

Abstract
Ether Oxazines or Xanthenes: Despite growing interest in applying single-molecule spectroscopy to study the molecular mechanisms of biological pathways in vitro and in vivo, the widely used, bright and photostable oxazine and xanthene fluorophores are still prepared by synthetic routes that are low yielding and give a mixture of products. Here, we demonstrate that recent Cu-coupling reactions can be exploited in a new synthetic strategy for the construction of oxazine and xanthene fluorophores that provides an avenue for gram-scale preparation of known fluorophores and facile exploration of new fluorophore analogs for improvements in photophysical properties.
Keywords: copper, cyclization, domino reaction, dyes/pigments, fluorescence
Recent advances in fluorescence spectroscopy have driven the demand for dyes with improved photophysical and fluorescence properties.[1] In addition to their development as cellular and single molecule imaging tools, engineered fluorescent dyes have also been developed as environmental sensors that can provide readouts of local viscosity, pH, solute concentration, and electrical potential.[2] Current and future developments in this field, especially those relevant to single-molecule and cellular imaging, depend on the synthesis of customized fluorescent dyes that emit in the red region of the visible spectrum, have high extinction coefficients and quantum yields, and display high photostability.[3]
Amongst our best dyes for these purposes are those from the oxazine and xanthene classes, exemplified by commercial compounds ATTO-655 and Alexa Fluor-594, respectively.[3a, 4] In the synthesis of derivatized or customized fluorophores, modification of commercially available dyes is often limited due either to cumbersome functionalization of the parent compounds or to prohibitive costs in obtaining sufficient quantities of dye for carrying out the necessary synthetic steps. Thus, the de novo synthesis of fluorescent dyes from basic organic building blocks is an essential aspect of technology development. Despite the obvious importance of these molecules and the evident need for improved synthetic methodologies, oxazines and xanthenes are still largely synthesized using methods reported decades or more ago that do not take advantage of the efficiencies of modern chemical transformations.[5]
Here, we report a novel and scalable synthetic approach to the assembly of the widely used oxazine and xanthene fluorophores through a common diaryl ether intermediate (Figure 1). Taking advantage of recent developments in transition metal catalysis, we constructed electronically activated diaryl ethers to serve as tethered nucleophiles, reacting with a range of substrates to undergo cyclization to cationic fluorescent compounds (Figure 1, our work). Final products were provided in good overall yields, were amenable to purification using standard silica-based normal phase flash chromatography, and, significantly, could be prepared on the gram scale.
Figure 1.
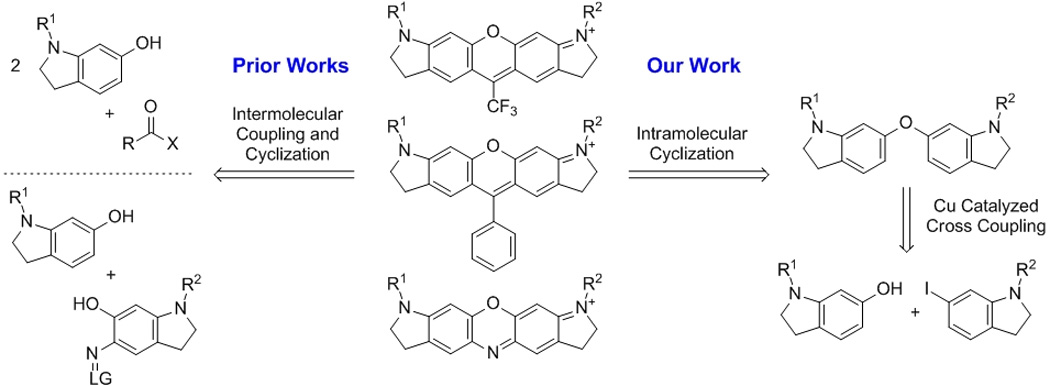
Retrosynthetic analysis for oxazine and xanthene fluorophores, highlighting the differences in approach between prior works and our work. LG = leaving group. X = H or RCO2–.
