

Summary
TAK1 is a MAP3K that mediates non-canonical TGF-β and BMP signaling. During the embryonic period, TAK1 is essential for cartilage and joint development as deletion of Tak1 in chondro-osteo progenitor cells leads to severe chondrodysplasia with defects in both chondrocyte proliferation and maturation. We have investigated the role of TAK1 in committed chondrocytes during early postnatal development. Using the Col2a1-CreERT2; Tak1f/f mouse model, we induced deletion of Tak1 at postnatal day 7 and characterized the skeletal phenotypes of these mice at 1 and 3 months of age. Mice with chondrocyte-specific Tak1 deletion exhibited severe growth retardation and reduced proteoglycan and type II collagen content in the extracellular matrix of the articular cartilage. We found reduced Col2a1 and Acan expression, but increased Mmp13 and Adamts5 expression, in Tak1-deficient chondrocytes along with reduced expression of the SOX trio of transcription factors, SOX9, SOX5 and SOX6. In vitro, BMP2 stimulated Sox9 gene expression and Sox9 promoter activity. These effects were reduced; however, following Tak1 deletion or treatment with a TAK1 kinase inhibitor. TAK1 affects both canonical and non-canonical BMP signal transduction and we found that both of these pathways contribute to BMP2-mediated Sox9 promoter activation. Additionally, we found that ATF2 directly binds the Sox9 promoter in response to BMP signaling and that this effect is dependent upon TAK1 kinase activity. These novel findings establish that TAK1 contributes to BMP2-mediated Sox9 gene expression and is essential for the postnatal development of normal growth plate and articular cartilages.
Key words: Chondrocyte, Cartilage, TAK1, SOX9, ATF2
Introduction
Development of the limb skeleton is a complex multistep process initiated when mesenchymal progenitor cells originating from the lateral plate mesoderm condense and differentiate into chondrocytes forming cartilaginous anlagen that serve as templates of bone formation (Mariani and Martin, 2003). Once committed to the chondrocyte lineage, these cells proliferate rapidly until those most central to the anlagen exit the cell cycle and undergo hypertrophy. Hypertrophic chondrocytes secrete factors that promote mineralization, degradation and vascular invasion of the cartilage matrix. Terminally hypertrophic chondrocytes undergo apoptosis and the remaining cartilage serves as a scaffold for bone matrix deposition and remodeling by invading osteoblasts and osteoclasts. On either side of the ossification center, continued chondrocyte proliferation and maturation establishes the growth plate cartilage and drives longitudinal bone growth. During the early postnatal period, a second ossification center develops in the epiphyses of the limbs separating the transient growth plate cartilage from the permanent articular cartilage (Kronenberg, 2003).
Articular cartilage first appears during embryogenesis at sites of joint formation, but continues to develop during the postnatal period of skeletal growth (Chan et al., 2012). This unique tissue is avascular and aneural with a high matrix to cell volume ratio and is divided into distinct zones varying in biochemical composition. The superficial zone, for example, is characterized by flattened chondrocytes expressing proteoglycan 4 (PRG4). Appositional growth of the articular cartilage tissue is thought to be due to the existence of a resident chondroprogenitor cell population also in this zone (Dowthwaite et al., 2004). The transitional, or intermediate, zone contains rounded chondrocytes expressing mainly type II collagen and aggrecan. The deep radial zone and zone of calcified cartilage contain larger, more mature, chondrocytes expressing proteins associated with chondrocyte hypertrophy such as type X collagen and alkaline phosphatase. Between these zones lies the tidemark, a basophilic line dividing the unmineralized and mineralized cartilage (Archer et al., 2003; Zuscik et al., 2008).
The SRY-related high mobility group (HMG) box transcription factor 9 (SOX9) is a master regulator of chondrogenesis and is expressed in both growth plate and articular chondrocytes (Ng et al., 1997; Salminen et al., 2001; Zhao et al., 1997). Deletion of Sox9 in limb mesenchymal progenitor cells prevents mesenchymal cell condensation and leads to the complete absence of cartilage and bone formation, whereas deletion in chondro-osteo progenitors, results in severe generalized chondrodysplasia (Akiyama et al., 2002). SOX9 is required for Sox5 and Sox6 expression and, together, SOX5, SOX6 and SOX9 (the SOX trio) coordinately regulate the expression of several cartilage matrix genes including Acan, Col2a1 and Col11a1 (Akiyama et al., 2002; Akiyama and Lefebvre, 2011; Bridgewater et al., 1998; Han and Lefebvre, 2008). During endochondral bone formation, Sox9 expression is downregulated in growth plate chondrocytes as they undergo maturation and begin to express markers of hypertrophy, such as Runx2, Col10a1 and Mmp13 (Dy et al., 2012; Leung et al., 2011). In articular cartilage, SOX9 is crucial for maintenance of the extracellular matrix as chondrocyte-specific postnatal deletion of Sox9 results in reduced matrix proteoglycan content probably as a result of increased ADAMTS5 expression (Henry et al., 2012).
BMP signaling is of key importance for regulation of Sox9 gene expression. Deletion of BMP type 1 receptor genes, Bmpr1a and Bmpr1b, in chondro-osteo progenitor cells leads to severe chondrodysplasia and loss of Sox9, Sox5 and Sox6 expression in chondrocytes (Yoon et al., 2005). Similarly, combined deletion of the BMP-specific transcription factors SMAD1, 5 and 8 results in severe chondrodysplasia and reduced Sox9 expression (Retting et al., 2009). During fracture healing, BMP2 induces the expression of Sox9, Sox5 and Sox6, and in limbs with deletion of Bmp2, cartilage callus formation is absent following fracture (Tsuji et al., 2006; Uusitalo et al., 2001). In vitro, BMP2 induces expression of Sox9 in C3H10T1/2 mesenchymal progenitor cells as well as in mouse embryonic fibroblasts (MEFs), although the precise mechanism by which it does so remains unclear (Pan et al., 2008; Zehentner et al., 1999).
In addition to the canonical SMAD-dependent signaling pathway, BMP receptors activate MAPK signaling via MAP3K, TGF-β-activated kinase 1 (TAK1) (Yamaguchi et al., 1995). Activated TAK1 directly phosphorylates MAP2K family members, MKK3 and MKK6 (Moriguchi et al., 1996). MKK3 and 6, in turn, phosphorylate and activate the p38 family of MAPKs affecting gene transcription through a number of transcription factors (Raingeaud et al., 1996). Deletion of Tak1 in chondro-osteo progenitor cells results in chondrodysplasia and a phenotype similar to that observed in mice lacking BMP type 1 receptors and SMADs (Gunnell et al., 2010; Shim et al., 2009). The similarity in phenotypes was attributed to the discovery that TAK1 also participates in the phosphorylation and activation of BMP-specific SMADs.
We have investigated the chondrocyte-specific role of TAK1 in early postnatal skeletal development and articular cartilage generation. Given the early postnatal lethality of the non-inducible Col2a1-Cre; Tak1f/f mice used in previous studies, we utilized the inducible Col2a1-CreERT2 transgene to specifically target chondrocytes during the postnatal period of development. Postnatal deletion of Tak1 resulted in severe growth retardation and a striking reduction in proteoglycan and type II collagen content in the extracellular matrix of the articular cartilage. Complementary in vivo and in vitro approaches demonstrated that BMP2–TAK1 signaling regulates the expression of the SOX trio of transcription factors. Altogether, the findings establish TAK1 signaling as a crucial regulatory pathway in postnatal cartilage development and as a novel effector of BMP2-mediated Sox9 expression.
