding aspect ratios (horizontal axis). The aspect ratio is the proportional relationship between the local height and local width of an insulin core where a higher percentage of insulin core aspect ratio indicates less uniform (more irregular) shapes. In both mouse genotypes these percentages fall as an exponential function of increasing aspect ratio, clarifying that insulin cores with lower aspect ratios are predominant in both mouse types although K8−/− has a higher proportion of insulin cores that are irregular (higher aspect ratio). Insulin vesicle core area (B) and aspect ratios (C) were calculated from 650 K8+/+ and 764 K8−/− insulin cores. (D) Pancreatic insulin content in K8+/+ (white bars) and K8−/− (black bars) are shown as mean+s.e.m. ng/mg pancreas weight from six mice per genotype. *P<0.05, ***P<0.001 compared with K8+/+.
K8−/− β-cells are resistant to the acute toxic effects of high-dose STZ, correlating with mislocalisation of the glucose transporter GLUT2
To assess whether keratins are important in acute β-cell stress, mice were subjected to a single high dose of the β-cell toxin STZ (200 mg/kg of body weight) and analysed for islet damage 48 hours after STZ treatment and hyperglycaemia at 28 and 48 hours after STZ. In K8+/+ mice, severe islet destruction (>50% β-cell loss) was observed in 60% of islets [Fig. 4A,B, as predicted from previous murine studies in normal mice (Cardinal et al., 2001)]. In contrast, the majority of K8−/− islets suffered limited β-cell damage, and significant islet destruction was seen in only 18% of islets (Fig. 4A,B). After 28 and 48 hours, STZ-induced a 6–7-fold and a 5-fold increase in blood glucose levels in K8−/− and K8+/+ mice, respectively, compared to the start of the experiment, but the difference was not statistically significant between the genotypes (not shown).
Fig. 4.

Decreased sensitivity to high-dose STZ-induced islet damage corresponds with basal GLUT2 mislocalisation in K8−/− mice. (A) Hematoxylin-eosin staining of pancreatic sections of untreated and high-dose STZ-treated K8+/+ and K8−/− mice 48 hours after STZ. Islet β-cells are almost entirely lost in K8+/+ mice, but better preserved in K8−/− mice after STZ treatment. Scale bar: 50 µm. (B) Percentage of islets in K8+/+ (white bar) versus K8−/− (black bar) mice with extensive β-cell loss after high-dose STZ. Bars represent means±s.e.m., n = 4 per group. *P<0.05 as calculated with the Student's t-test. (C) Confocal images of immunostaining for GLUT2 (green), insulin (red) and cell nuclei (blue) in islet β-cells from untreated and high-dose STZ-treated K8+/+ and K8−/− mice, 48 hours after STZ. The white arrows exemplify sites of distinctly membrane-proximal GLUT2 localisation in untreated K8+/+ β-cells and the increased cytoplasmic GLUT2 in K8−/− β-cells. The white arrowheads exemplify the apoptotic-looking β-cell nuclei in STZ-treated K8+/+ mice and intact nuclei in STZ-treated K8−/− β-cells. Scale bar: 20 µm. (D) The peak immunofluorescence intensity for GLUT2 between adjacent β-cell nuclei was assessed by quantification from confocal images. K8−/− GLUT2 peak immunofluorescence intensity over a distance between the nuclei of two adjacent β-cells is on average lower compared with K8+/+ indicating decreased GLUT2 at the cell–cell interfaces. The bars represent mean± s.e.m. values of the GLUT2 fluorescence peak (or maximum amplitude) calculated for 30 K8+/+ and 30 K8−/− β-cell pairs (see examples in supplementary material Fig. S4), n = 4 mice per genotype. (E) Relative GLUT2 levels determined by western blotting in the cytoplasmic fraction from crude islet lysates normalised to Hsc70 for K8+/+ (white bars) and K8−/− (black bars) mice (n = 4 per group). (F) Western blots from total pancreas lysates showing GLUT2, K8 and the loading control Hsc70 protein bands for three untreated K8+/+ and K8−/− mice (1–3). The difference in relative GLUT2 levels after normalisation with Hsc70 were statistically insignificant, measuring 0.80±0.07 in K8+/+ and 0.71±0.03 in K8−/− mice (mean±s.e.m.)
STZ is taken up by β-cells through the glucose transporter GLUT2, and mice with dysfunctional GLUT2 are insensitive to STZ (Hosokawa et al., 2001; Stolarczyk et al., 2007). Given that the response to acute STZ toxicity is reduced in K8−/− mice, we assessed whether this might be due to aberrant GLUT2 localisation or expression. GLUT2 was localised only to the plasma membrane in K8+/+ β-cells, but was more dispersed throughout the cytoplasm in K8−/− β-cells of untreated mice (Fig. 4C; supplementary material Fig. S3). Co-staining of GLUT2 with the membrane marker E-cadherin or with insulin demonstrated a close colocalisation of GLUT2 and E-cadherin in K8+/+ β-cells at the plasma membrane, whereas in K8−/− β-cells GLUT2 was also distributed over a wider area, correlating with the localisation of insulin in the cytoplasmic regions (supplementary material Fig. S3). This analysis also shows that the location of the peak of cellular GLUT2 fluorescence intensity, which in K8+/+ β-cells is at the plasma membrane (supplementary material Fig. S3), is 30% decreased in K8−/− (Fig. 4D), suggesting that K8−/− actual GLUT2 plasma membrane levels are decreased. Our findings also imply that the GLUT2 redistribution is not a general phenomenon for membrane proteins after K8 inactivation because E-cadherin (supplementary material Fig. S3) and F-actin (not shown) are, similar to in K8+/+ mice, tightly localised to the membrane. Western blot analysis of isolated β-cell membrane and cytosolic fractions confirmed the aberrant localisation of GLUT2 in K8−/− β-cells; GLUT2 levels normalised to the cytosolic marker Hsc70 were significantly higher in the K8−/− cytosolic fraction compared to K8+/+ (Fig. 4E). No major GLUT2 difference was seen in the membrane fraction, which contains plasma membrane and internal membranes (not shown), nor in total pancreas GLUT2 protein levels (Fig. 4F). After high-dose STZ-treatment, K8−/− mice appeared to retain more GLUT2 expression in β-cells, compared to K8+/+ mice (Fig. 4C). The immunofluorescence staining moreover shows better preservation of nuclear morphology and more homogenous insulin staining in K8−/− compared to K8+/+ β-cells (Fig. 4C), verifying the islet histology shown in Fig. 4A. The aggravated nuclear damage in K8+/+ mice after high-dose STZ was further confirmed by staining for phosphorylated histone 2 (H2AX), which is induced by double-stranded DNA breaks (supplementary material Fig. S4). GLUT2 mislocalisation in K8−/− mice therefore might limit acute STZ-induced β-cell responses.
