

Abstract
Background
The prevalence of Coronary artery disease (CAD) in India has increased considerably over the past few years and could become the number one killer disease if interventions are not done. Factor V Leiden (FVL) mutation and FII G20210A polymorphism are two recently described genetic factors with a propensity towards venous thrombosis. This warrants the investigations for thrombophilia in myocardial infarction patients in India.
Methods
The study cohort consisted of 51 patients aged below 50 years presenting with acute coronary syndromes. In both patient group and normal individuals the major risk factors Protein C deficiency, Protein S deficiency, anticardiolipin antibodies, Fibrinogen and Lipoprotein [a] were studied. Factor V Leiden (FVL) G1691A mutation in both control and patient group was looked by using Polymerase chain reaction (PCR) followed by sequencing of the PCR products.
Results
Our results indicated significantly higher levels of anticardiolipin antibodies and fibrinogen in the patients and absence of FVL (G1691A) mutation in our study cohort. One of the patients (H5) showed insertion of an extra A nucleotide in exon 10 of the Factor V gene resulting in frame shift mutation in this patient.
Conclusion
The results of present study showed absence of FVL mutation in our population. However, there is a need to confirm the above findings on patients from different populations from different parts of the country. The insertion of an extra A in exon 10 in the patient needs to be ascertained to confirm that it is one of its kinds or is prevalent in the population.
Keywords: Coronary artery disease, Factor V Leiden mutation, Polymerase chain reaction
1. Introduction
Arterial thrombosis is a central pathologic mechanism contributing to myocardial infarction and stroke, together the leading causes of death in developed countries. Considerable progress has been made over the past 10 years in identifying important genetic risk factors underlying predisposition to venous thrombosis.1
The mechanism of both blood coagulation and the anticoagulant pathways have been explained, and several genetic risk factors for venous thrombosis have been identified. These genetic risk factors affect the natural anticoagulant mechanisms and result in a hypercoagulable state due to an imbalance between procoagulant and anticoagulant forces. Thrombosis is a lifelong risk, and thrombotic events tend to occur when one or more of the circumstantial risk factors come into play. It is a typical multifactorial disease, with the pathogenesis involving both environmental and genetic mechanisms.2 The three most common genetic thrombophilias known to predispose to venous thrombosis are Factor V G1691A (FV Leiden) (FVL), Methylenetetrahydrofolate reductase (MTHFR) mutation and Factor II (FII) gene mutation. FVL, FII G20210A and MTHFR C677T mutations are currently the most common known genetic risk factors for venous thrombosis among Caucasians.3–5
In FII G20210A, a G–A transition at position 20210 of the 3′untranslated region of the Factor II gene has been found to be associated with increased prothrombin level and a 3-fold increase in the risk for venous thrombosis in heterozygotes. The homozygous state for the C–T transition at position 677 of MTHFR gene is associated with hyperhomocysteinemia which predisposes to thrombosis.6–8 Another risk factor in MTHFR gene is known as A1298C mutation.9 The contribution of mutations in the prothrombin (FII G20210A), methylenetetrahydrofolate reductase (C677T) genes and Factor V Leiden (FVL) to the pathogenesis of arterial thrombosis remains controversial.
However, similar insight into the genetic component of arterial thrombosis predisposition has not materialized, despite considerable effort. Though there is a significant genetic component to both forms of thrombosis, there appears to be little overlap among the known genetic risk factors for venous thrombosis and those predisposing to arterial thrombosis. In FVL G1691A, arginine is substituted by glutamine at amino acid residue in 506th position of coagulation Factor V due to this substitution, Factor Va becomes resistant to degradation by activated Protein C, resulting in a 3–5-fold increase in the risk of venous thromboembolism in heterozygous individuals.10,11 Another risk factor in Factor V gene is FV H1299R.4,9 In a study in northern part of India FVL and prothrombin G20210A mutations were observed with an allele frequency 2%,12 therefore, in the present study the prevalence of FVL (G1691A) mutation an important genetic cause of thromboembolism was studied in subjects of Rayalaseema region of India.