Results
Tamoxifen induces efficient chondrocyte-specific gene deletion in Col2a1-CreERT2; R26R+/− and Col2a1-CreERT2; Tak1f/f mice
To assess the postnatal recombination efficiency of the Col2a1-CreERT2 transgene, we administered tamoxifen (100 mg/kg) by intraperitoneal injection to Col2a1-CreERT2; R26R+/− and R26R+/− mice beginning at postnatal day (P)7 every other day for a total of three doses. Recombination was assessed by determining β-galactosidase expression through X-gal staining in knee joint sections of 1-month- and 3-month-old mice. X-gal staining was present only in the chondrocytes of mice expressing Cre recombinase. Staining was observed in over 80% of articular chondrocytes and over 60% of growth plate chondrocytes in Col2a1-CreERT2; R26R+/− tamoxifen-treated 1-month-old mice and in greater than 50% of articular chondrocytes in 3-month-old mice (Fig. 1A). Staining was greatly reduced (less than 50%) in the growth plate chondrocytes of 3-month-old mice. Tamoxifen-induced Tak1 deletion was also examined by TAK1 immunohistochemistry of 1-month-old Tak1f/f and Col2a1-CreERT2; Tak1f/f mice (Fig. 1B).
Fig. 1.
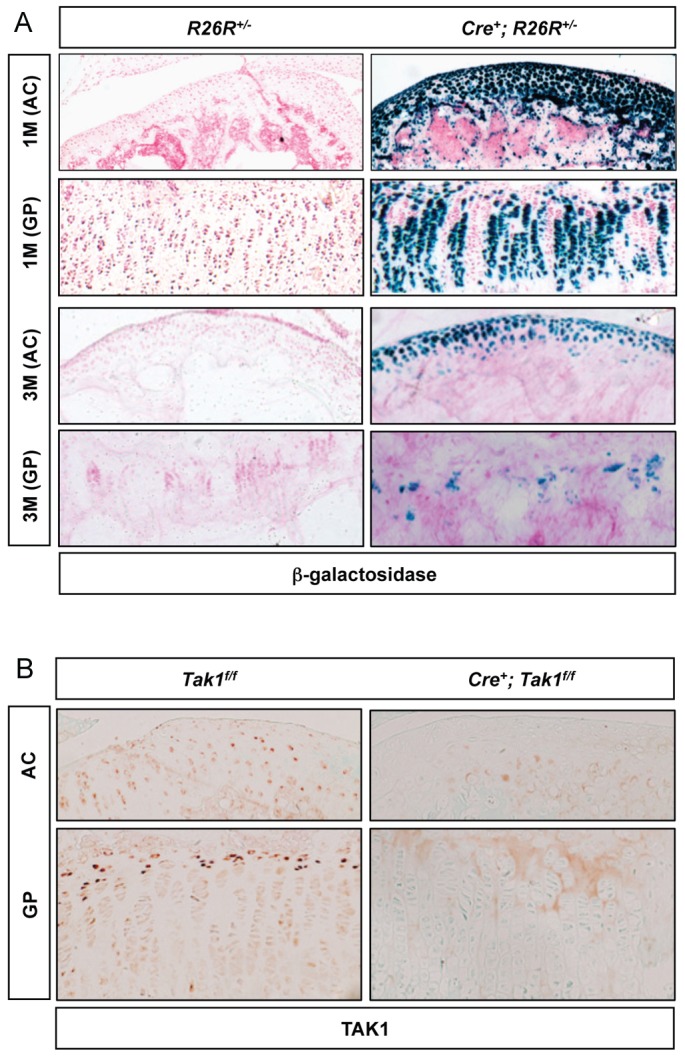
Tamoxifen induces efficient chondrocyte-specific gene deletion in Col2a1-CreERT2; R26R+/− and Col2a1-CreERT2; Tak1f/f mice. (A) β-galactosidase staining of knee joint sections from Col2a1-CreERT2; ROSA26R (Cre+; R26R) mice and Cre-negative control littermates (R26R) injected with tamoxifen (100 mg/kg body weight, intraperitoneally) three times, every other day, beginning at 1 week of age. The mice were killed at 1 (top two panels) or 3 (bottom two panels) months of age. (B) TAK1 immunohistochemistry of knee joint sections from Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) injected with tamoxifen as described above and sacrificed at 1 month of age. AC, articular cartilage; GP, growth plate cartilage.
Postnatal deletion of Tak1 in chondrocytes results in growth retardation, disorganization of the growth plate cartilage, and decreased proteoglycan content of the articular cartilage
Tamoxifen-induced Tak1 deletion at P7 in Col2a1-CreERT2; Tak1f/f transgenic mice results in severe growth retardation evident at 1 month of age compared with Cre-negative control Tak1f/f littermates, as shown in radiographs (Fig. 2A, upper panel: anterior-posterior view; lower panel: lateral view). To further assess cartilage development, knee joint sections were stained with Alcian Blue (Fig. 2B). The proximal tibia of the Col2a1-CreERT2; Tak1f/f transgenic mice has an irregular shape and a smaller secondary ossification center. This is probably due to a delay in the onset of secondary ossification center formation suggested by the presence of immature chondrocytes at the periphery of the ossified region. The growth plates of the TAK1-deficient mice have disorganized chondrocyte columns with a reduced number of chondrocytes (Fig. 2C, lower panels). Additionally, in vivo and in vitro BrdU labeling experiments revealed a decrease in chondrocyte proliferation upon Tak1 gene deletion (supplementary material Fig. S1A,B). At 3 months of age, the overall size of the Col2a1-CreERT2; Tak1f/f mice is not so dissimilar to that of their Cre-negative control littermates (supplementary material Fig. S2A). This is probably due to the initial escape of many highly proliferative growth plate chondrocytes from Tak1 deletion that, by 3 months of age, make up most of the growth plate as suggested by the ROSA26 reporter studies (Fig. 1A). The most striking phenotype of the Tak1-deficient mice at both 1 month and 3 months of age, however, is the lack of Alcian Blue staining in the unmineralized region of the articular cartilage, indicating a decrease in proteoglycan content within this region (Fig. 2C, upper panels; supplementary material Fig. S2B,C, upper panels).
Fig. 2.
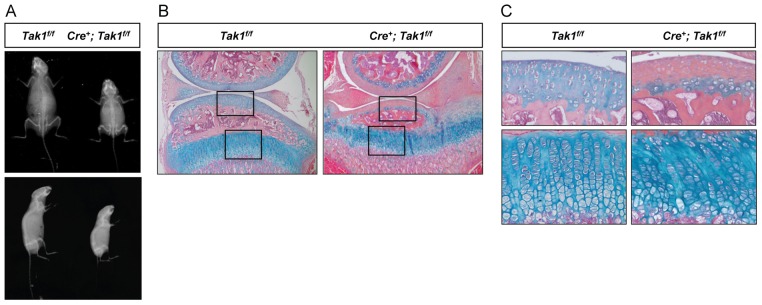
Postnatal chondrocyte-specific deletion of Tak1 results in growth retardation, growth plate disorganization and reduced proteoglycan content in the articular cartilage. (A) Radiographic analyses of 1-month-old Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) following injection with tamoxifen at 1 week of age. Anterior-posterior (top panels) and lateral (bottom panels) radiographic images are shown. (B) Alcian Blue, Hematoxylin and Orange G staining of knee joint sections from 1-month-old Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) injected with tamoxifen at 1 week of age (original magnification: 5×). (C) High magnification images (20×) of the articular cartilage (upper panels) or growth plate cartilage (lower panels) from corresponding boxed regions in B.
TAK1 is required for normal articular cartilage extracellular matrix production
Articular cartilage extracellular matrix (ECM) production was assessed over time in the Col2a1-CreERT2; Tak1f/f transgenic mice following recombination, using Alcian Blue staining and type II collagen immunostaining of tissue sections obtained from the mouse knee joint. Sustained loss of proteoglycan content was observed at 1 month and 3 months of age in mice with postnatal chondrocyte-specific Tak1 gene deletion (Fig. 3A). Similarly, immunostaining of articular cartilage showed that deletion of Tak1 reduces type II collagen content in 1-month- and 3-month-old mice (Fig. 3B). Gene expression analyses were carried out using mRNA directly isolated from articular cartilage tissue of 1-month-old control mice and mice with postnatal chondrocyte-specific Tak1 gene deletion. Tamoxifen-treated Col2a1-CreERT2; Tak1f/f mice exhibited reduced expression of the Sox9 and Col2a1 genes. In addition, expression levels of the aggrecanase gene, Adamts5, and the matrix metalloproteinase gene, Mmp13, were increased in Tak1-deficient articular cartilage (Fig. 3C). Expression levels of Col10a1 and Col1a1 were unchanged. Altogether, the findings show that TAK1 regulates the expression of genes that are both anabolic and catabolic for articular cartilage extracellular matrix. The overall effect of postnatal Tak1 gene deletion in chondrocytes is consistent with a reduction in articular cartilage extracellular matrix production.