As part of our ongoing research program to develop improved technologies for in vivo super resolution imaging, we sought to prepare derivatives of the commonly utilized oxazine ATTO-655.[4b] Due to the expense associated with obtaining large quantities of this dye and the lack of commercial availability of other desirable analogs, we set out to synthesize the oxazines following previously described methods.[5b,c] These prior works rely on coupling a pair of aminophenol derivatives, one of which is substituted with an electrophilic nitroso or diazo functionality, by heating the two components in an acidic medium (Figure 1, prior works). Though the aminophenol intermediates are readily prepared, the final coupling and cyclization reactions frequently result in low yields (~15%) and necessitate the use of preparative scale reverse phase high performance liquid chromatography (prep-HPLC) in order to obtain material of satisfactory purity. In our hands, the quantities obtained by this method proved insufficient to reasonably carry out the remaining steps in the synthesis of our final targets, and we concluded that scaling up the process to obtain the desired quantities was impractical. As a result, we concluded that the existing approach was not a viable synthetic route and chose to pursue alternative synthetic strategies.
Within the oxazine core structure, we identified a previously unexplored diaryl ether disconnection, which is exploited in the restrosynthetic analysis shown in Figure 1. We envisioned that a step-wise coupling and cyclization process would proceed in higher overall yield when compared to the classical coupling strategy and, more importantly, would yield fewer byproducts in the final step. Thus, isolation and purification could be carried out using standard flash chromatography techniques amenable to gram-scale operations. In pursuit of this synthetic strategy, it was necessary to examine methods for synthesizing the diaryl ether in a versatile and robust fashion.
In addition to the more traditional dihydroquinoline and tetrahydroquinoline derivatives, we also chose to explore an indoline-based scaffold to study this synthetic sequence. The precursor indoles provide a large number of building blocks from which to start, and we were also interested in the photophysical properties of the resulting oxazine products, which are not well described for the indoline class. Preparation of the indoline coupling partners proved to be straightforward by Gribble reduction and alkylation of the commercially available indole starting materials (Supplemental Scheme 1).[6]
The Ullmann ether synthesis poses a potential route to the diaryl ether structural motif by copper(I) promoted reaction between phenols and aryl halides.[7] Although attractive in theory, the classical reaction conditions typically employ strong base, stoichiometric quantities of copper, and heating at temperatures in excess of 200 °C. Additionally, the reaction is known to be highly substrate dependent and, when successful, commonly results in only modest yields. To overcome these limitations and eliminate the harsh reaction conditions, we chose to explore a more contemporary method that utilizes palladium catalysis.[8] Unexpectedly, coupling between phenol 1c and its corresponding triflate resulted predominantly in reaction at C-5 of the indoline in a Heck-type manner, producing the biaryl phenol derivative as the major product (Supplemental Scheme 2). Thus, the palladium catalyzed coupling reaction does not appear to be a useful method for synthesizing diaryl ethers when electron rich carbon nucleophiles, such as those that exist in our system, are present.
We next turned to recent developments in ligand-assisted copper catalyzed coupling reactions.[9] Based on work reported by Buchwald and coworkers, we found that the coupling between phenol 1b and 3-iodoaniline (2a) provided the diaryl ether 3b in 82% yield when carried out in the presence of catalytic copper iodide (10 mol %) and 2-picolinic acid (20 mol %) at 85 °C in DMSO (Table 1, entry 2). This catalytic system offers the advantage of orthogonality with the aniline functional group. As opposed to aryl iodides, aryl bromides were found to be very poor substrates in our system, with nearly no product formation observed even at elevated temperatures (180 °C) and increased catalyst loadings (50 mol %). Nonetheless, aryl iodides were readily prepared from the corresponding commercially available aryl bromides by employing a copper promoted halogen exchange reaction.[10] Couplings carried out according to the aforementioned conditions provided a range of substituted diaryl ethers in good yields (Table 1). Following these coupling reactions, anilines 3a and 3b were transformed to their dihydroquinoline derivatives via a modified Skraup reaction and subsequently alkylated and reduced where appropriate (see SI).
Table 1.