Delayed hyperglycaemia but increased exocrine damage after multiple low-dose STZ treatment in K8−/− mice
As a chronic endocrine stress and diabetes model, we analysed the effect of K8 deletion on diabetes incidence and pancreatic damage after multiple low-dose STZ treatment. At 2 weeks after STZ, no difference in K8−/− and K8+/+ diabetes incidence was noted (Fig. 5A; 50% of K8+/+ and K8−/− animals developed diabetes). However, blood glucose levels were marginally but significantly lower in K8−/− mice compared with K8+/+ at 1 week after STZ (Fig. 5B). The level of insulitis was also similar in K8+/+ and K8−/− mice after low-dose STZ, although a trend (P = 0.089) towards less insulitis in K8−/− mice was observed 2 weeks after STZ (not shown). By 5 weeks after STZ-treatment (but not after 2 weeks, not shown) extensive exocrine pancreatic changes were observed in K8−/− mice, with loss of acinar cell structure, atrophy, lipids, necrosis, oedema and vacuoles, whereas usually mild and occasional exocrine changes were seen in K8+/+ mice (Fig. 5C; Table 1). Taken together, loss of K8 modestly delays the onset of low-dose STZ-induced hyperglycaemia, but does not protect against diabetes development and exacerbates long-term exocrine pancreatic damage.
Fig. 5.

Delayed hyperglycaemia but increased exocrine injury in K8−/− mice after low-dose STZ treatment. (A) Diabetes incidence is shown for K8+/+ (black solid line) and K8−/− (broken grey line) mice after five low-dose STZ i.p. injections during the first 5 days (arrows). n = 13 K8+/+ and 11 K8−/− mice. (B) Mean±s.e.m. blood glucose levels at the start of the experiments, and at 1 and 2 weeks after low-dose STZ injections in K8+/+ (white bars) and K8−/− (black bars) mice. n = 13 K8+/+ and 11 K8−/− mice. *P<0.05, **P<0.01 calculated with one-way ANOVA and Bonferroni's posthoc test. (C) Representative haematoxylin-eosin staining of the exocrine pancreatic damage 5 weeks after low-dose STZ treatment in K8+/+ and K8−/− mice. Extensive oedema can be seen in the K8−/− pancreas. v, vacuoles, the black arrow indicates hyperplasia, the arrowhead shows atrophy and I, islet, which in this picture also contains some inflammatory cells. Scale bar: 200 µm.
Table 1. Damage scores for the exocrine pancreas in K8+/+ and K8−/− mice shows increased damage in K8−/− 5 weeks after low-dose STZ treatment.
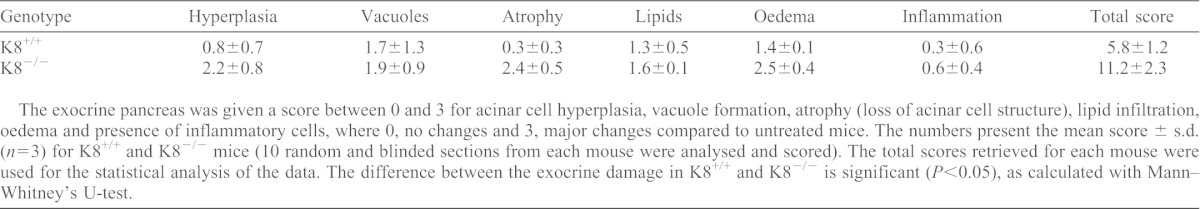
The exocrine pancreas was given a score between 0 and 3 for acinar cell hyperplasia, vacuole formation, atrophy (loss of acinar cell structure), lipid infiltration, oedema and presence of inflammatory cells, where 0, no changes and 3, major changes compared to untreated mice. The numbers present the mean score ± s.d. (n = 3) for K8+/+ and K8−/− mice (10 random and blinded sections from each mouse were analysed and scored). The total scores retrieved for each mouse were used for the statistical analysis of the data. The difference between the exocrine damage in K8+/+ and K8−/− is significant (P<0.05), as calculated with Mann–Whitney's U-test.
β-cell keratin expression is increased during chronic islet stress in NOD mice and in multiple low-dose STZ diabetes
To evaluate whether β-cell keratin expression is regulated by chronic cell stress, keratin expression was examined in two models of diabetes and/or islet injury: NOD mice and multiple low-dose STZ-treated normal K8+/+ mice. In both diabetes models, analysed by confocal imaging with identical settings, K8 was clearly upregulated in the remaining insulin positive β-cells in the islets but not in the exocrine compartment (Fig. 6A). The keratin upregulation in both models involves a dramatic increase in the density of the β-cell K8 filament network (Fig. 6B). Hence, β-cell keratins are upregulated in chronic islet stress. K18, however, was not visibly upregulated in low-dose STZ-treated K8−/− mice (Fig. 6C), indicating that K8 is essential for efficient upregulation of β-cell keratins in chronic stress and diabetes.
Discussion
K8 and K18 are the main keratins in simple-type epithelia and the only keratins that have previously been described in the endocrine pancreas of adult mice under basal conditions (Bouwens, 1998). The presence of K18 in K8−/− islets observed in this study both by immunofluorescence staining and biochemical methods, ascertains the presence of another type II keratin in addition to K8, given the obligate heteropolymeric nature of keratins (Coulombe and Omary, 2002). Accordingly, low levels of type II K7 colocalise with K18 in K8+/+ and K8−/− islets. This is in contrast to acinar cells of the exocrine pancreas and adult hepatocytes, where K8 is the only type II keratin, leading to K18 absence in K8−/− mice (Baribault et al., 1993; Toivola et al., 2000a). The scarcity of keratin filaments in K8−/− mouse islets and the apparent lack of upregulation upon islet stress, make the K8−/− mouse a useful model for studying the role of islet keratins under physiological conditions and in diabetes.