Fig. 3.

Postnatal articular chondrocyte expression of TAK1 is required for the development of a normal articular cartilage ECM. (A) Alcian Blue, Hematoxylin and Orange G staining of femoral articular cartilage sections from 1-month-old (top panels; 1M) and 3-month-old (bottom panels; 3M) Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) injected with tamoxifen at 1 week of age (original magnification: 20×). (B) Type II collagen immunohistochemistry of femoral articular cartilage sections from 1-month-old Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) injected with tamoxifen at 1 week of age (original magnification: 20×). (C) Quantitative real time RT-PCR analyses for the indicated genes using articular cartilage tissue isolated from 1-month-old Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) injected with tamoxifen at 1 week of age. *P<0.05, Student's t-test.
Expression of the SOX trio of transcription factors is regulated by TAK1
Because the transcription factor SOX9 induces Col2a1, Col9a1 and Acan expression, we examined whether TAK1 signaling regulates the expression of SOX9 in chondrocytes. We performed immunohistochemistry for SOX9 on articular and growth plate cartilage sections from tamoxifen-treated Cre-negative control Tak1f/f and Col2a1-CreERT2; Tak1f/f mice. TAK1 deficiency caused a decrease in SOX9 protein levels in both articular and growth plate chondrocytes (Fig. 4A; supplementary material Fig. S2D). Primary chondrocytes were then isolated from Tak1f/f mice and infected with adenovirus encoding GFP (Ad-GFP, control cultures), adenovirus encoding Cre recombinase (Ad-Cre, loss-of-function cultures) or adenovirus encoding TAK1 (Ad-TAK1, gain-of-function cultures). Western blotting revealed that SOX9 expression is reduced in TAK1-deficient chondrocytes and is increased in chondrocytes overexpressing TAK1 (Fig. 4B). SOX5 and SOX6, along with SOX9, were previously shown to act as master regulators of the chondrocyte phenotype (Akiyama and Lefebvre, 2011). Both SOX5 and SOX6 protein levels were regulated by TAK1 in a pattern similar to SOX9 (Fig. 4B). The expression of the Sox5, Sox6 and Sox9 genes were increased in chondrocytes with Tak1 overexpression and reduced in chondrocytes deleted of Tak1 (Fig. 4C). These findings suggest that TAK1 modulates the expression of the SOX trio of transcription factors through transcriptional regulation. Additionally, Col2a1, Acan and Col9a1 expression levels were positively regulated by TAK1, whereas TAK1 inhibited Adamts5 and Mmp13 expression levels (Fig. 4D), consistent with gene expression data obtained from the articular cartilage of 1-month-old Cre-negative control Tak1f/f and Col2a1-CreERT2; Tak1f/f mice (Fig. 3C).
Fig. 4.

TAK1 positively regulates expression of the SOX trio of transcription factors in chondrocytes. (A) SOX9 immunohistochemistry of articular (AC) and growth plate (GP) cartilage sections from 1-month-old Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) injected with tamoxifen at 1 week of age. (B–D) Primary sternal chondrocytes from Tak1f/f mice were infected with adenovirus encoding GFP (Ad-GFP), Cre-recombinase (Ad-Cre) or TAK1 (Ad-TAK1). Forty-eight hours later, cells were harvested for western blotting (B) of the indicated proteins or quantitative real-time RT-PCR analyses for the genes involved in signaling (C) or extracellular matrix metabolism (D). *P<0.05, one-way ANOVA followed by Dunnett's test.
TAK1 signaling contributes to the regulation of Sox9 expression by BMP2
TAK1 was previously shown to participate in SMAD-dependent BMP signaling in chondrocytes (Gunnell et al., 2010; Shim et al., 2009). Consistent with these reports, we observed a reduction in levels of phosphorylated SMAD1, 5 and 8 in both articular and growth plate cartilage sections from 1-month-old Col2a1-CreERT2; Tak1f/f mice when compared with sections from Cre-negative control Tak1f/f mice (Fig. 5A). Similarly, in vitro deletion of Tak1 in primary sternal chondrocytes resulted in approximately 50% reduction in BMP2-mediated phosphorylation of SMAD1, 5 and 8 (Fig. 5C). BMP2 signaling also resulted in phosphorylation of p38, a MAP kinase downstream of TAK1, and the transcription factor ATF2. Following the deletion of Tak1 in vitro, phosphorylation of p38 and ATF2 was undetectable (Fig. 5C). Consistently, levels of phosphorylated ATF2 were dramatically reduced in articular and growth plate cartilage sections from 1-month-old Col2a1-CreERT2; Tak1f/f mice when compared with sections from Cre-negative control Tak1f/f mice (Fig. 5B).
Fig. 5.

TAK1 positively regulates BMP2-mediated Sox9 gene expression. Phospho-SMAD1, 5 and 8 (A) and phospho-ATF2 (B) immunohistochemistry of articular (AC) and growth plate (GP) cartilage sections from 1-month-old Col2a1-CreERT2; Tak1f/f (Cre+; Tak1f/f) mice and Cre-negative control littermates (Tak1f/f) injected with tamoxifen at 1 week of age. (C–E) Primary sternal chondrocytes from Tak1f/f mice were infected with adenovirus encoding GFP (Ad-GFP) or Cre-recombinase (Ad-Cre). Forty-eight hours later, cells were starved of serum for 12 hours and then treated with vehicle or BMP2 (100 ng/ml) for either 30 minutes (C) or for 24 hours (D,E). Total protein was harvested from the cultures for western blotting of the indicated proteins (C,E). Total RNA was harvested from the cultures for quantitative real-time RT-PCR analysis of Sox9 gene expression (D). *P<0.01, one-way ANOVA followed by Newman-Keuls post test.
Prior work has established that BMP signaling regulates Sox9 gene expression during endochondral bone formation (Yoon et al., 2005). We, therefore, performed a series of experiments to determine whether TAK1 is involved in Sox9 regulation by BMP2. Primary sternal chondrocytes from Tak1f/f mice were isolated and infected with control adenovirus, Ad-GFP, or adenovirus encoding Cre recombinase, Ad-Cre. Although BMP2 treatment of control chondrocytes stimulated Sox9 gene expression, deletion of Tak1 inhibited the expression of Sox9 following BMP2 treatment (Fig. 5D). Protein levels of SOX9 were similarly suppressed following BMP2 treatment of the Tak1-deficient cultures (Fig. 5E). Additionally, we treated the chondrogenic rat chondrosarcoma (RCS) cell line with BMP2 in the presence or absence of TAK1 kinase inhibitor, 5Z-7-oxozeanol (TI-2), and found that inhibition of TAK1 kinase activity completely suppressed BMP2-mediated Sox9 gene expression, consistent with our results in primary chondrocytes (supplementary material Fig. S3A,B). Because TAK1 can function as an effector of both BMP- and TGF-β-mediated MAPK signaling, we performed similar experiments to test whether TGF-β–TAK1 signaling could also induce Sox9 gene expression in chondrocytes. Using either RCS cells or wild-type primary sternal chondrocytes, we treated cells with TGF-β1 in the presence or absence of the TAK1 kinase inhibitor, TI-2. Treatment of chondrocytes with TGF-β1, however, did not induce Sox9 expression in either chondrogenic cell type (supplementary material Fig. S4A,B).