Copper(I) catalyzed couplings between phenols and aryl iodides to furnish diaryl ethers.[a]
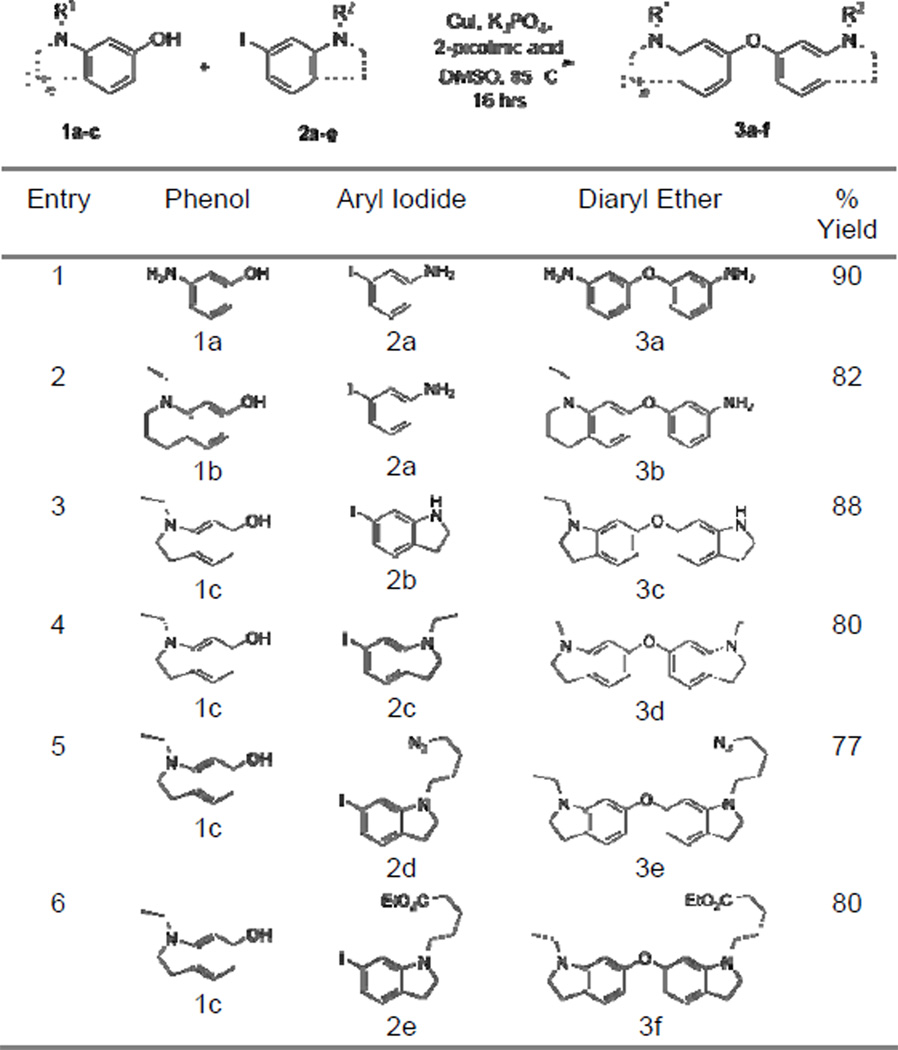 |
Conditions: 1.2 eq. phenol, 10 mol % copper(I) iodide, 20 mol % 2-picolinic acid, 2.0 eq. K3PO4, DMSO, 85 °C, 24 hrs.
Having established a reliable means of synthesizing the critical diaryl ether intermediates, we next explored conditions for converting these compounds to their corresponding oxazine dyes. This transformation was readily accomplished by reaction of diaryl ether 3d with one equivalent of 4-nitrobenzene diazonium tetrafluoroborate, followed by heating the corresponding diazene 4d with p-toluenesulfonic acid (TsOH) to 65 °C in ethanol (Scheme 1). The latter process proceeded with near-quantitative conversion and following trivial silica based flash chromatography to remove the liberated p-nitroaniline, we obtained the oxazine-tosylate salt in high purity and 90% yield. Implementing this strategy, several substituted oxazine dyes with various spectroscopic properties were synthesized in good to excellent yields from the corresponding diazene compounds (Table 2). Of note, over one gram of oxazine 6f was prepared by this method in 94% yield, demonstrating the scalability of our approach (Table 2, entry 6). Additionally, 6g, the sulfonated analog of 6c resembling commercially available ATTO-655 was synthesized in good yield by this method.
Scheme 1.

Reaction sequence for conversion of diaryl ethers to oxazine dyes.
Table 2.