The results presented here show that K8 inactivation leads to reduced fasting blood glucose levels and resistance to hyperglycaemia after glucose challenge. Correspondingly, K8−/− mice have increased sensitivity to insulin, demonstrated by lower blood glucose levels after insulin injection. K8 inactivation also modestly but significantly delays low-dose STZ-induced hyperglycaemia. Taken together, this suggests that keratins are involved in the regulation of blood glucose levels. The lower blood glucose levels in K8−/− mice are, however, not due to increased basal or glucose stimulated serum insulin levels. By contrast, K8−/− has a decreased glucose stimulated serum insulin response and lower amount of pancreatic insulin per unit weight. The abnormal glucose regulation in K8−/− mice might thus be related to factors such as insulin action, glucose uptake, glycogen storage or its use in other K8-containing organs. This is supported by abnormal clustering of glycogen granules in K8−/− mouse hepatocytes shown by electron microscopy (Toivola et al., 1998) and periodic-acid–Schiff (PAS) staining (our unpublished data). In addition, it was recently reported in an elegant study, that K8- and K18-knockdown hepatocytes in culture have increased glucose uptake as well as increased glycogen formation that is further amplified by insulin (Mathew et al., 2013). Taken together, these findings suggest that hepatocyte keratins also have a central role in the control of systemic glucose regulation. A relationship between glucose metabolism in muscle cells (expressing low levels of K8 and K19) and abnormal blood glucose regulation in K8−/− mice can also not be ruled out (Stone et al., 2007).
The differences in β-cells reported here between K8+/+ and K8−/− mice, in regards to GLUT2 localisation, morphology of insulin vesicles, as well as the upregulation of keratins in diabetic mice, indicate that keratins are necessary for β-cell intracellular organisation. The disruption of insulin vesicles in K8−/− β-cells might specify a function for keratin networks in the organisation, maturation or release of insulin vesicles, analogous to the functions of microtubules and actin filaments (Lacy et al., 1972; Orci et al., 1972). Actin is thought to restrict insulin release under basal conditions, although it might aid insulin secretion in β-cells with low granularity (Li et al., 1994). Microtubules are involved in the transport of insulin vesicles to the membrane-proximal actin filaments and are necessary for sustained nutrient-induced (second-phase) insulin release (Farshori and Goode, 1994; Wang and Thurmond, 2009). The mislocalisation of GLUT2 in K8−/− mice shown in our study (i.e. an increased cytoplasmic localisation, and probably decreased plasma membrane localisation) further suggests that keratins might have a function in the targeting of β-cell proteins. Keratin-dependent targeting of glucose transporters to the appropriate sub-cellular locations is also evidenced by the mislocalisation of GLUT1 and GLUT3 in yolk sack endothelial cells of keratin type II knockout mice (Vijayaraj et al., 2009) and mildly increased cytoplasmic GLUT2 in K8- and K18-null hepatocytes (Mathew et al., 2013). Similarly to these studies, the results herein demonstrate that overall glucose transporter protein levels are unchanged, although their cellular localisation is altered. This could imply that changes in GLUT2 in the absence of keratins would encompass subtle differences in membrane-proximal localisation or shifts within specific plasma membrane domains (Orci et al., 1989) inaccessible to STZ.
The β-cells in K8−/− mice displayed a remarkable resistance to acute high-dose STZ-induced injury and a slightly delayed onset of hyperglycaemia after low-dose STZ treatment, supporting the idea that K8−/− mice are protected in two regimens of this diabetes model. This resistance was unexpected considering the well-established IF cell stress functions in many different cell types (Toivola et al., 2010). The reduced or delayed response to STZ in K8−/− mice is probably caused by mislocalisation of GLUT2, because STZ is taken up by the β-cells through this transporter. GLUT2 is abundant in murine β-cells and has a high capacity for glucose uptake. Hence, a significant dysfunction in GLUT2 is required to alter insulin secretion from β-cells (Leturque et al., 2009). This might partly explain the high glucose tolerance and normal basal serum insulin levels, despite GLUT2 mislocalisation in K8−/− β-cells. Although GLUT2 is not rate-limiting for the glucose response under normal circumstances, it is possible that GLUT2 mislocalisation in K8−/− β-cells limits or delays STZ uptake, leading to a reduced response to high-dose STZ and delayed hyperglycaemia after low-dose STZ treatment in K8−/− mice. It is also possible that K8−/− endocrine cell protection relates to similar cytoprotective mechanisms to those that have been described for the exocrine pancreas, which in K8−/− mice is highly resistant to injury in several non-chronic experimental pancreatitis models (Toivola et al., 2009; Toivola et al., 2000a). This exocrine cytoprotection is related to the dramatic upregulation of the injury-response protein regeneration protein 2 (REG2) in the K8−/− mouse acinar cells under basal conditions (Zhong et al., 2007).
Diabetes development in NOD mice and multiple low-dose chronic STZ treatment of K8+/+ mice (2 and 5 weeks after STZ) resulted in a robust K8 upregulation specifically in the remaining β-cells. This is consistent with the reported K8 upregulation after glucose stimulation in vitro (Ahmed and Bergsten, 2005) and with the stress behaviour of keratins in other cell types, including stress- and regeneration-related keratin upregulation as summarised in Toivola et al. (Toivola et al., 2010). This suggests that keratins play a role in islet stress and injury, or recovery thereafter, which needs to be addressed in further studies. Interestingly, K8 acetylation and phosphorylation, which affect filament organisation and solubility, were recently found to be responsive to glucose, further supporting the hypothesis that keratins are dynamic in these contexts (Snider et al., 2013). Keratin upregulation was not observed in the diabetic K8−/− mice at 2 or 5 weeks after low-dose STZ, indicating that K7 cannot effectively compensate for K8 in K8−/− mice during conditions of chronic stress and/or diabetes. Prolonged stress (5 weeks after low-dose STZ) moreover resulted in extensive injury in the exocrine pancreas of K8−/− but not K8+/+ mice, giving further support to a keratin function in the pancreas during chronic stress. On the basis of these findings, a further examination of the role of keratins as potential modifiers in type II diabetes blood glucose regulation and β-cell function might prove interesting.