ATF2 binds to the Sox9 promoter to regulate gene expression following BMP2 treatment
To determine whether TAK1 is involved in activation of the Sox9 promoter, we transfected RCS cells with a luciferase reporter construct containing an ∼1.0 kb segment of the murine Sox9 promoter (bases −1 to −967 relative to the transcriptional start site). BMP2 treatment induced activation of the Sox9 promoter more than sevenfold (Fig. 6A). Treatment of the cultures with the TAK1 kinase inhibitor, however, resulted in more than a 50% reduction in BMP2-stimulated Sox9 promoter activity (Fig. 6A). In a similar experiment, treatment of cells with TGF-β1 did not induce activation of the Sox9 promoter (supplementary material Fig. S4C).
Fig. 6.

Non-canonical BMP signaling positively regulates Sox9 transcription by promoter activation. (A) Luciferase reporter assay using lysates from RCS cells transfected with a luciferase reporter construct containing a 1.0 kb fragment of the murine Sox9 promoter. The cells were serum starved for 12 hours and then treated with either vehicle, 5Z-7-oxozeanol (TI-2; 3 µM) alone, BMP2 (100 ng/ml) alone, or TI-2 and BMP-2 in combination for 8 hours. *P<0.05, one-way ANOVA followed by Newman-Keuls test. (B) Reporter assay using lysates from RCS cells co-transfected with the Sox9 luciferase reporter and control siRNA or siRNA targeting Smad1 or Atf2. The cells were serum starved for 12 hours and then treated with vehicle or BMP2 for 8 hours. *P<0.05, one-way ANOVA followed by Newman-Keuls test; #P<0.05, one-way ANOVA followed by Newman-Keuls test compared with the BMP2-treated, control siRNA group. (C) Schematic of the Sox9 promoter region showing the putative ATF2-binding site relative to the Sox9 open reading frame (ORF) and transcriptional start site (arrow) as well as the location of the primer set used for PCR amplification. (D) ChIP assay with cell lysates from primary sternal chondrocytes serum starved for 12 hours and then treated with vehicle, TI-2 (3 µM) alone, BMP2 (100 ng/ml) alone, or TI-2 and BMP2 in combination for 90 minutes.
To determine the relative contribution of canonical (SMAD-dependent) and non-canonical (MAPK-dependent) signaling on the regulation of Sox9 gene expression by BMP2, RCS cells were transfected with either SMAD1 siRNA or ATF2 siRNA, or with both siRNAs together, and a Sox9 promoter reporter assay performed (Fig. 6B). Alone, SMAD1 siRNA (39%) and ATF2 siRNA (43%) both significantly reduced BMP2-mediated activation of the Sox9 promoter (Fig. 6B). Greater repression (65%) of Sox9 promoter activity, however, was observed when SMAD1 and ATF2 siRNAs were used in combination (Fig. 6B).
Analysis of the murine Sox9 promoter demonstrated a putative ATF2-binding site located from −861 to −848 base pairs (Fig. 6C). Chromatin immunoprecipitation (ChIP) analysis of the Sox9 promoter was performed in primary sternal chondrocytes using an anti-ATF2 antibody to precipitate DNA fragments from control and BMP2-treated cultures in the presence or absence of the TAK1 kinase inhibitor. Immunoprecipitated DNA was amplified with primers specific for sequences located within the Sox9 promoter. Gel electrophoresis demonstrated the presence of a 153 base pair amplified fragment of the Sox9 promoter in chondrocytes treated with BMP2 only and immunoprecipitated with the anti-ATF2 antibody, consistent with ATF2 binding to the Sox9 promoter (Fig. 6C; supplementary material Fig. S5). In contrast, the amplified DNA fragment was absent in cultures treated with control medium or with BMP2 in combination with the TAK1 kinase inhibitor as well as in control and BMP2-treated cultures incubated with normal rabbit IgG (Fig. 6C). These findings suggest that BMP2 regulates Sox9 gene expression through a mechanism that includes the TAK1–ATF2 signaling pathway.
Discussion
The work presented here demonstrates for the first time a crucial role for TAK1 in regulation of postnatal growth plate and articular cartilage development. Using the Col2a1-CreERT2 transgene in combination with the Tak1f/f allele, we deleted Tak1 specifically in committed chondrocytes of postnatal mice. Conditional deletion of Tak1 in this manner resulted in severe growth retardation and disorganization of the growth plate cartilage with defects in chondrocyte proliferation. The most striking and surprising finding was the diminished proteoglycan content of the articular cartilage matrix. Further in vivo and in vitro investigations led to the identification of a role for TAK1 in regulation of the gene expression of several cartilage ECM components as well as that of the SOX trio of transcription factors. Specifically, we identified Sox9 as a novel transcriptional target of ATF2, uncovering a role for TAK1–ATF2 signaling in direct regulation of Sox9 gene expression.
Previous studies demonstrated a requirement for TAK1 in chondrocytes during embryonic development (Gunnell et al., 2010; Shim et al., 2009). Using the non-inducible Col2a1-Cre or Prx1-Cre transgenes, it was discovered that Tak1 gene deletion in cartilage results in chondrodysplasia due to defects in chondrocyte proliferation and maturation as well as survival. These mice exhibited long bone phenotypes similar to several other BMP loss-of-function mouse models suggesting that TAK1 plays a very important role as a downstream effector of the BMP signaling pathway during embryonic development of the appendicular skeleton (Bandyopadhyay et al., 2006; Retting et al., 2009; Storm et al., 1994; Yoon et al., 2005). The Cre transgenes used in these studies are active at very early stages of endochondral bone development. Gene recombination occurs in chondro-osteo progenitor cells in the case of the Col2a1-Cre transgene and in limb mesenchymal stem cells in the case of the Prx1-Cre transgene. Interestingly, the severity of the phenotypes, particularly with regard to proliferation and survival, progressively decreased over time (Gunnell et al., 2010). Whether this was due to a compensatory effect, or suggests that the role of TAK1 becomes less critical with skeletal maturity was unclear. In the study presented here, we deleted Tak1 specifically in chondrocytes at 1 week of age and observed severe growth retardation evident at 1 month of age suggesting that TAK1 remains important in maintaining postnatal skeletal growth. We observed a smaller secondary ossification center surrounded by many immature chondrocytes suggesting that deletion at 1 week, the time when the secondary ossification center is just beginning to form, delays formation, consistent with the findings of Shim and colleagues (Shim et al., 2009). Finally, we observed decreased proliferation of TAK1-deficient chondrocytes both in vivo and in vitro, also consistent with previous findings. Our analysis of the growth plate cartilage, therefore, establishes that TAK1 plays a similarly essential role in growth plate chondrocytes during postnatal development as it does during embryonic development. However, we uncovered a novel role for TAK1 in the development of the articular cartilage that was precluded by embryonic or very early postnatal lethality in previous studies.
Although derived from a common precursor and sharing cellular and molecular features, growth plate and articular chondrocytes have marked functional differences (Yamane et al., 2007). Longitudinal bone growth is dependent upon the proliferation, maturation and turnover of a population of growth plate chondrocytes. In contrast, articular chondrocytes have a relatively stable phenotype and maintain an exquisitely organized matrix that is highly resistant to long-term compression and shear forces (Becerra et al., 2010). Development of the articular cartilage continues throughout adolescence in humans and in mice (Chan et al., 2012). Our studies established that TAK1 has an essential role in the postnatal development of the articular cartilage extracellular matrix. The articular cartilage of mice with chondrocyte-specific Tak1 deficiency had reduced type II collagen and proteoglycan content as determined by immunostaining and Alcian Blue staining, respectively. This was consistent with the finding of reduced expression levels of the Col2a1, Col9a1 and Acan genes in Tak1-deficient articular cartilage tissue and primary sternal chondrocytes. These genes are unique to chondrocytes and are required for normal articular cartilage function. Type II collagen is the most abundant constituent of articular cartilage, accounting for ∼50% of the dry weight of this tissue (Becerra et al., 2010). Type IX collagen is covalently cross-linked to the surface of the type II collagen fibrils and interconnects the type II collagen matrix with other components of the extracellular matrix, including fibronectin, matrilin 3, and cartilage oligomeric protein (COMP) (Parsons et al., 2011). Aggregating proteoglycans are necessary for boundary lubrication of the articular surface and their negative charge is the basis of high osmotic pressure that provides resistance to compressive loads (Seror et al., 2011).