2. Materials and methods
2.1. Study population
Fifty-one consecutive patients aged below 50 years having acute coronary syndromes (diagnosed by ECG abnormalities and elevated cardiac enzymes) like unstable angina and myocardial infarction (STEMI/NSTEMI), admitted in the Department of Cardiology, Sri Venkateswara Institute of Medical Sciences hospital during the period August 2005–July 2008 were recruited into the study cohort. Patients of chronic stable angina, patients with other co-morbid conditions (renal, lung, liver, CNS, pregnancy and other systemic illness) and patients unwilling to participate in the study were excluded. Fifty age and sex matched voluntary blood donors without documented risk factors were recruited into the control group. An informed consent was taken from each of the patients and controls following ethical guidelines of the 1975 declaration of Helsinki. This study was approved by the Institutional Ethical Committee of Sri Venkateswara Institute of Medical Sciences, Tirupati, Andhra Pradesh, India.
2.2. Baseline assessment
History, physical examination and cardiovascular examinations were done in all patients. Risk factors like diabetes mellitus, hypertension, smoking, obesity and family history of premature coronary artery disease were assessed. BMI >25 kg/m2 was considered as obese in the present study.13
2.3. Laboratory measurements
Ten ml of peripheral venous blood was drawn from each patient after stabilization. Three ml of blood was transferred into a vial containing 3.2% tri-sodium citrate solution and the separated plasma was used for the estimation of Protein C and Protein S levels by ELISA technique (using Diagnostica Stago, France). Two ml of blood was transferred into a vial containing heparin and the separated plasma was used for the estimation of fibrinogen by Immunoturbidimetry method (using Tulip Diagnostics, India). Three ml of blood was transferred into a plain vial and the separated serum was used for the estimation of Lipoprotein [a] (Lp [a]) by Immunoturbidimetry method (using Daiichi Chemicals, Japan) and anticardiolipin antibodies (aCL) by ELISA method (using Hycor Biochemical laboratories, USA). The remaining blood was transferred into a vial containing EDTA (0.05 M) which was used for DNA analysis. The separated DNA samples were stored at −20 °C till the assays were done. Fasting lipid profile (total cholesterol, HDL-cholesterol, triglycerides) was done within 24 h for all patients using enzymatic kits on Beckman auto analyzer. LDL-Cholesterol (LDLC) was computed with the Friedewald formula.14 The cut-off values were 200 mg/dl for total cholesterol (TC), 130 mg/dl for LDLC, 40 mg/dl for HDL-Cholesterol (HDLC) and 150 mg/dl for triglycerides.
2.4. Genetic analysis of Factor V
The genomic DNA was extracted from all the blood samples following the standard protocol mentioned by Sambrook and Russel.15 This DNA was used as template for amplification of the exon 10 of the Factor V gene.
2.5. Polymerase chain reaction
Primers for Factor V were designed from the total Factor V gene sequence released in the database Genbank Accession No. Z99572.16 Factor V gene amplification was carried out by polymerase chain reaction using Master cycler gradient thermo cycler (Eppendorf, Germany) with forward primer: 5′-ACCCACAGAAAATGATGCCCAG-3′ and reverse primer: 5′-TGCCCCATTATTTAGCCAGGAG-3′, under the conditions – 5 min at 92 °C for initial denaturation; 40 s at 66 °C for annealing; 60 s at 72 °C for primer extension and final extension of 10 min at 72 °C. Further analysis of PCR products were carried out through Single Strand Conformation Polymorphism (SSCP).17 The PCR products which showed considerable variations in the SSCP gels were sequenced following dye termination method adopted from Sanger Institute at a commercial laboratory (Xcelris Labs Ltd., Ahmedabad, India) and were compared with the existing sequence of Factor V gene in the database.
2.6. Statistical analysis
Descriptive statistics include mean and standard error of mean (SEM) for continuous variables and proportions for categorical variables were calculated. The strength of the association between traditional and thrombotic risk factors, acute coronary syndromes at young age was estimated by calculating the odds ratios and 95% confidence intervals. Some of the continuous variables showed skewness and hence a non-parametric test, Mann–Whitney U test, was used to compare means. Categorical variables were compared by using Chi-square test. Association between variables was ascertained by using linear regression analysis. Relation between dependent variables and independent variables was found with logistic regression analysis. All computation and statistical analysis were done using SPSS version 13.0, SPSS Inc. Chicago, USA.
