

Abstract
WNT signaling is critical in most aspects of skeletal development and homeostasis, and antagonists of WNT signaling are emerging as key regulatory proteins with great promise as therapeutic agents for bone disorders. Here we show that Sost and its paralog Sostdc1 emerged through ancestral genome duplication and their expression patterns have diverged to delineate non-overlapping domains in most organ systems including musculoskeletal, cardiovascular, nervous, digestive, reproductive and respiratory. In the developing limb, Sost and Sostdc1 display dynamic expression patterns with Sost being restricted to the distal ectoderm and Sostdc1 to the proximal ectoderm and the mesenchyme. While Sostdc1–/– mice lack any obvious limb or skeletal defects, Sost–/– mice recapitulate the hand defects described for Sclerosteosis patients. However, elevated WNT signaling in Sost–/–; Sostdc1–/– mice causes misregulation of SHH signaling, ectopic activation of Sox9 in the digit 1 field and preaxial polydactyly in a Gli1- and Gli3-dependent manner. In addition, we show that the syndactyly documented in Sclerosteosis is present in both Sost–/– and Sost–/–; Sostdc1–/– mice, and is driven by misregulation of Fgf8 in the AER, a region lacking Sost and Sostdc1 expression. This study highlights the complexity of WNT signaling in skeletal biology and disease and emphasizes how redundant mechanism and non-cell autonomous effects can synergize to unveil new intricate phenotypes caused by elevated WNT signaling.
Keywords: WNT signaling, Sost, Sclerostin, Sostdc1, Shh, Limb formation, Polydactyly, syndactyly
Introduction
Gene duplication is at the center of evolutionary diversification and represents a dominant contributor to biological innovation. The process of gene duplication is the main mechanism by which paralogous genes with redundant functions emerge, and represents a means of protecting an organism against deleterious mutations (Hoffmann et al., 2010; Moleirinho et al., 2011). At the same time, genes that are not critical (where a critical gene is described by a lethal embryonic phenotype) are more likely to evolve under less stringent selective pressure, and in the case of duplicated genes, maintain some partial functional redundancy. The gene encoding Sclerostin or Sost is located on human chromosome 17 and its protein sequence was found to be 55% similar to a homologous gene, Sostdc1, located on chromosome 7 (Fig. 1). In humans and mouse models Sost deficiency causes Sclerosteosis, a rare autosomal recessive disorder, characterized by generalized hyperostosis of the axial and appendicular skeleton (Balemans et al., 2001; Collette et al., 2012). Due to its highly specialized null phenotype, and abundant transcription in bone, primarily osteocyte-derived, sclerostin was originally described as a protein exclusively secreted by osteocytes that functions as a negative regulator of bone formation (van Bezooijen et al., 2005; Winkler et al., 2003), through antagonizing the BMP signaling pathway; later was found to also bind to LRP5/6 co-receptors and antagonize WNT signaling (Kusu et al., 2003; Li et al., 2005; Semenov et al., 2005; ten Dijke et al., 2008; van Bezooijen et al., 2007b; Winkler et al., 2003).
Fig. 1.

Sost–Sostdc1 evolutionary relationship. An overview of SOSTDC1 (left) and SOST (right) evolution, created by tracking the SOSTDC1 and SOST gene loci through representative vertebrate genomes (not to scale) from (A) Euteleostomi, (B) Tetrapoda and (C) Mammals clades. Predicted orthologs (where annotation is not available) are shown with gray dotted lines. Genes are shown as arrowheads, with their gene symbols above. Genes that are not conserved/likely to be poorly annotated are represented in white. The direction of the arrowhead indicates the relative transcriptional orientation and the relevant genome coordinates indicated on the left and right respectively.
Similar to Sost, its paralog Sostdc1 (Sost domain-containing protein 1; aka Sostl, USAG-1, Wise, ectodin) has been described as a WNT antagonist (Ahn et al., 2010), as well as an inhibitor of BMP signaling (Lintern et al., 2009; Murashima-Suginami et al., 2008). SOSTDC1 has been shown to be expressed in the kidney (Blish et al., 2010; Turk et al., 2009), lung (Zhang et al., 2012), the developing tooth bud of ferrets (Jarvinen et al., 2009), and SNPs in SOSTDC1 have been associated with a low bone-mass phenotype in Chinese women, consistent with a possible role in maintaining functions of the musculoskeletal system (He et al., 2011). Sostdc1-deficient mice display severe teeth defects characterized by enlarged enamel knots, altered cusp patterns, fused molars, and extra teeth (supernumerary incisors) (Kassai et al., 2005; Munne et al., 2009). In addition, Sostdc1 has been shown to be highly expressed in distal convoluted tubules and connecting tubules in the kidney (Tanaka et al., 2008) and Sostdc1-deficient mice were shown to be resistant to tubular injury in an acute renal failure and interstitial fibrosis rodent model, revealing that Sostdc1 may influence the progression of kidney disease (Yanagita et al., 2006). Although Sost and Sostdc1 have been studied primarily from the perspectives of bone mass and kidney response to injury, respectively, here we show that these genes are broadly expressed in the mouse during development and adulthood, and we dissect their shared roles during limb development.