In addition to reducing expression of cartilage matrix genes, Tak1 deletion resulted in increased expression of matrix metalloproteinases and aggrecanases that catabolize the collagen matrix and target proteoglycan degradation, respectively. Chondrocytes in articular cartilage tissue and in isolated cell culture had increased expression of Mmp13 and Adamts5 upon deletion of Tak1. Although these proteins are necessary for normal cartilage homeostasis, increased levels result in tissue catabolism and are associated with the development of osteoarthritis (Struglics and Hansson, 2012; Zhen et al., 2008). In contrast, mice with deletion of Adamts5 or Mmp13 have reduced joint degeneration following injury compared with wild-type mice (Glasson et al., 2005; Li et al., 2011; Majumdar et al., 2007; Wang et al., 2013). While we did not observe any histopathological signs of osteoarthritis in our chondrocyte-specific Tak1-deficient mice, we cannot rule out that over time, loss of TAK1 may lead to development of the disease. Indeed, mice with postnatal disruption of components from either the TGF-β or BMP signaling pathways develop phenotypes reminiscent of osteoarthritis. For example, global deletion of Smad3, expression of a dominant-negative TGF-β type II receptor (TβRII) in skeletal tissue, and chondrocyte-specific loss of TβRII (Col2a1-CreERT2; Tgfbr2f/f) all result in osteoarthritis due to aberrant hypertrophy of articular chondrocytes evident by ectopic expression of Col10a1 in the articular cartilage (Serra et al., 1997; Shen et al., 2013; Yang et al., 2001). Additionally, loss of Bmpr1a expression in developing synovial joints leads to erosion of the articular cartilage with loss of Col2a1 and Acan gene expression (Rountree et al., 2004). Finally, aged mice expressing a dominant-negative p38 MAP kinase, a downstream target of TAK1, are reported to develop osteoarthritic lesions in knee joint articular cartilage (Namdari et al., 2008). Whether Tak1 deficiency will also result in insufficient maintenance of the articular cartilage leading eventually to osteoarthritis will require deletion of Tak1 after skeletal maturity and characterization of cartilage phenotypes at time points later than those addressed in the current study. This will be a focus of future work.
Interestingly, the articular cartilage phenotype of the TAK1-deficient mice appeared very similar to that of mice with postnatal deletion of Sox9 specifically in chondrocytes using the Agc1-CreERT2 knock-in allele (Henry et al., 2012). We, therefore, investigated Sox9 as a potential downstream target of TAK1. We observed a decrease in expression of Sox9 in articular cartilage tissue as well as in primary sternal chondrocytes deficient for Tak1. Conversely, expression of SOX9 was increased upon overexpression of TAK1 in chondrocyte cultures. The changes in Sox5 and Sox6 expression paralleled Sox9, consistent with the known role of SOX9 as a regulator of these factors (Akiyama et al., 2002). In combination, SOX5, SOX6 and SOX9 have been named the SOX trio for their cooperative role in regulating the chondrocyte phenotype and, specifically, Acan and Col2a1 gene expression (Akiyama and Lefebvre, 2011; Han and Lefebvre, 2008; Ikeda et al., 2004). Although chondrocyte-specific loss of SOX9 or TAK1 both lead to decreased proteoglycan content as well as decreased Acan and Col2a1 transcript levels in articular cartilage, only the TAK1-deficient mice exhibited a decrease in type 2 collagen protein levels. The concomitant increase in Mmp13 gene expression upon loss of TAK1 probably accounts for the loss of type 2 collagen protein and must be due to roles of TAK1 in regulation of Mmp13 gene expression independent of SOX9.
Given that deletion of Tak1 in chondrocytes leads to reduced Sox9 gene expression and phenotypes similar to those due to loss of BMP signaling, we investigated the possibility that TAK1 is a downstream effector of BMP-mediated Sox9 gene expression. It is well established that BMP signaling regulates Sox9 expression during development, but the precise mechanism by which it does so is unclear (Yoon and Lyons, 2004; Yoon et al., 2005; Zehentner et al., 1999). It was suggested previously that the non-canonical signaling pathway may be involved when Pan and colleagues showed that intact p38 MAP kinase signaling is required for BMP2-mediated chondrogenic differentiation of and Sox9 expression in mouse embryonic fibroblasts (Pan et al., 2008). Our in vitro experiments confirm that BMP2 induces Sox9 promoter activation and gene expression in committed chondrocytes and further show that this effect is dependent upon TAK1 signaling. In the absence of Tak1, or upon inhibition of TAK1 kinase activity, BMP2-induced Sox9 gene and protein expressions were reduced. Additionally, we show that full activation of the Sox9 promoter by BMP2 requires both SMAD1 and ATF2. Determining whether these factors bind the promoter independently or within the same molecular complex requires further analysis. We did, however, identify an ATF2-binding site within the Sox9 promoter and only in the presence of BMP2 were we able to successfully immunoprecipitate this region of the chromatin with an antibody against ATF2. Furthermore, inhibition of TAK1 kinase activity prevented binding suggesting that non-canonical TAK1–ATF2 signaling is required for full activation of the Sox9 promoter by BMP2. Interestingly, global deletion of Atf2 results in some of the same phenotypes as chondrocyte-specific deletion of Tak1, namely reduced chondrocyte proliferation and growth plate disorganization, consistent with ATF2 as a downstream target of TAK1 in regulation of the chondrocyte phenotype (Reimold et al., 1996).
Although TAK1 is known to be an important mediator of TGF-β signaling in other cell types, our data presented here and in previous publications (Gunnell et al., 2010; Shim et al., 2009) suggest that TAK1 is primarily a mediator of BMP signaling in the committed chondrocyte. As described in detail above, chondrocyte-specific deletion of Tak1 closely resembles that of other chondrocyte-specific BMP loss-of-function models. Chondrocyte-specific loss of TGF-β signaling, however, has little effect on long bone development, but results in an osteoarthritis-like phenotype postnatally as a result of excessive articular chondrocyte hypertrophy (Baffi et al., 2004; Shen et al., 2013). Here, we observed no changes indicative of excessive chondrocyte hypertrophy. Interestingly, in vitro deletion of Tak1 in chondrocytes resulted in downregulation of basal levels of the BMP target gene Id1, but no change in TGF-β target gene PAI1 (supplementary material Fig. S4D), again highlighting the importance of TAK1 in the maintenance of canonical BMP signaling in committed chondrocytes. We show here that this signaling is clearly important in promoting transcription of Sox9. It is reported that TGF-β is also important in regulation of SOX9, but at the post-translational level and in mesenchymal progenitor cells (Furumatsu et al., 2005). Of note, loss of Tak1 or Tgfbr2 in limb mesenchymal progenitor cells using the Prx1-Cre transgene, results in some similar phenotypes, among them fused joints, suggesting that TAK1 is a key player downstream of TGF-β in this specific cell population (Gunnell et al., 2010; Longobardi et al., 2012; Seo and Serra, 2007). The role of TAK1 in BMP and TGF-β signaling is, therefore, quite complex and seemingly different depending upon cell type. Therefore, although we cannot rule out that TAK1 may be involved in other signaling pathways in the chondrocyte, our data presented here provide evidence that it is essential for BMP signaling.
In summary, our findings further establish TAK1 as a crucial regulator of growth plate and articular cartilage development. Postnatal loss of TAK1 results in a growth plate cartilage phenotype reminiscent of BMP loss-of-function and similar to that when Tak1 is deleted at very early stages of embryonic skeletal development. Surprisingly, we discovered that TAK1 reciprocally regulates genes responsible for cartilage synthesis and breakdown, leading to a marked decrease of proteoglycan and type II collagen content in unmineralized articular cartilage upon loss of TAK1 in postnatal articular chondrocytes. Also novel is our finding that TAK1–ATF2 signaling regulates BMP-mediated Sox9 transcription. Future studies will focus on investigating the potential role of TAK1 in articular cartilage maintenance and disease as well as the mechanism by which TAK1 regulates Mmp13 gene expression.