3. Results
In our study consisting of 51 patients of acute coronary syndrome, 35 (69%) had anterior wall infarction (AWMI), 9 (17%) had inferior wall infarction (IWMI) and 7 (14%) had unstable angina (USA). In the present study the following risk factors were observed in subjects: 26 (51%) patients were smokers, 13 (25%) were diabetic, 17 (33%) were hypertensive, 34 (67%) were having dyslipidemia, 17 (33%) were obese and 2 (4%) patients were reported to have family history of CAD. In the present study 86% of the patients were males. The demographic and clinical information of the patients with acute coronary syndromes was summarized in the Table 1. Among the thrombotic profile that we have studied Protein C was found to be deficient in 18% of the cases, Protein S was deficient in 14% of the cases. The levels of Lp [a] were found to be high in 16%, fibrinogen was high in 55% of cases and aCL were positive in 43% of cases. Multiple thrombotic risk factors were found in 37% of the cases and 4% in controls. The lipid profile of patients of this study is summarized in Table 2.
Table 1.
Demographic and clinical information of patients with acute coronary syndrome.
| Risk factor | ACS patients [N = 51] |
|---|---|
| Age | |
| 35 and below | 40% |
| Above 35 | 60% |
| Gender | |
| Male | 86% |
| Female | 14% |
| Diagnosis | |
| AWMI | 35 (69%) |
| IWMI | 9 (17%) |
| USA | 7 (14%) |
| Hypertension | 17 (33%) |
| Diabetes mellitus | 13 (25%) |
| Smoking | 26 (51%) |
| Dyslipidemia | 34 (67%) |
| Obesity | 17 (33%) |
| Family history | 2 (4%) |
ACS, Acute coronary syndromes; AWMI, Anterior wall myocardial infarction; IWMI, Inferior wall myocardial infarction; USA, Unstable angina.
Table 2.
Blood biochemistry of patients with acute coronary syndrome.
| Risk factor | Male |
Female |
p-value |
|---|---|---|---|
| Mean ± SEM | Mean ± SEM | ||
| Total cholesterol (mg/dL) | 193.79 ± 8.88 | 190.00 ± 11.80 | 0.877 |
| HDLC (mg/dL) | 46.55 ± 1.14 | 48.00 ± 3.14 | 0.661 |
| Triglycerides (mg/dL) | 156.93 ± 11.68 | 114.25 ± 22.16 | 0.195 |
| LDLC (mg/dL) | 116.17 ± 8.63 | 119.15 ± 5.83 | 0.899 |
| Haemoglobin (gm/dL) | 13.53 ± 0.32 | 9.49 ± 0.78 | <0.001** |
| Serum creatinine (mg/dL) | 1.05 ± 0.04 | 0.81 ± 0.05 | 0.029* |
| Serum urea (mg/dL) | 25.85 ± 1.50 | 28.00 ± 3.81 | 0.611 |
HDLC, High density lipoprotein cholesterol; LDLC, Low density lipoprotein cholesterol.
* Indicates p-value <0.05.
** Indicates p-value <0.001.
A total of 19 patients (37%) had the presence of more than one thrombotic marker in combination. Out of these, 12 had the presence of two markers in combination and 7 had three thrombotic markers in combination. Only 2 controls had two thrombotic markers in combination. Combined thrombotic risk factors were significant in cases in comparison to controls (p < 0.001). Protein C levels were found to be deficient in cases as compared to controls [(Mean ± SEM) 82.81 ± 2.90 vs 96.80 ± 3.00 respectively; p = 0.001] whereas aCL levels were found to be higher in cases as compared to controls [10.66 ± 0.83 vs. 2.71 ± 0.55 respectively; p < 0.001]. Fibrinogen levels were also found to be higher in cases as compared to controls [432.39 ± 32.67 vs 295.12 ± 17.58 respectively; p < 0.001]. No difference was found in the levels of Protein S and Lp [a]. Among the studied thrombotic risk factors, aCL (IgG) was found to be highly associated with the risk of acute coronary syndrome (OR 1.5; 95% CI, 1.2–1.7; P < 0.001), followed by fibrinogen (OR 1.005; 95% CI, 1.002–1.009; P = 0.004) and then by Protein C (OR 0.95; 95% CI, 0.92–0.99; P = 0.03) (Table 3).
Table 3.