Synthesis of substituted oxazine dyes.[a]
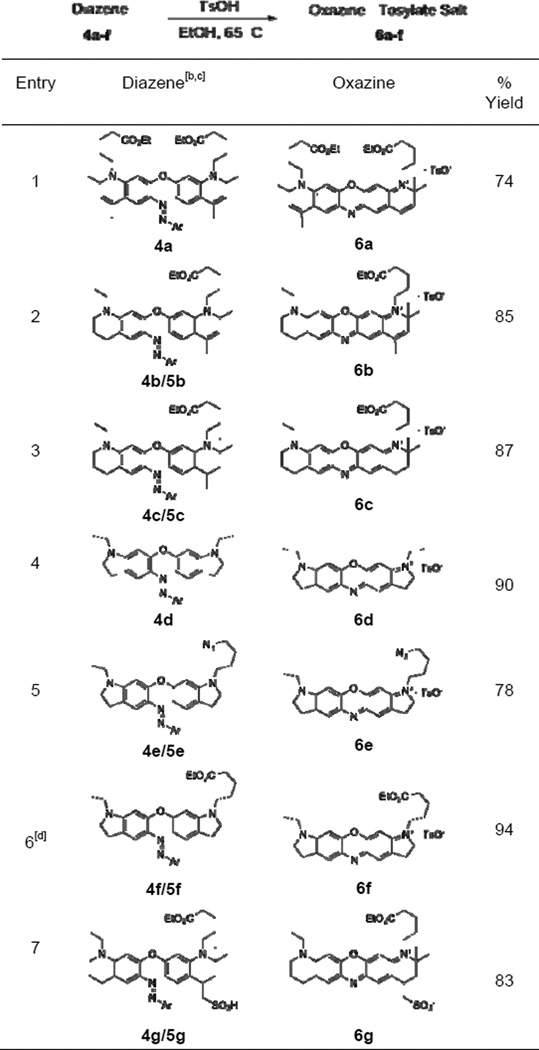 |
Conditions: 3.0 eq TsOH, EtOH, 65 °C, 4–8 hrs.
Ar = p-NO2Ph.
Diazenes were obtained and used as regioisomeric mixtures, which cyclize to a single oxazine product.
Reaction conducted on the gram-scale.
To further expand the scope of this methodology, we examined other popular fluorescent dyes for similar diaryl ether disconnections and identified xanthene dyes, exemplified by rhodamine and rosamine, as potential targets (Figure 1). The classical method for synthesizing these aminoxanthenes calls for heating two equivalents of an aminophenol with one equivalent of a carboxylic acid anhydride or aldehyde under acidic conditions (ZnCl2 or H2SO4) often at high temperatures (Figure 1, prior works).[5a] When this strategy is employed with derivatized analogs, the reactions typically result in modest to poor yields and make isolation of the product difficult by conventional purification methods.[11] We sought to prepare these fluorescent dyes from our diaryl ethers in an analogous manner to the synthesis of the oxazine dyes. After screening several Lewis acid catalysts and reaction conditions, we found that Ga(OTf)3 catalyzed the tandem Friedel-Crafts acylation-cyclization reaction between the aforementioned diaryl ethers and aromatic acid chlorides to provide the corresponding xanthene dyes in modest to good yields with substantial recoverable starting material (Table 3).[12]
Table 3.
Tandem catalytic Friedel-Crafts acylation/cylization reaction for the synthesis of xanthene fluorophores.[a]
 |
Conditions: 8 eq acid chloride, 15 mol % Ga(OTf)3, MeNO2, 60°C, 4Å mol sieves, 16 hrs.
B.R.S.M = % yield based on recovered starting material.
No Lewis acid catalyst required, 2.2 eq of TFAA in DCM at RT for 12 hrs.
5 eq of the acid chloride was used.
Reaction conducted on the gram-scale.
Supplementing the reaction with additional Ga(OTf)3 did not lead to further product formation, suggesting that a byproduct formed in the course of the reaction was inhibiting forward progression. Possibly, this was due to substrate inactivation by HCl, which is generated in the course of the Friedel-Crafts acylation and from hydrolysis of the acid chloride (water is generated in the cyclization step). Interestingly, in the case of the p-Me2N derivative (Table 3, entry 3), nearly full consumption of starting material was observed, suggesting that the dimethylamino group may be capable of buffering the reaction to allow for increased conversion. Higher conversions of starting material could be obtained by re-subjecting the crude product mixture (following workup) to the initial reaction conditions.