Materials and Methods
Mouse strains
Tak1f/f mice and ROSA26R mice were crossed with Col2a1-CreERT2 transgenic mice to generate Col2a1-CreERT2; Tak1f/f mice and Col2a1-CreERT2; ROSA26R reporter mice, respectively (Chen et al., 2007; Xie et al., 2006). Tamoxifen (100 mg/kg) was administered to transgenic mice by injection into the peritoneum beginning at P7 every other day for a total of three doses. Animals were maintained in accordance with the ‘NIH Guide for the Care and Use of Laboratory Animals’. The institutional committee on animal care approved the experimental protocol.
Cell isolation and culture
Primary sternal chondrocytes were isolated from 3-day-old Tak1f/f mice as previously described (Gunnell et al., 2010) and cultured in DMEM, high glucose (Life Technologies, Grand Island, NY, USA) containing 10% FBS and 1% penicillin and streptomycin (Life Technologies, Grand Island, NY, USA). Ad5-CMV-GFP and Ad5-CMV-Cre viruses were purchased from Baylor Vector Development Laboratory (Houston, TX, USA). Ad5-CMV-TAK1 was purchased from Vector Biolabs (Philadelphia, PA, USA). The primary chondrocytes were infected for 48 hours and then harvested immediately for RNA and protein isolation. In experiments with BMP2 or TGF-β1 treatment, adenoviral-infected cultures were serum starved for 12 hours prior to the addition of BMP2 (100 ng/ml; R&D Systems, Minneapolis, MN, USA) or TGF-β1 (5 ng/ml) to the culture medium. Rat chondrosarcoma (RCS) cells were cultured in the same medium as primary chondrocytes (Mukhopadhyay et al., 1995). For the TAK1 kinase inhibition experiments, the TAK1 inhibitor (TI-2), 5Z-7-oxozeanol, (LLZ1640-2; Bioaustralis, Smithfield, NSW, Australia) was added to cultures 30 minutes prior to BMP2 or TGF-β1 treatment (Ninomiya-Tsuji et al., 2003).
Western blotting
Western blotting was performed as previously described (Gunnell et al., 2010). The antibodies used included those recognizing TAK1, phosphorylated (phospho-) SMAD1/5/8, phospho-p38, total p38, phospho-ATF2, total ATF2 (Cell Signaling Technology, Danvers, MA, USA), total SMAD1/5/8, SOX9, SOX5, SOX6 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and β-actin (Sigma-Aldrich, St. Louis, MO, USA).
Luciferase reporter assays
A 967 bp fragment (bases −1 to −967) of the mouse Sox9 promoter was amplified by PCR (forward primer: 5′-attcacgcgttcaaccccggagtagttttg-3′, reverse primer: 5′-cgacttccagctcagggtct-3′) and placed into a pGL3 plasmid containing the firefly luciferase gene (Promega, Madison, WI, USA). RCS cells were either co-transfected directly with the Sox9 luciferase reporter construct and the SV40-Renilla reporter construct using Fugene-HD (Roche, Indianapolis, IN, USA) or, first, transfected with siRNA targeting Smad1 (Life Technologies, Grand Island, NY, USA) and/or siRNA targeting Atf2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) using Lipofectamine™ RNAiMAX (Life Technologies, Grand Island, NY, USA). For experiments utilizing siRNA, cells were transfected with the luciferase reporter constructs 24 hours following transfection of siRNA. In both cases, 24 hours after transfection of the luciferase reporter constructs, cells were serum-starved overnight and then treated with TI-2 (3 µM), BMP2 (100 ng/ml), or TGF-β1 (5 ng/ml). Luciferase activity was measured 8 hours later using the Dual-Promoter Luciferase Assay Kit (Promega, Madison, WI, USA) and firefly luciferase was normalized to Renilla luciferase.
Articular chondrocyte mRNA isolation
1-week-old Col2a1-CreERT2; Tak1f/f transgenic mice and Cre-negative littermate control mice were injected with tamoxifen and killed at 1 month of age. Both femur heads were exposed, and the articular cartilage was separated from the underlying subchondral bone using sterile tweezers. The tissue was immediately frozen in liquid nitrogen, Trizol reagent (Life Technologies, Grand Island, NY, USA) was added, the tissue homogenized, and the manufacturer's protocol for RNA isolation performed. Five mice were used in each group.
Quantitative real-time RT-PCR
Total RNA from cultured cells was isolated using the RNeasy kit (Qiagen, Valencia, CA, USA). Total RNA (1 µg) from cells or articular tissues was reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Real-time PCR was performed on a Rotor-Gene 6000 real-time DNA amplification system (Qiagen, Valencia, CA, USA) using the PerfeCTa SYBR Green SuperMix (Quanta BioSciences, Inc., Gaithersburg, MD, USA) according to the manufacturer's instructions. Supplementary material Table S1 includes a list of primers used.
Histology and immunohistochemistry
For X-gal staining, mouse knee joint samples were harvested and fixed in 0.25% glutaraldehyde, decalcified, and embedded for frozen sectioning and staining as previously described (Chen et al., 2007). For all other histology and immunohistochemistry, knee joint samples were harvested and fixed in 4% paraformaldehyde, decalcified, dehydrated and embedded in paraffin. Mid-sagittal sections were cut (5 µm) and Alcian Blue, Hematoxylin and Orange G Eosin stainings were performed for visualization of cartilage and bone. For immunohistochemisty, sections were de-paraffinized and rehydrated. Antigen retrieval was performed in 0.01 M sodium citrate buffer in a pressure chamber followed by quenching of endogenous peroxidase activity in 3% H2O2 for 20 minutes. Blocking of nonspecific antibody binding sites was performed with a 1∶20 dilution of normal goat serum for 20 minutes. Sections were incubated overnight with a 1∶200 dilution of anti-TAK1 antibody, a 1∶200 dilution of anti-SOX9 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), a 1∶100 dilution of anti-collagen type II antibody (Rockland Immunochemicals, Gilbertsville, PA, USA), a 1∶200 dilution of phospho-SMAD1/5/8 antibody, or a 1∶200 dilution of anti-phospho-ATF2 antibody (Cell Signaling Technology, Danvers, MA, USA). Sections were rinsed in phosphate-buffered saline (PBS) three times for 5 minutes each and incubated at room temperature with a 1∶200 dilution (anti-COL2A1 and anti-SOX9) or a 1∶1000 dilution (anti-TAK1, anti-phospho-SMAD1/5/8, and anti-P-ATF2) of biotinylated goat anti-rabbit secondary antibody for 30 minutes. After a final rinse with PBS, antigen was detected following application of horseradish-peroxidase-conjugated streptavidin and a 5-minute incubation with the Romulin AEC Chromogen Kit (Biocare Medical, Concord, CA, USA). Slides were counterstained with Hematoxylin or Fast Green.
Chromatin immunoprecipitation assay
Primary sternal chondrocyte cultures were serum starved for 12 hours and then treated with vehicle (DMSO), TI-2 alone, BMP2 alone or TI-2 and BMP2 in combination for 90 minutes prior to performing chromatin immunoprecipitation (ChIP). Chromatin isolation and immunoprecipitation (IP) was performed with the MAGnify Chromatin Immunoprecipitation System following the manufacturer's protocol (Life Technologies, Grand Island, NY, USA) and an anti-ATF2 antibody or normal rabbit IgG (Cell Signaling Technology, Danvers, MA, USA). The region of the Sox9 promoter containing the putative ATF2-binding site was amplified by PCR before or after immunoprecipitation (forward primer: 5′-ctacgaggggcgtcagagta-3′, reverse primer: 5′-ggggaatcaatgaaaaccaa-3′). Amplified DNA was separated by gel electrophoresis using 1% agarose.