Thrombotic risk factors in cases and controls.
| Thrombotic risk factors | Cases |
Controls |
p-value |
|---|---|---|---|
| Mean ± SEM | Mean ± SEM | ||
| Protein C deficiency (%) | 82.81 ± 2.90 | 96.80 ± 3.00 | 0.001** |
| Protein S deficiency (%) | 94.47 ± 4.06 | 97.33 ± 3.52 | 0.596 |
| aCL-IgG (GPL U/mL) | 10.66 ± 0.83 | 2.71 ± 0.55 | <0.001** |
| Lp [a] (mg/dL) | 14.50 ± 1.70 | 13.20 ± 1.34 | 0.551 |
| Fibrinogen (mg/dL) | 432.39 ± 32.67 | 295.12 ± 17.58 | <0.001** |
| Combined thrombophilia | 1.31 ± 0.90 | 0.34 ± 0.55 | <0.001** |
Mann–Whitney U test was used for non-parametric variables. aCL, anticardiolipin antibodies; Lp [a], Lipoprotein [a].
** Indicates p-value <0.001.
Chromosomal DNA was isolated from both the cases and controls for genetic analysis. The present protocol adopted, yielded good amount of DNA with least shearing. The extracted DNA was used for amplification of exon 10 of coagulation Factor V (FV) gene. The PCR amplified products both in the patients and in controls (Fig. 1) were further analyzed on SSCP gels in order to identify any variation in the nucleotide sequence. The SSCP mobility pattern of exon in the patient showed distinct variation from the normal. These results were further analyzed by subjecting these PCR products to dye terminating sequencing at Xcelris labs Ltd, Ahmedabad. The sequences obtained were deposited in the GenBank, National centre for Biotechnology Information (NCBI), Bethesda, Maryland, USA. Accession numbers are GQ354888, GQ354889, GQ354890, GQ354891 and GQ354892.
Fig. 1.
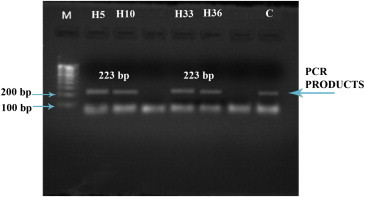
1% Agarose gel showing PCR products of Exon 10 of Factor V: 1% agarose gel showing PCR products (223bp) amplified from the isolated DNA samples. Lane M: Molecular size marker; Lanes H5, H10, H33, H36: PCR products of patients; Lane C: PCR product of control.
All the sequences of the exon 10 of FV along with control were analyzed to find any mutation in the sequence. The SSCP analysis and DNA sequencing showed the absence of FVL mutation in the case cohort as well as in the controls. In all the patients the sequences matched with existing FV gene sequence with no distinct variation either in the gene sequence or in the protein sequence. Although, the study group was small yet absence of FVL (G1691A) which is in consistent in this part of country therefore this variant cannot be considered as independent risk factor or as a predictor for CAD. However one of the patients (H5) with unstable angina had a distinct mutation in exon 10 of the FV gene where one A nucleotide was inserted resulting in a frame shift mutation in FV gene (Fig. 2).
Fig. 2.
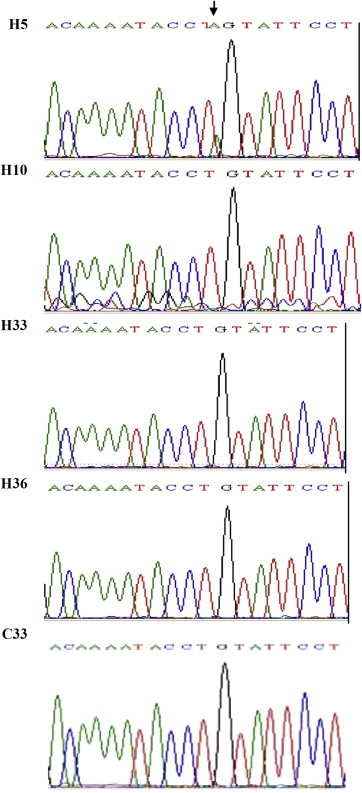
Chromatogram of the exon 10: Chromatogram of the sequence showing mutated region compared to normal sequences. The arrow-marked region shows new mutation in the exon 10.
4. Discussion
Venous thromboembolism is a major medical problem, affecting 1–5 individuals per 1000 annually.6,18 In the past 50 years, the molecular bases of both blood coagulation and the anticoagulant pathways have been explained, and several genetic risk factors for venous thrombosis have been identified. These genetic risk factors affect the natural anticoagulant mechanisms and result in a hypercoagulable state due to an imbalance between procoagulant and anticoagulant forces. It is a typical multifactorial disease, with the pathogenesis involving both environmental and genetic mechanisms.18 In the present study the prevalence of FVL mutation (G1691A) an important genetic cause of thromboembolism was evaluated along with other major risk factors, Protein C, Protein S, aCL, fibrinogen and Lp [a] in subjects from Rayalaseema region of India.