The results in this study present novel evidence for keratin proteins as modulators in the endocrine pancreas and for their involvement in maintenance of systemic glucose levels. The range of differences observed in K8−/− mice under basal conditions and during experimental diabetes (Table 2) suggests that islet keratins are dynamic structures with multifaceted functions in the β-cells, the mechanisms of which need further study. These observations open up the question as to whether naturally occurring keratin mutations in the human population similarly affect β-cell organisation, and whether such mutations could affect the health and stress tolerance of the endocrine pancreas.
Table 2. Summary of the blood glucose regulation and Islet of Langerhans phenotypes in K8+/+ and K8−/− mice.
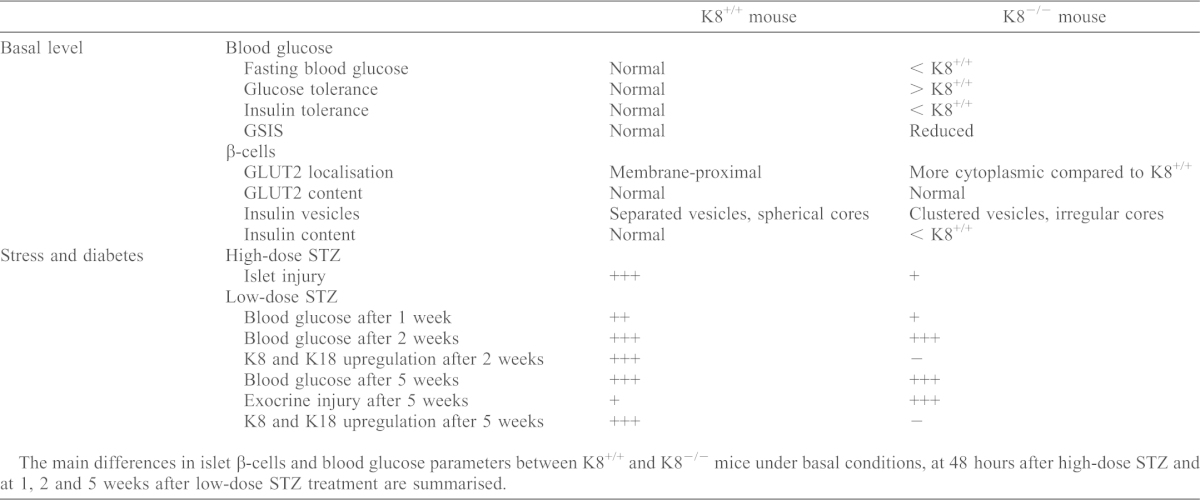
The main differences in islet β-cells and blood glucose parameters between K8+/+ and K8−/− mice under basal conditions, at 48 hours after high-dose STZ and at 1, 2 and 5 weeks after low-dose STZ treatment are summarised.
Materials and Methods
Experimental animals
Sex- and age-matched K8+/+ and K8−/− mice (Baribault et al., 1994) of FVB/n background and female non-obese diabetic (NOD) mice were used in this study. All animals were bred and raised at the Central Animal Laboratory at the University of Turku. K8−/− mice were generated, bred and genotyped as described previously (Baribault et al., 1994; Zhong et al., 2007). Diabetes was confirmed in the NOD mice by blood glucose measurements 2–4 days prior to experiments. All animals were used for experiments at 4–7 months of age and were killed by CO2 inhalation. Animal experiments were approved by the National Animal Experiment Board and conformed to the regulations set by The Finnish Act on Animal Experimentation.
Fasting blood glucose measurements, glucose tolerance and insulin tolerance tests
Fasting blood glucose was measured after overnight fasting of K8+/+ and K8−/− mice, using a hand-held glucose monitor (Contour, Bayer, Basel, Switzerland). Glucose tolerance tests were performed by fasting mice overnight and then challenging them with 2 g/kg of body weight, intraperitoneal (i.p.) glucose injection (Sigma-Aldrich). Blood glucose was measured before and at 30-minute intervals, up to 150 minutes, after glucose injections. Insulin tolerance tests were performed by fasting mice for 4 hours, injecting them with 0.75 U/kg of body weight, i.p. insulin (Humalog, Lilly, Houten, The Netherlands) and monitoring blood glucose before and at the indicated intervals up to 2 hours after insulin challenge.
Insulin enzyme-linked immunosorbent assay
K8+/+ and K8−/− mice were fasted for 4 hours and then administered glucose (2 g/kg of body weight, i.p.). Blood was collected from the submandibular vein of the mice with Golden rod lancets (MEDIpoint, Mineola, NY, USA) before and 10 minutes after glucose injection. Serum was separated by centrifugation after the blood had been allowed to clot. For measurement of insulin levels from pancreas samples, the acid-ethanol method was used to extract insulin from the pancreas. Pancreata were collected in acid-ethanol, containing 1.5% vol/vol concentrated HCl (Sigma Aldrich) in 70% ethanol (20 µl solvent/mg pancreas tissue) and homogenised using a TissueRuptor homogenizer (Qiagen, Hilden, Germany). The homogenate was incubated under constant stirring at 4°C overnight. Samples were then centrifuged at 13,000 g for 30 minutes and supernatants were collected for insulin measurements. The insulin concentrations in the serum and in pancreas samples were determined according to the manufacturer's recommendations, using a Mouse Ultrasensitive insulin enzyme-linked immunosorbent assay (ELISA) kit (Alpco, Salem, NH, USA).
STZ treatments
STZ (Sigma-Aldrich) was dissolved in a buffer containing 50 mM sodium citrate (Sigma-Aldrich) in PBS (pH 4.5) and administered immediately i.p. to mice as a single high dose (200 mg/kg of body weight) or in multiple low doses (40 mg/kg of body weight/day for 5 consecutive days). The animals were fasted 4 hours before the first injection. Blood glucose levels were measured from tail vein blood before STZ administration and at 28 and 48 hours after STZ for high-dose experiments. Blood glucose monitoring for low-dose STZ experiments were performed daily between day 7–14 and thereafter once a week. Animals were considered diabetic when two consecutive measurements exceeded 14 mmol/l (Engkilde et al., 2010) Animals were killed and samples collected 48 hours after high-dose STZ treatment and 15 or 35 days after low-dose STZ.