Of note, this synthetic strategy allows for the synthesis of asymmetrically functionalized xanthene dyes, which are not accessible by the classical coupling strategy. Additionally, several dyes were prepared in significantly higher yields when compared to traditional syntheses. For example, compound 7b, an asymmetric xanthene, was synthesized in 83% yield simply by reaction of the diaryl ether with 2.2 equivalents of trifluoroacetic anhydride at room temperature without a Lewis acid catalyst. By comparison, rhodamine 700, a similar dye possessing the trifluoromethyl substituent at the meso carbon, is reported to be prepared in only 5% yield from aminophenols.[13] Although this current work is limited to the synthesis of rosamines with fully substituted anilines, it may be possible to synthesize mono- or non-alkylated analogs by minor modification of this synthetic approach.
Figure 2 shows the absorption and fluorescence spectra for several of the oxazine and xanthene dyes synthesized. Notably, the increased conjugation of oxazines 6a and 6b results in significant red-shifted absorption and fluorescence. Similarly, xanthenes 7b, 7e and 7f possessing electron withdrawing side chains also display red-shifted spectra. The dimethylamino-substituted rosamine 7c displays pH dependent fluorescence, with a near 30-fold increase in fluorescence intensity in acidic conditions (data not shown). Spectral properties of the dyes in aqueous solution are provided in Table 4.
Figure 2.

(a) Absorbance and (b) fluorescence spectra of various oxazine and xanthene derivatives. Spectra were obtained in H2O, with the exception of 7c, which was obtained in aqueous 50 mM HCl.
Table 4.
Spectral properties of fluorescent dyes in H2O.[a]
| Dye | λmax abs (nm) |
εmax (M−1 cm−1) |
λfluor (nm) |
Fwhm[b] (nm) |
Φf |
|---|---|---|---|---|---|
| 6a | 703 | 50,000 | 717 | 42 | 0.08 |
| 6b | 682 | 69,000 | 696 | 44 | 0.09 |
| 6c | 664 | 67,000 | 681 | 45 | 0.24 |
| 6d | 648 | 66,000 | 661 | 43 | 0.11 |
| 6g | 663 | 97,000 | 677 | 43 | 0.20 |
| 7a | 553 | 58,000 | 576 | 49 | 0.19 |
| 7b | 616 | 61,000 | 643 | 90 | 0.08 |
| 7c | 559 | 79,000 | 585 | 53 | 0.14 |
| 7d | 555 | 60,000 | 575 | 47 | 0.21 |
| 7e | 579 | 68,000 | 602 | 44 | 0.27 |
| 7f | 581 | 57,000 | 600 | 42 | 0.24 |
Measurements were made in H2O, with the exception of 7c, which was measured in aqueous 50 mM HCl.
Full-width at half-maximum height.
In summary, we have established a high-yielding, scalable synthetic route for the preparation of the widely used oxazine and xanthene based fluorescent dyes from a common diaryl ether intermediate. Compared to previously existing synthetic methodologies, our work provides a versatile means for preparing these fluorescent dyes and eliminates the need for tedious and expensive purifications. Following this synthetic approach, a number of oxazine and xanthene fluorophores were synthesized and characterized. With proper synthetic planning, we believe this to be a general and widely applicable approach to the synthesis of derivatized oxazine and xanthene dyes, and may facilitate the development of novel fluorophores and probes with unique properties.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (GM090126, GM087519, GM091804). A.V.A. is supported by Columbia University College of Physicians and Surgeons Medical Scientist Training Program (NIH T32 GM07367). We are grateful to Dr. D. Romanini for editing the manuscript, Professor S.A. Snyder and R.L. Gonzalez for helpful discussions, and Professor W. Min for graciously offering the use of his spectroscopy equipment.