Among the studied thrombotic risk factors aCL and fibrinogen levels were significantly elevated in patients compared with controls. Also, Protein C levels were found to be associated with the risk of acute coronary syndrome.19–21 Lp [a] is composed of low density lipoprotein particle linked to apolipoprotein B-100 which also stimulates the secretion of Plasminogen activator inhibitor (PAI) which interferes with fibrinolysis and thereby causes atherosclerotic plaque in the arteries.22 However, in the present study Lp [a] was not significantly elevated in patients as compared to controls.
The role of FVL as a major genetic risk factor associated with CAD has been proved in Western population.2,4,7 However, its prevalence in India is highly variable in different populations. A study in the North Indian population showed clear absence of FVL in North Indian population.23 However, in another study low prevalence of FVL was observed in North Indian population.12 Whereas, a study in Tamilian population in Southern part of India showed no significant role of FVL and FII Single Nucleotide Polymorphisms (SNP).24 In our study also absence of FVL mutation was observed which is consistent with other reports in this part of the country. Interesting results were observed in one patient (H5) who presented with unstable angina showed insertion of A nucleotide in exon 10 of the Factor V gene resulting in frame shift mutation in this patient (Fig. 2). Although this is a new mutation only observed in one patient, it further needs to be ascertained that this SNP is only confined to this patient or present in the family and in turn distributed in the population.
5. Conclusion
The inherited thrombophilia is caused by several factors of them Factor V Leiden (FVL) and prothrombin mutations are thought to be most frequent in occurrence. However, contribution of these factors varies from population to population. The prevalence of FVL in Indian population is quiet interesting although its low occurrence in northern part of India is well documented. However, in southern part its relevance is not significantly proven. In consistence with the observation in the present study we did not find presence of FVL in our study cohort. Although, the study group was primarily consisted of patients of middle aged and young patients which was very small yet the study showed higher levels of anticardiolipin antibodies and fibrinogen, Protein C levels were deficient compared to controls and low levels of lipoprotein [a] and Protein S. Occurrence of a novel insertional mutation A in exon 10 of Factor V gene in a patient may not be that significant in terms of the study group however if the same study is exploited in a large group may through new insights in the involvement of thromboembolic factors in ACS.
Limitations
Sample size of this study is small.
Financial support
None.
Conflicts of interest
All authors have none to declare.
Acknowledgement
None.
References
- 1.Crowther M.A., Kelton J.G. Congenital thrombophilic states associated with venous thrombosis: a qualitative overview and proposed classification system. Ann Intern Med. 2003;138:128–134. doi: 10.7326/0003-4819-138-2-200301210-00014. [DOI] [PubMed] [Google Scholar]
- 2.Renaud C., Tardy-Poncet B., Presles E., Chabrier S. Low prevalence of coagulation F2 and F5 polymorphisms in mothers and children in a large cohort of patients with neonatal arterial ischemic stroke. Br J Haematol. 2010;150:709–712. doi: 10.1111/j.1365-2141.2010.08259.x. [DOI] [PubMed] [Google Scholar]
- 3.Gerhardt A., Scharf R.E., Beckmann M.W. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med. 2000;342:374–380. doi: 10.1056/NEJM200002103420602. [DOI] [PubMed] [Google Scholar]
- 4.Auro K., Alanne M., Kristiansson K. Combined effects of thrombosis pathway gene variants predict cardiovascular events. PLoS Genet. 2007;3:e120. doi: 10.1371/journal.pgen.0030120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koksal V., Baris I., Etlik O. Primer-engineered multiplex PCR-RFLP for detection of MTHFR C677T, prothrombin G20210A and factor V Leiden mutations. Exp Mol Pathol. 2007;83:1–3. doi: 10.1016/j.yexmp.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 6.März W., Nauck M., Wieland H. The molecular mechanisms of inherited thrombophilia. Z Kardiol. 2000;89:575–586. doi: 10.1007/s003920070206. [DOI] [PubMed] [Google Scholar]