Pancreas histology
Pancreata from untreated (control) mice and STZ-treated mice were dissected and fixed in paraformaldehyde (4% vol./vol. in PBS, pH 7.4) for preparation of hematoxylin-eosin stained paraffin-embedded sections. Islet damage was scored from 0–3, where 0 was no apparent damage, 1 was loss of ≤25% of islet cells, 2 was 25–50% islet cell loss, 3 was ≥50% islet cell loss. Insulitis was scored from 0–3 (Alam et al., 2011) where 0 was no infiltration, 1 was peri-insulitis, 2 was infiltration covering half the islet and 3 was full insulitis. A minimum of 40 islets per pancreas were scored. A mean insulitis score for each pancreas was calculated by dividing the sum of individual islet scores with the number of islets analysed in that pancreas. Exocrine pancreatic damage was assessed in a blinded fashion, as described (Hashimoto et al., 2000), by histological scoring from 0–3 for hyperplasia, vacuoles, atrophy, lipids, oedema and inflammation, where 0 was negligible exocrine changes, 1 was mild to moderate changes in <10% exocrine pancreas, 2 was 10–25% of the exocrine areas mildly or moderately damaged and 3 was extensive damage in >25% of the exocrine pancreas. A mean score for each type of damage was given based on analysis of 20 sections per pancreas and these scores were added up to give a final exocrine damage score between 0 and 18 for each mouse.
Immunofluorescence staining and analysis
Pancreata were dissected and frozen in tissue-embedding compound (Tissue-Tek, Sakura Finetek, Alphen, The Netherlands). Cryosections 7 µm thick were cut and fixed with acetone at −20°C for 10 minutes (except for anti-E-cadherin staining, where samples were fixed first with methanol, followed by acetone and anti-tubulin fixed with methanol; each step at −20°C for 10 minutes). The following primary antibodies were used for tissue stainings: mouse anti-K7 (RCK 105) and mouse anti-K20 (Progen Biotechnic, Heidelberg, Germany), rat anti-K8 (Troma I) and rat anti-K19 (Troma III; Developmental Studies Hybridoma Bank, NIH, USA), rabbit anti-K18 (Ab4668) (Ku et al., 2004a), goat anti-insulin, rabbit anti-insulin (Santa Cruz Biotechnonology, Dallas, Texas, USA), rabbit anti-glucagon, Alexa-Fluor-488-conjugated mouse anti-E-cadherin (BD Pharmingen, San Jose, CA, USA), mouse anti-phospho-Histone H2A.X (Ser139), Alexa-Fluor-488–phalloidin (Invitrogen, Eugene, OR, USA), mouse anti-β-tubulin (Sigma Aldrich, St. Louis, MO, USA) and rabbit anti-GLUT2 (Millipore, Temecula, CA, USA). Cell nuclei were counterstained with DRAQ5 (eBioscience, San Diego, CA, USA). Secondary antibodies used were: donkey anti-rabbit conjugated to Alexa Fluor 546, donkey anti-goat conjugated to Alexa Fluor 488, donkey anti-rat conjugated to Alexa Fluor 488 and goat anti-mouse conjugated to Alexa Fluor 488 (Molecular Probes, Eugene, Oregon, USA). The sections were mounted with ProLong gold antifade reagent (Invitrogen, Paisley, UK) and analysed using a SP5 confocal microscope (Leica). Quantitative analysis of the subcellular localisation of GLUT2, E-cadherin and insulin from confocal images was performed using the LAS AF lite confocal software (Leica) by drawing line regions of interest between the nuclei of adjacent β-cells and measuring the fluorescence intensity along the line for the co-stained proteins. Peak fluorescence intensity (maximum amplitude) values were recorded for GLUT2.
Islet isolation
Collagenase P (Roche, Mannheim, Germany) at a concentration of 1.5 mg/ml in Hanks' Balanced Salt Solution (HBSS) (Sigma-Aldrich, Saint Louis, MO, USA) supplemented with 0.1% BSA and 100 U/ml Penicillin-Streptomycin, pH 7.4, was injected into the pancreas of K8+/+ and K8−/− mice through the common bile duct, after which the pancreata were dissected, injected with additional collagenase solution and incubated at 37°C for 16 minutes. The digested pancreata were loosely shaken and digestion was stopped by adding cold HBSS. The pancreas digest was filtered, washed in HBSS and mixed with 27% Ficoll PM 400 (Amersham Biosciences, Uppsala, Sweden) in HBSS supplemented with 0.2% BSA, 100 U/ml Penicillin-Streptomycin and 10 mM Hepes, pH 7.4. The 27% Ficoll solution was overlaid with 25%, 23% and 11% Ficoll solutions and centrifuged at 800 g for 19 minutes, at 4°C. The islets were collected from the upper Ficoll layer interfaces, washed with HBSS, stained with dithiozone (Sigma-Aldrich) and hand-picked with a pipette under an M60 stereo microscope (Leica).
High-salt extraction
High-salt extraction (HSE) was carried out according to Ku et al. (Ku et al., 2004b), to enrich the keratin fraction in the isolated islet cells from K8+/+ and K8−/− mouse pancreas and in liver samples from K8+/+ mice, which were used as a positive control for K8 and K18. In brief, Triton X-100 (Sigma-Aldrich) was added to the freshly isolated islets (roughly 100–140 islets pooled from three mice, per sample), vortexed, incubated on ice for 5 minutes and then centrifuged for 10 minutes at 13,000 rpm to repellet. The islet samples were homogenised manually in high-salt buffer [10 mM Tris-HCl, 140 mM NaCl, 1.5 M KCl, 5 mM EDTA, 0.5% Triton X100, 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) and complete protease inhibitor cocktail (Roche)], incubated for 30 minutes at 4°C, centrifuged for 10 minutes at 13,000 rpm and washed twice. Samples were then prepared for western blotting, by adding 30 µl Laemmli sample buffer (30% glycerol, 20% SDS, 0.1875 M Tris-HCl, 0.015% Bromphenol Blue and 3% β-mercaptoethanol) to each sample and heating the samples for 4 minutes at 95°C.