In vivo chondrocyte proliferation analysis
Mice were injected intraperitoneally with 5-bromo-2′-deoxyuridine (BrdU; Invitrogen, Carlsbad, CA, USA) at a dose of 0.1 ml/100 g body weight. Three hours later, mice were sacrificed and hindlimbs fixed, decalcified, dehydrated, embedded into paraffin, and sectioned at 5 µm. BrdU-labeled cells were detected by performing immunohistochemistry for BrdU with the Invitrogen BrdU Staining Kit (Invitrogen, Carlsbad, CA, USA) containing a biotinylated monoclonal anti-BrdU antibody.
In vitro chondrocyte proliferation analysis
Primary sternal chondrocytes were isolated from 3-day-old Tak1f/f mice and subsequently infected with Ad5-CMV-GFP, Ad5-CMV-Cre or Ad5-CMV-TAK1. After 72 hours, cells were labeled with 10 µM BrdU for 4 hours prior to harvest. Cells were harvested and analyzed for BrdU incorporation using the Cell Proliferation ELISA, BrdU Kit (Roche, Indianapolis, IN, USA).
Statistics
Data are presented as the means ± s.e.m. Statistical significance was determined by Student's t-tests or one-way ANOVA followed by Dunnett's test or Neuman-Keuls test as indicated in the figure legends; P-values of less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
The authors thank Dr Michael Schneider and Dr Di Chen for providing mouse strains, and Dr Benoit de Crombrugghe for providing the RCS cell line. The authors appreciate the technical support from the Center for Musculoskeletal Research Histology Core members Ryan Tierney, Sarah Mack and Kathy Maltby for assistance with the tissue sample processing.
Footnotes
Author contributions
L.G., R.J.O. and J.H.J. conceived and designed the study. M.J.Z. and M.J.H. participated in study design. L.G., T.S., Y.D. and D.M.H. performed data collection and analysis. L.G., M.J.Z., E.M.S., M.J.H., R.J.O. and J.H.J. interpreted the data. L.G., R.J.O. and J.H.J. drafted the manuscript. M.J.H., E.M.S. and J.H.J. edited the manuscript.
Funding
This work was supported by the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases [grant numbers P50 AR054041 and R01 AR053717 to R.J.O., P30 AR061307 to E.M.S., R01 AR063071 to M.J.H.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.135483/-/DC1
References
- Akiyama H., Lefebvre V. (2011). Unraveling the transcriptional regulatory machinery in chondrogenesis. J. Bone Miner. Metab. 29, 390–395 10.1007/s00774-011-0273-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Chaboissier M. C., Martin J. F., Schedl A., de Crombrugghe B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828 10.1101/gad.1017802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer C. W., Dowthwaite G. P., Francis-West P. (2003). Development of synovial joints. Birth Defects Res. C Embryo Today 69, 144–155 10.1002/bdrc.10015 [DOI] [PubMed] [Google Scholar]
- Baffi M. O., Slattery E., Sohn P., Moses H. L., Chytil A., Serra R. (2004). Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev. Biol. 276, 124–142 10.1016/j.ydbio.2004.08.027 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A., Tsuji K., Cox K., Harfe B. D., Rosen V., Tabin C. J. (2006). Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2, e216 10.1371/journal.pgen.0020216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra J., Andrades J. A., Guerado E., Zamora-Navas P., López-Puertas J. M., Reddi A. H. (2010). Articular cartilage: structure and regeneration. Tissue Eng. Part B Rev. 16, 617–627 10.1089/ten.teb.2010.0191 [DOI] [PubMed] [Google Scholar]
- Bridgewater L. C., Lefebvre V., de Crombrugghe B. (1998). Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J. Biol. Chem. 273, 14998–15006 10.1074/jbc.273.24.14998 [DOI] [PubMed] [Google Scholar]
- Chan E. F., Harjanto R., Asahara H., Inoue N., Masuda K., Bugbee W. D., Firestein G. S., Hosalkar H. S., Lotz M. K., Sah R. L. (2012). Structural and functional maturation of distal femoral cartilage and bone during postnatal development and growth in humans and mice. Orthop. Clin. North Am. 43, 173–185v. (v.) 10.1016/j.ocl.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Lichtler A. C., Sheu T. J., Xie C., Zhang X., O'Keefe R. J., Chen D. (2007). Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis 45, 44–50 10.1002/dvg.20261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowthwaite G. P., Bishop J. C., Redman S. N., Khan I. M., Rooney P., Evans D. J., Haughton L., Bayram Z., Boyer S., Thomson B. et al. (2004). The surface of articular cartilage contains a progenitor cell population. J. Cell Sci. 117, 889–897 10.1242/jcs.00912 [DOI] [PubMed] [Google Scholar]
- Dy P., Wang W., Bhattaram P., Wang Q., Wang L., Ballock R. T., Lefebvre V. (2012). Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell 22, 597–609 10.1016/j.devcel.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furumatsu T., Tsuda M., Taniguchi N., Tajima Y., Asahara H. (2005). Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 280, 8343–8350 10.1074/jbc.M413913200 [DOI] [PubMed] [Google Scholar]
- Glasson S. S., Askew R., Sheppard B., Carito B., Blanchet T., Ma H. L., Flannery C. R., Peluso D., Kanki K., Yang Z. et al. (2005). Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434, 644–648 10.1038/nature03369 [DOI] [PubMed] [Google Scholar]
- Gunnell L. M., Jonason J. H., Loiselle A. E., Kohn A., Schwarz E. M., Hilton M. J., O'Keefe R. J. (2010). TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J. Bone Miner. Res. 25, 1784–1797 10.1002/jbmr.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Lefebvre V. (2008). L-Sox5 and Sox6 drive expression of the aggrecan gene in cartilage by securing binding of Sox9 to a far-upstream enhancer. Mol. Cell. Biol. 28, 4999–5013 10.1128/MCB.00695-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry S. P., Liang S., Akdemir K. C., de Crombrugghe B. (2012). The postnatal role of Sox9 in cartilage. J. Bone Miner. Res. 27, 2511–2525 10.1002/jbmr.1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T., Kamekura S., Mabuchi A., Kou I., Seki S., Takato T., Nakamura K., Kawaguchi H., Ikegawa S., Chung U. I. (2004). The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 50, 3561–3573 10.1002/art.20611 [DOI] [PubMed] [Google Scholar]
- Kronenberg H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332–336 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- Leung V. Y., Gao B., Leung K. K., Melhado I. G., Wynn S. L., Au T. Y., Dung N. W., Lau J. Y., Mak A. C., Chan D. et al. (2011). SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 7, e1002356 10.1371/journal.pgen.1002356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Anemaet W., Diaz M. A., Buchanan S., Tortorella M., Malfait A. M., Mikecz K., Sandy J. D., Plaas A. (2011). Knockout of ADAMTS5 does not eliminate cartilage aggrecanase activity but abrogates joint fibrosis and promotes cartilage aggrecan deposition in murine osteoarthritis models. J. Orthop. Res. 29, 516–522 10.1002/jor.21215 [DOI] [PubMed] [Google Scholar]
- Longobardi L., Li T., Myers T. J., O'Rear L., Ozkan H., Li Y., Contaldo C., Spagnoli A. (2012). TGF-β type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Dev. Cell 23, 71–81 10.1016/j.devcel.2012.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar M. K., Askew R., Schelling S., Stedman N., Blanchet T., Hopkins B., Morris E. A., Glasson S. S. (2007). Double-knockout of ADAMTS-4 and ADAMTS-5 in mice results in physiologically normal animals and prevents the progression of osteoarthritis. Arthritis Rheum. 56, 3670–3674 10.1002/art.23027 [DOI] [PubMed] [Google Scholar]
- Mariani F. V., Martin G. R. (2003). Deciphering skeletal patterning: clues from the limb. Nature 423, 319–325 10.1038/nature01655 [DOI] [PubMed] [Google Scholar]