- 7.Friedline J.A., Ahmad E., Garcia D. Combined Factor V Leiden and prothrombin genotyping in patients presenting with thromboembolic episodes. Arch Pathol Lab Med. 2001;125:105–111. doi: 10.5858/2001-125-0105-CFVLAP. [DOI] [PubMed] [Google Scholar]
- 8.Carp H., Salomon O., Seidman D., Dardik R., Rosenberg N., Inbal A. Prevalence of genetic markers for thrombophilia in recurrent pregnancy loss. Hum Reprod. 2002;17:1633–1637. doi: 10.1093/humrep/17.6.1633. [DOI] [PubMed] [Google Scholar]
- 9.Coulam C.B., Wallis D., Weinstein J., DasGupta D.S., Jeyendran R.S. Comparison of thrombophilic gene mutations among patients experiencing recurrent miscarriage and deep vein thrombosis. Am J Reprod Immunol. 2008;60:426–431. doi: 10.1111/j.1600-0897.2008.00640.x. [DOI] [PubMed] [Google Scholar]
- 10.Dahlbaeck B., Carlsson M., Svensson P.J. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koster T., Rosendaal F.R., de Ronde H., Briet E., Vandenbroucke J.P., Bertina R.M. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden thrombophilia study. Lancet. 1993;342:1503–1506. doi: 10.1016/s0140-6736(05)80081-9. [DOI] [PubMed] [Google Scholar]
- 12.Kumar S.I., Kumar A., Srivastava S., Saraswat V., Aggarwal R. Low frequency of factor V Leiden and prothrombin G20210A mutations in patients with hepatic venous outflow tract obstruction in northern India: a case-control study. Indian J Gastroenterol. 2005;24:211–215. [PubMed] [Google Scholar]
- 13.International Obesity Task Force (on behalf of the Steering Committee) Western Pacific Region Health Communications Australia Pty Limited; Sydney, Australia: 2002. The Asia-Pacific Perspective: Redefining Obesity and Its Treatment. [Google Scholar]
- 14.Friedewald W.T., Levy R.J., Fredrickson D.S. The concentration of LDL-cholesterol in plasma without the preparative ultracentrifuge. Clin Chem. 1972;8:499. [PubMed] [Google Scholar]
- 15.Sambrook J., Russel W.D. 3rd ed. Cold Spring Harbor Laboratory; New York: 2001. Molecular Cloning. [Google Scholar]
- 16.Bird C. Homo sapiens. Coagulation Factor V (FV) on chromosome 1q23. ACCESSION Z99572. http://www.ncbi.nlm.nih.gov/entrez/view.
- 17.Sunnucks P., Wilson A.C., Behereqaray L.B., Zenger K., French J., Taylor A.C. SSCP is not so difficult: the application and utility of single-stranded conformation polymorphism in evolutionary biology and molecular ecology. Mol Ecol. 2000;9:1699–1710. doi: 10.1046/j.1365-294x.2000.01084.x. [DOI] [PubMed] [Google Scholar]
- 18.Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112:19–27. doi: 10.1182/blood-2008-01-077909. [DOI] [PubMed] [Google Scholar]
- 19.Mishra M.N., Kalra R., Gupta M.K. Antiphospholipid antibodies in young myocardial infarction patients. Indian J Biochem Biophys. 2007;44:481–484. [PubMed] [Google Scholar]
- 20.Jose J., Selvakumar D., Selvakumar R., Kanagasapabathy A.S., Jeyaseelan L. Plasma fibrinogen – an independent risk factor for ischaemic heart disease. Indian Heart J. 1998;50:45–48. [PubMed] [Google Scholar]
- 21.Ganapathyraman L., Shanthi P., Baba Krishnan K., Madhvan M., Lakshmikanthan C. Protein C levels in ischaemic heart disease. Indian Heart J. 1996;48:125–127. [PubMed] [Google Scholar]
- 22.Gambhir J.K., Kaur H., Gambhir D.S., Prabhu K.M. Lipoprotein (a) as an independent risk factor for coronary artery disease in patients below 40 years of age. Indian Heart J. 2000;52:411–415. [PubMed] [Google Scholar]
- 23.Gupta N., Khan F., Tripathi M. Absence of factor V Leiden (G1691A) mutation, FII G20210A allele in coronary artery disease in North India. Indian J Med Sci. 2003;57:535–542. [PubMed] [Google Scholar]
- 24.Angeline T., Bentley H.A., Hawk A.B. Prevalence of the Factor V G1691A and the Factor II/prothrombin G20210A gene polymorphisms among Tamilians. Exp Mol Pathol. 2005;79:9–13. doi: 10.1016/j.yexmp.2005.03.003. [DOI] [PubMed] [Google Scholar]