Subcellular fractionation of isolated islets
Islets for subcellular fractionation were isolated from K8+/+ and K8−/− mouse pancreata using a simplified islet isolation protocol: pancreata were digested in 1.5 mg/ml collagenase P (Sigma-Aldrich) dissolved in Dulbecco's Modified Eagle's Medium (DMEM) (Sigma-Aldrich) for 20 minutes, at 37°C. The digest was vortexed and filtered through a metal sieve and diluted with DMEM. Islets were washed three times by sedimentation at room temperature for 5 minutes, to eliminate a large fraction of the acinar cells. The remaining islet-enriched digest was homogenised using a 25G needle and syringe in a hypotonic lysis buffer containing 10 mM Tris-HCl and 1 mM EDTA, pH 7.4, supplemented with a protease inhibitor cocktail (Roche). After incubation on ice for 20 minutes, the lysate was centrifuged at 10,000 g for 10 minutes at 4°C to pellet nuclear and mitochondrial fractions and cell debris. The supernatant was further centrifuged at 100,000 g for 1 hour at 4°C and the supernatant, containing the cytosolic fraction, was collected. The pellet containing the membrane fraction was washed in the lysis buffer and re-centrifuged at 100,000 g for 45 minutes, at 4°C.
Western blotting
Whole pancreata, dissected from K8+/+ and K8−/− mice and snap-frozen in liquid nitrogen, and freshly isolated pancreatic islets were homogenised on ice with homogenisation buffer (0.187 M Tris-HCl, 3% SDS and 5 mM EDTA, pH 6.8) (Ku et al., 2004b). Protein concentration in whole-pancreas lysate samples was determined using a BSA protein assay reagent kit (Thermo-Scientific, Rockford, IL USA) according to manufacturer's instructions. The samples were diluted in Laemmli sample buffer, heated for 4 minutes at 95°C and loaded on an SDS-polyacrylamide gel (15 µg protein/sample of whole-pancreas lysates, roughly ten isolated islets in the islet total lysate sample, and 10 µl of islet HSE samples and subcellular fractionation lysates). Rabbit anti-GLUT2 (Millipore), rat anti-K8 (Developmental Studies Hybridoma Bank, NIH, USA), rabbit anti-K18 (Toivola et al., 1997), rabbit anti-E-cadherin (Cell Signaling, Danvers, MA, USA), rat anti-K18 and rat anti-Hsc70 (Stressgen Bioreagents, Ann Arbor, MI, USA) primary antibodies as well as anti-rabbit IgG (Promega Biosciences, San Luis Obispo, CA, USA) and anti-rat IgG (GE Healthcare, Little Chalfont, UK) secondary antibodies were used. The blots were incubated with ECL or ECL+ developing solutions (GE Healthcare) and exposed to X-ray film (Super RX, Fuji Corporation), whereafter individual bands were quantified using ImageJ software (Schneider et al., 2012).
Transmission electron microscopy
Pancreata were dissected, cut into 1–2 mm2 pieces and fixed with 5% glutaraldehyde in 0.16 M s-collidine buffer, pH 7.4, for preparation of Epon-embedded tissue blocks (Fluka Chemicals) according to standard procedures (Toivola et al., 2000b). Sections of 70-nm thickness were placed on copper grids (Leica) and imaged at 10,000× using a JEM 1200EX transmission electron microscope (Jeol). Insulin vesicle core area and aspect ratios were calculated from electron microscopy images adjusted for brightness and contrast, to optimise ocular clarity between darker electron-dense (insulin core) and lighter areas, using ImageJ. Thresholding, step-wise erosion, particle analysis and manual correction tools were applied to the images to differentiate the insulin cores from other electron-dense non-vesicle core structures. For aspect ratio analysis, the proportional relationship between the local height and local width of an insulin core was calculated.
Statistical analysis
Statistical calculations were performed using GraphPad PRISM and Student's t-test, Mann–Whitney's U test, or one-way or two-way ANOVA and Bonferroni post-hoc tests as appropriate.
Supplementary Material
Acknowledgments
We thank Helena Saarento (Åbo Akademi University) and the personnel at the Central Animal laboratory at the University of Turku for skilful technical assistance. Hélène Baribault (Amgen, CA, USA) is acknowledged for providing the K8−/− mouse strain and John Eriksson (Åbo Akademi University) is acknowledged for providing the rabbit anti-K18 antibody.
Footnotes
Author contributions
C.M.A. designed and performed experiments, analysed data and wrote the manuscript, J.S.G.S. designed and performed experiments, analysed data and reviewed the manuscript, E.N.D. and S.M.K performed experiments and analysed data, G.T. performed the initial blood glucose experiments and reviewed and edited the manuscript, P.A. analysed data and reviewed and edited the manuscript, M.B.O. designed the initial blood glucose experiments, reviewed and edited the manuscript and contributed to discussion, A.H. contributed with transgenic mice, reviewed and edited the manuscript and contributed to discussion, and D.M.T. designed the study, performed experiments, and contributed to the writing and editing of the manuscript.
Funding
This work was funded by The Finnish Cultural Foundation and Jalmari and Rauha Ahokas Foundation (to C.M.A.); The Finnish Diabetes Research Foundation (to D.M.T., A.H.); The Academy of Finland (to D.M.T., A.H.); Juselius Foundation (to D.M.T.); European Union Framework 7 Infrastructure Reflection Group Marie Curie Action (to D.M.T.); Liv och hälsa Foundation (to D.M.T.); The Centre of Excellence for Cell Stress and Molecular Aging at Åbo Akademi University (to D.M.T.); Päivikki and Sakari Sohlberg Foundation (to A.H.); and the Department of Veterans Affairs; and National Institutes of Health [grant number DK47918 to M.B.O.]. Deposited in PMC for release after 12 months.
supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.132795/-/DC1
References
- Ahmed M., Bergsten P. (2005). Glucose-induced changes of multiple mouse islet proteins analysed by two-dimensional gel electrophoresis and mass spectrometry. Diabetologia 48, 477–485 10.1007/s00125-004-1661-7 [DOI] [PubMed] [Google Scholar]
- Alam C., Bittoun E., Bhagwat D., Valkonen S., Saari A., Jaakkola U., Eerola E., Huovinen P., Hänninen A. (2011). Effects of a germ-free environment on gut immune regulation and diabetes progression in non-obese diabetic (NOD) mice. Diabetologia 54, 1398–1406 10.1007/s00125-011-2097-5 [DOI] [PubMed] [Google Scholar]
- Baribault H., Price J., Miyai K., Oshima R. G. (1993). Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 7, 1191–1202 10.1101/gad.7.7a.1191 [DOI] [PubMed] [Google Scholar]
- Baribault H., Penner J., Iozzo R. V., Wilson-Heiner M. (1994). Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 8, 2964–2973 10.1101/gad.8.24.2964 [DOI] [PubMed] [Google Scholar]
- Bertelli E., Regoli M., Orazioli D., Bendayan M. (2001). Association between islets of Langerhans and pancreatic ductal system in adult rat. Where endocrine and exocrine meet together? Diabetologia 44, 575–584 10.1007/s001250051663 [DOI] [PubMed] [Google Scholar]
- Blessing M., Rüther U., Franke W. W. (1993). Ectopic synthesis of epidermal cytokeratins in pancreatic islet cells of transgenic mice interferes with cytoskeletal order and insulin production. J. Cell Biol. 120, 743–755 10.1083/jcb.120.3.743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwens L. (1998). Cytokeratins and cell differentiation in the pancreas. J. Pathol. 184, 234–239 [DOI] [PubMed] [Google Scholar]
- Cardinal J. W., Margison G. P., Mynett K. J., Yates A. P., Cameron D. P., Elder R. H. (2001). Increased susceptibility to streptozotocin-induced beta-cell apoptosis and delayed autoimmune diabetes in alkylpurine-DNA-N-glycosylase-deficient mice. Mol. Cell. Biol. 21, 5605–5613 10.1128/MCB.21.16.5605-5613.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe P. A., Omary M. B. (2002). ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 14, 110–122 10.1016/S0955-0674(01)00301-5 [DOI] [PubMed] [Google Scholar]
- Engkilde K., Buschard K., Hansen A. K., Menné T., Johansen J. D. (2010). Prevention of diabetes in NOD mice by repeated exposures to a contact allergen inducing a sub-clinical dermatitis. PLoS ONE 5, e10591 10.1371/journal.pone.0010591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J. E., Dechat T., Grin B., Helfand B., Mendez M., Pallari H. M., Goldman R. D. (2009). Introducing intermediate filaments: from discovery to disease. J. Clin. Invest. 119, 1763–1771 10.1172/JCI38339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshori P. Q., Goode D. (1994). Effects of the microtubule depolymerizing and stabilizing agents Nocodazole and taxol on glucose-induced insulin secretion from hamster islet tumor (HIT) cells. J. Submicrosc. Cytol. Pathol. 26, 137–146 [PubMed] [Google Scholar]
- Grimm-Günter E. M., Revenu C., Ramos S., Hurbain I., Smyth N., Ferrary E., Louvard D., Robine S., Rivero F. (2009). Plastin 1 binds to keratin and is required for terminal web assembly in the intestinal epithelium. Mol. Biol. Cell 20, 2549–2562 10.1091/mbc.E08-10-1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T., Yamada T., Yokoi T., Sano H., Ando H., Nakazawa T., Ohara H., Nomura T., Joh T., Itoh M. (2000). Apoptosis of acinar cells is involved in chronic pancreatitis in Wbn/Kob rats: role of glucocorticoids. Pancreas 21, 296–304 10.1097/00006676-200010000-00012 [DOI] [PubMed] [Google Scholar]
- Hosokawa M., Dolci W., Thorens B. (2001). Differential sensitivity of GLUT1- and GLUT2-expressing beta cells to streptozotocin. Biochem. Biophys. Res. Commun. 289, 1114–1117 10.1006/bbrc.2001.6145 [DOI] [PubMed] [Google Scholar]
- Kim S., Coulombe P. A. (2010). Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat. Rev. Mol. Cell Biol. 11, 75–81 10.1038/nrm2818 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Fu H., Omary M. B. (2004a). Raf-1 activation disrupts its binding to keratins during cell stress. J. Cell Biol. 166, 479–485 10.1083/jcb.200402051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Toivola D. M., Zhou Q., Tao G-Z., Zhong B., Omary M. B. (2004b). Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 78, 489–517 [DOI] [PubMed] [Google Scholar]
- Ku N. O., Toivola D. M., Strnad P., Omary M. B. (2010). Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat. Cell Biol. 12, 876–885 10.1038/ncb2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P. E., Walker M. M., Fink C. J. (1972). Perifusion of isolated rat islets in vitro. Participation of the microtubular system in the biphasic release of insulin. Diabetes 21, 987–998 [DOI] [PubMed] [Google Scholar]
- Leturque A., Brot-Laroche E., Le Gall M. (2009). GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am. J. Physiol. 296, E985–E992 10.1152/ajpendo.00004.2009 [DOI] [PubMed] [Google Scholar]
- Li G., Rungger-Brändle E., Just I., Jonas J. C., Aktories K., Wollheim C. B. (1994). Effect of disruption of actin filaments by Clostridium botulinum C2 toxin on insulin secretion in HIT-T15 cells and pancreatic islets. Mol. Biol. Cell 5, 1199–1213 10.1091/mbc.5.11.1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin T. M., Vijayaraj P., Leube R. E. (2007). Structural and regulatory functions of keratins. Exp. Cell Res. 313, 2021–2032 10.1016/j.yexcr.2007.03.005 [DOI] [PubMed] [Google Scholar]
- Marceau N., Schutte B., Gilbert S., Loranger A., Henfling M. E., Broers J. L., Mathew J., Ramaekers F. C. (2007). Dual roles of intermediate filaments in apoptosis. Exp. Cell Res. 313, 2265–2281 10.1016/j.yexcr.2007.03.038 [DOI] [PubMed] [Google Scholar]
- Mathew J., Loranger A., Gilbert S., Faure R., Marceau N. (2013). Keratin 8/18 regulation of glucose metabolism in normal versus cancerous hepatic cells through differential modulation of hexokinase status and insulin signaling. Exp. Cell Res. 319, 474–486 10.1016/j.yexcr.2012.11.011 [DOI] [PubMed] [Google Scholar]