- Moriguchi T., Kuroyanagi N., Yamaguchi K., Gotoh Y., Irie K., Kano T., Shirakabe K., Muro Y., Shibuya H., Matsumoto K. et al. (1996). A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J. Biol. Chem. 271, 13675–13679 10.1074/jbc.271.23.13675 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay K., Lefebvre V., Zhou G., Garofalo S., Kimura J. H., de Crombrugghe B. (1995). Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-alpha 1(II) collagen gene. J. Biol. Chem. 270, 27711–27719 10.1074/jbc.270.46.27711 [DOI] [PubMed] [Google Scholar]
- Namdari S., Wei L., Moore D., Chen Q. (2008). Reduced limb length and worsened osteoarthritis in adult mice after genetic inhibition of p38 MAP kinase activity in cartilage. Arthritis Rheum. 58, 3520–3529 10.1002/art.23999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng L. J., Wheatley S., Muscat G. E., Conway-Campbell J., Bowles J., Wright E., Bell D. M., Tam P. P., Cheah K. S., Koopman P. (1997). SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev. Biol. 183, 108–121 10.1006/dbio.1996.8487 [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J., Kajino T., Ono K., Ohtomo T., Matsumoto M., Shiina M., Mihara M., Tsuchiya M., Matsumoto K. (2003). A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J. Biol. Chem. 278, 18485–18490 10.1074/jbc.M207453200 [DOI] [PubMed] [Google Scholar]
- Pan Q., Yu Y., Chen Q., Li C., Wu H., Wan Y., Ma J., Sun F. (2008). Sox9, a key transcription factor of bone morphogenetic protein-2-induced chondrogenesis, is activated through BMP pathway and a CCAAT box in the proximal promoter. J. Cell. Physiol. 217, 228–241 10.1002/jcp.21496 [DOI] [PubMed] [Google Scholar]
- Parsons P., Gilbert S. J., Vaughan-Thomas A., Sorrell D. A., Notman R., Bishop M., Hayes A. J., Mason D. J., Duance V. C. (2011). Type IX collagen interacts with fibronectin providing an important molecular bridge in articular cartilage. J. Biol. Chem. 286, 34986–34997 10.1074/jbc.M111.238188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J., Whitmarsh A. J., Barrett T., Dérijard B., Davis R. J. (1996). MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 16, 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold A. M., Grusby M. J., Kosaras B., Fries J. W., Mori R., Maniwa S., Clauss I. M., Collins T., Sidman R. L., Glimcher M. J. et al. (1996). Chondrodysplasia and neurological abnormalities in ATF-2-deficient mice. Nature 379, 262–265 10.1038/379262a0 [DOI] [PubMed] [Google Scholar]
- Retting K. N., Song B., Yoon B. S., Lyons K. M. (2009). BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136, 1093–1104 10.1242/dev.029926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rountree R. B., Schoor M., Chen H., Marks M. E., Harley V., Mishina Y., Kingsley D. M. (2004). BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2, e355 10.1371/journal.pbio.0020355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen H., Vuorio E., Säämänen A. M. (2001). Expression of Sox9 and type IIA procollagen during attempted repair of articular cartilage damage in a transgenic mouse model of osteoarthritis. Arthritis Rheum. 44, 947–955 [DOI] [PubMed] [Google Scholar]
- Seo H. S., Serra R. (2007). Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 310, 304–316 10.1016/j.ydbio.2007.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seror J., Merkher Y., Kampf N., Collinson L., Day A. J., Maroudas A., Klein J. (2011). Articular cartilage proteoglycans as boundary lubricants: structure and frictional interaction of surface-attached hyaluronan and hyaluronan—aggrecan complexes. Biomacromolecules 12, 3432–3443 10.1021/bm2004912 [DOI] [PubMed] [Google Scholar]
- Serra R., Johnson M., Filvaroff E. H., LaBorde J., Sheehan D. M., Derynck R., Moses H. L. (1997). Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol. 139, 541–552 10.1083/jcb.139.2.541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J., Li J., Wang B., Jin H., Wang M., Zhang Y., Yang Y., Im H. J., O'Keefe R., Chen D. (2013). Deletion of the Type II TGF-β receptor gene in articular chondrocytes leads to a progressive OA-like phenotype in mice. Arthritis Rheum doi: 10.1002/art.38122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J. H., Greenblatt M. B., Xie M., Schneider M. D., Zou W., Zhai B., Gygi S., Glimcher L. H. (2009). TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 28, 2028–2041 10.1038/emboj.2009.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm E. E., Huynh T. V., Copeland N. G., Jenkins N. A., Kingsley D. M., Lee S. J. (1994). Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 368, 639–643 10.1038/368639a0 [DOI] [PubMed] [Google Scholar]
- Struglics A., Hansson M. (2012). MMP proteolysis of the human extracellular matrix protein aggrecan is mainly a process of normal turnover. Biochem. J. 446, 213–223 10.1042/BJ20120274 [DOI] [PubMed] [Google Scholar]
- Tsuji K., Bandyopadhyay A., Harfe B. D., Cox K., Kakar S., Gerstenfeld L., Einhorn T., Tabin C. J., Rosen V. (2006). BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 38, 1424–1429 10.1038/ng1916 [DOI] [PubMed] [Google Scholar]
- Uusitalo H., Hiltunen A., Ahonen M., Gao T. J., Lefebvre V., Harley V., Kähäri V. M., Vuorio E. (2001). Accelerated up-regulation of L-Sox5, Sox6, and Sox9 by BMP-2 gene transfer during murine fracture healing. J. Bone Miner. Res. 16, 1837–1845 10.1359/jbmr.2001.16.10.1837 [DOI] [PubMed] [Google Scholar]
- Wang M., Sampson E. R., Jin H., Li J., Ke Q. H., Im H. J., Chen D. (2013). MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15, R5 10.1186/ar4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M., Zhang D., Dyck J. R., Li Y., Zhang H., Morishima M., Mann D. L., Taffet G. E., Baldini A., Khoury D. S. et al. (2006). A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc. Natl. Acad. Sci. USA 103, 17378–17383 10.1073/pnas.0604708103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. (1995). Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270, 2008–2011 10.1126/science.270.5244.2008 [DOI] [PubMed] [Google Scholar]
- Yamane S., Cheng E., You Z., Reddi A. H. (2007). Gene expression profiling of mouse articular and growth plate cartilage. Tissue Eng. 13, 2163–2173 10.1089/ten.2006.0431 [DOI] [PubMed] [Google Scholar]
- Yang X., Chen L., Xu X., Li C., Huang C., Deng C. X. (2001). TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 153, 35–46 10.1083/jcb.153.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon B. S., Lyons K. M. (2004). Multiple functions of BMPs in chondrogenesis. J. Cell. Biochem. 93, 93–103 10.1002/jcb.20211 [DOI] [PubMed] [Google Scholar]
- Yoon B. S., Ovchinnikov D. A., Yoshii I., Mishina Y., Behringer R. R., Lyons K. M. (2005). Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. USA 102, 5062–5067 10.1073/pnas.0500031102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehentner B. K., Dony C., Burtscher H. (1999). The transcription factor Sox9 is involved in BMP-2 signaling. J. Bone Miner. Res. 14, 1734–1741 10.1359/jbmr.1999.14.10.1734 [DOI] [PubMed] [Google Scholar]
- Zhao Q., Eberspaecher H., Lefebvre V., De Crombrugghe B. (1997). Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 209, 377–386 [DOI] [PubMed] [Google Scholar]
- Zhen E. Y., Brittain I. J., Laska D. A., Mitchell P. G., Sumer E. U., Karsdal M. A., Duffin K. L. (2008). Characterization of metalloprotease cleavage products of human articular cartilage. Arthritis Rheum. 58, 2420–2431 10.1002/art.23654 [DOI] [PubMed] [Google Scholar]
- Zuscik M. J., Hilton M. J., Zhang X., Chen D., O'Keefe R. J. (2008). Regulation of chondrogenesis and chondrocyte differentiation by stress. J. Clin. Invest. 118, 429–438 10.1172/JCI34174 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.