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Tinnefeld P, Sauer M. Angew. Chem. Int. Ed. 2005;4:2642–2671. doi: 10.1002/anie.200300647. [DOI] [PubMed] [Google Scholar]; b) Huang B, Bates M, Zhuang X. Ann. Rev. Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fernàndez-Suàrez M, Ting AY. Nat. Rev. Mol. Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 2.a) Haidekker MA, Brady TP, Lichlyter D, Theodorakis EA. J. Am. Chem. Soc. 2006;128:398–399. doi: 10.1021/ja056370a. [DOI] [PubMed] [Google Scholar]; b) Charier S, Ruel O, Baudin J-B, Alcor D, Allemand J-F, Meglio A, Jullien L. Angew. Chem. Int. Ed. 2004;43:4785–4788. doi: 10.1002/anie.200460557. [DOI] [PubMed] [Google Scholar]; c) Grynkiewicz G, Poenie M, Tsien RY. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]; d) Yu F, Li P, Li G, Zhao G, Chu T, Han K. J. Am. Chem. Soc. 2011;133:11030–11033. doi: 10.1021/ja202582x. [DOI] [PubMed] [Google Scholar]; e) Fluhler E, Burnham VG, Loew LM. Biochemistry. 1985;24:5749–5755. doi: 10.1021/bi00342a010. [DOI] [PubMed] [Google Scholar]
- 3.a) Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Nat. Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fölling J, Belov V, Kunetsky R, Medda R, Schönle A, Egner A, Eggeling C, Bossi M, Hell SW. Angew. Chem. Int. Ed. 2007;46:6266–6270. doi: 10.1002/anie.200702167. [DOI] [PubMed] [Google Scholar]
- 4.a) Heilemann M, van de Linde S, Mukherjee A, Sauer M. Angew. Chem. Int. Ed. 2009;48:6903–6908. doi: 10.1002/anie.200902073. [DOI] [PubMed] [Google Scholar]; b) Wombacher R, Heidbreder M, van de Linde S, Sheetz MP, Heilemann M, Cornish VW, Sauer M. Nat. Methods. 2010;7:717–719. doi: 10.1038/nmeth.1489. [DOI] [PubMed] [Google Scholar]
- 5.a) Beija M, Afonso CAM, Martinho JMG. Chem. Soc. Rev. 2009;38:2410–2433. doi: 10.1039/b901612k. [DOI] [PubMed] [Google Scholar]; b) Kanitz A, Hartmann H. Eur. J. Org. Chem. 1999;4:923–930. [Google Scholar]; c) Pauff SM, Miller SC. Org. Lett. 2011;13:6196–6199. doi: 10.1021/ol202619f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gribble GW, Lord PD, Skotnicki J, Dietz SE, Eaton JT, Johnson JL. J. Am. Chem. Soc. 1974;96:7812–7814. [Google Scholar]
- 7.a) Ullmann F, Sponagel P. Ber. Dtsch. Chem. Ges. 1905;38:2211–2212. [Google Scholar]; b) Sawyer JS. Tetrahedron. 2000;56:5045–5065. [Google Scholar]
- 8.Burgos CH, Barder TE, Huang X, Buchwald SL. Angew. Chem. Int. Ed. 2006;45:4321–4326. doi: 10.1002/anie.200601253. [DOI] [PubMed] [Google Scholar]
- 9.Maiti D, Buchwald SL. J. Am. Chem. Soc. 2009;131:17423–17429. doi: 10.1021/ja9081815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klapars A, Buchwald SL. J. Am. Chem. Soc. 2002;124:14844–14845. doi: 10.1021/ja028865v. [DOI] [PubMed] [Google Scholar]
- 11.Mitronova GY, Belov VN, Bossi ML, Wurm CA, Meyer L, Medda R, Moneron G, Bretschneider S, Eggeling C, Jakobs S, Hell SW. Chem. Eur. J. 2010;16:4477–4488. doi: 10.1002/chem.200903272. [DOI] [PubMed] [Google Scholar]
- 12.a) Olah GA, Farooq O, Farnia SMF, Olah JA. J. Am. Chem. Soc. 1988;110:2560–2565. [Google Scholar]; b) Kobayashi S, Komoto I, Matsuo J-i. Adv. Synth. Catal. 2001;343:71–74. [Google Scholar]; c) Prakash GKS, Mathew T, Olah GA. Acc. Chem. Res. 2012;45:565–577. doi: 10.1021/ar2002039. [DOI] [PubMed] [Google Scholar]
- 13.Clunas S, Strory JMD, Rickard JE, Horsley D, Harrington CR, Wischik CM. WO2010/067078. 2010
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.