- Omary M. B. (2009). “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J. Clin. Invest. 119, 1756–1762 10.1172/JCI39894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary M. B., Coulombe P. A., McLean W. H. (2004). Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 351, 2087–2100 10.1056/NEJMra040319 [DOI] [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Tao G. Z., Toivola D. M., Liao J. (2006). “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem. Sci. 31, 383–394 10.1016/j.tibs.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Strnad P., Hanada S. (2009). Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Invest. 119, 1794–1805 10.1172/JCI37762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L., Gabbay K. H., Malaisse W. J. (1972). Pancreatic beta-cell web: its possible role in insulin secretion. Science 175, 1128–1130 10.1126/science.175.4026.1128 [DOI] [PubMed] [Google Scholar]
- Orci L., Thorens B., Ravazzola M., Lodish H. F. (1989). Localization of the pancreatic beta cell glucose transporter to specific plasma membrane domains. Science 245, 295–297 10.1126/science.2665080 [DOI] [PubMed] [Google Scholar]
- Oriolo A. S., Wald F. A., Ramsauer V. P., Salas P. J. (2007). Intermediate filaments: a role in epithelial polarity. Exp. Cell Res. 313, 2255–2264 10.1016/j.yexcr.2007.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Hobbs R. P., Coulombe P. A. (2013). The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr. Opin. Cell Biol. 25, 47–56 10.1016/j.ceb.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M., Lane E. B. (2007). Intermediate filaments and stress. Exp. Cell Res. 313, 2244–2254 10.1016/j.yexcr.2007.04.023 [DOI] [PubMed] [Google Scholar]
- Schneider C. A., Rasband W. S., Eliceiri K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubart U. K., Fields K. L. (1984). Identification of a calcium-regulated insulinoma cell phosphoprotein as an islet cell keratin. J. Cell Biol. 98, 1001–1009 10.1083/jcb.98.3.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer J., Bowden P. E., Coulombe P. A., Langbein L., Lane E. B., Magin T. M., Maltais L., Omary M. B., Parry D. A., Rogers M. A. et al. (2006). New consensus nomenclature for mammalian keratins. J. Cell Biol. 174, 169–174 10.1083/jcb.200603161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider N. T., Leonard J. M., Kwan R., Griggs N. W., Rui L., Omary M. B. (2013). Glucose and SIRT2 reciprocally mediate the regulation of keratin 8 by lysine acetylation. J. Cell Biol. 200, 241–247 10.1083/jcb.201209028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolarczyk E., Le Gall M., Even P., Houllier A., Serradas P., Brot-Laroche E., Leturque A. (2007). Loss of sugar detection by GLUT2 affects glucose homeostasis in mice. PLoS ONE 2, e1288 10.1371/journal.pone.0001288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone M. R., O'Neill A., Lovering R. M., Strong J., Resneck W. G., Reed P. W., Toivola D. M., Ursitti J. A., Omary M. B., Bloch R. J. (2007). Absence of keratin 19 in mice causes skeletal myopathy with mitochondrial and sarcolemmal reorganization. J. Cell Sci. 120, 3999–4008 10.1242/jcs.009241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek P., Kasper M., Karsten U. (1990). Expression of cytokeratin 19 during human liver organogenesis. Liver 10, 59–63 10.1111/j.1600-0676.1990.tb00436.x [DOI] [PubMed] [Google Scholar]
- Strnad P., Paschke S., Jang K. H., Ku N. O. (2012). Keratins: markers and modulators of liver disease. Curr. Opin. Gastroenterol. 28, 209–216 10.1097/MOG.0b013e3283525cb8 [DOI] [PubMed] [Google Scholar]
- Styers M. L., Kowalczyk A. P., Faundez V. (2005). Intermediate filaments and vesicular membrane traffic: the odd couple's first dance? Traffic 6, 359–365 10.1111/j.1600-0854.2005.00286.x [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Goldman R. D., Garrod D. R., Eriksson J. E. (1997). Protein phosphatases maintain the organization and structural interactions of hepatic keratin intermediate filaments. J. Cell Sci. 110, 23–33 [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Omary M. B., Ku N. O., Peltola O., Baribault H., Eriksson J. E. (1998). Protein phosphatase inhibition in normal and keratin 8/18 assembly-incompetent mouse strains supports a functional role of keratin intermediate filaments in preserving hepatocyte integrity. Hepatology 28, 116–128 10.1002/hep.510280117 [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Baribault H., Magin T., Michie S. A., Omary M. B. (2000a). Simple epithelial keratins are dispensable for cytoprotection in two pancreatitis models. Am. J. Physiol. 279, G1343–G1354 [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Ku N. O., Ghori N., Lowe A. W., Michie S. A., Omary M. B. (2000b). Effects of keratin filament disruption on exocrine pancreas-stimulated secretion and susceptibility to injury. Exp. Cell Res. 255, 156–170 10.1006/excr.1999.4787 [DOI] [PubMed] [Google Scholar]
- Toivola D. M., Tao G. Z., Habtezion A., Liao J., Omary M. B. (2005). Cellular integrity plus: organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 15, 608–617 10.1016/j.tcb.2005.09.004 [DOI] [PubMed] [Google Scholar]
- Toivola D., Ostrowski S., Baribault H., Magin T., Ramsingh A., Omary M. (2009). Keratins provide virus-dependent protection or predisposition to injury in coxsackievirus-induced pancreatitis. Cell Health Cytoskelet. 1, 51–65 10.2147/CHC.S5792 [DOI] [Google Scholar]
- Toivola D. M., Strnad P., Habtezion A., Omary M. B. (2010). Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 20, 79–91 10.1016/j.tcb.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraj P., Kröger C., Reuter U., Windoffer R., Leube R. E., Magin T. M. (2009). Keratins regulate protein biosynthesis through localization of GLUT1 and -3 upstream of AMP kinase and Raptor. J. Cell Biol. 187, 175–184 10.1083/jcb.200906094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Thurmond D. C. (2009). Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 122, 893–903 10.1242/jcs.034355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B., Strnad P., Toivola D. M., Tao G. Z., Ji X., Greenberg H. B., Omary M. B. (2007). Reg-II is an exocrine pancreas injury-response product that is up-regulated by keratin absence or mutation. Mol. Biol. Cell 18, 4969–4978 10.1091/mbc.E07-02-0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.