We have recently shown that in addition to functioning as a WNT antagonist in the adult bone, Sost also plays a critical role as a negative regulator of WNT signaling in the developing limb. A less common human phenotype described for sclerosteosis patients is the occasional presence of hand defects at birth. These abnormalities are primarily characterized by syndactyly [asymmetric cutaneous or bony syndactyly of the index and middle fingers (digits 2 and 3)] and radial deviation of the digits, with hypoplasia and nail dysplasia (symmetric or asymmetric; most commonly associated with the index finger) (Hamersma et al., 2003; Itin et al., 2001; Sugiura and Yasuhara, 1975).
Moreover, using a genetic approach we have previously demonstrated that over-expression of human SOST from a bacterial artificial chromosome (BAC) perturbs anterior–posterior and proximal–distal patterning of the developing limb. These transgenic mice showed a wide range of limb defects including fused, split, missing bones and whole digits and the severity of the limb defects were shown to be dose-dependent. We also showed that Sost-deficiency rescued significant aspects of the Lrp6–/– skeletal phenotypes supporting the view that SOST gain-of-function impairs limb patterning by inhibiting WNT signaling through the LRP5/6 co-receptors (Collette et al., 2010).
Because of the evolutionary relationship between Sost and Sostdc1 as well as their common molecular roles as WNT-, and possibly BMP- antagonists, we have examined the shared and unique functions of these paralogs, in single and double knockout mice. Initially, we describe in detail, both the embryonic and adult tissue distribution of these transcripts through the use of LacZ-knock-in alleles. We find both genes to have dynamic and complex expression patterns during embryonic and limb development, and are often expressed in adjacent tissues or cell types. In the adult mouse, we find Sostdc1 to be more widely distributed than Sost; however significant expression of Sost was detected in non-skeletal tissues. In general, when these genes are expressed in the same organ system, they are present in non-overlapping expression domains, suggesting that these genes have evolved different sub-specializations within the signaling pathways they regulate, as a function of their cellular location.
In particular, we focused our analysis on the characterization of their shared roles during limb development. Herein, we show that Sost deficient mice recapitulate the hand defects described for sclerosteosis patients (Itin et al., 2001), at a frequency of 4%, while Sostdc1–/– lack any skeletal patterning defects; they do display mild ventralization characterized by pigmentation and hair growth on the ventral side of the autopod. We also find that consistent with their site of gene expression in the developing limb, Sost–/–; Sostdc1–/– mice exhibit preaxial polydactyly, detected visually as early as E11.5, indicating that Sost and Sostdc1 play partially redundant and complementary roles in the developing limb. Through a combination of in situ marker and micro-array gene expression analysis we show that the combined absence of Sost and Sostdc1 interferes with components of WNT, BMP, SHH, FGF and TGFb signaling to produce several limb abnormalities that include: preaxial polydactyly, syndactyly, dorsalization, radial deviation and nail dysplasia. In particular we show that the preaxial polydactyly is driven by misregulation of SHH signaling, where the Shh and Gli1 expression domains are elevated and expanded anteriorly, while Gli3 expression levels are reduced. Grem1 expression is missing in the anterior mesenchyme of the limb bud where the duplicated digits form, and Hoxd13 is ectopically expressed. We conclude that Sox9 ectopic activation in the digit 1 field is promoted by the misexpression of Gli1 transcription factor, which has been previously shown to control Sox9 transcription, and by the lack of Gli3-dependent gene repression. We also examined the underlying causes of the observed syndactyly, and found Fgf8 levels to be elevated, and BMP4 and BMP7 to be absent in the AER; the Fgf8 AER expression domain is expanded proximally and disorganized which resulted in a reduction in interdigital apoptosis in regions corresponding to the observed syndactyly. Thus, the absence of Sost and Sostdc1 in the limb disrupts the epithelial–mesenchymal communication required for proper limb patterning in these compound mutants, in vivo.
Materials and methods
Mouse strains and embryos
Sost–/– and Sostdc1–/– mice were generated by replacing the open reading frame with the LacZ reporter as previously described (Collette et al., 2010; Tanaka et al., 2010). Sost–/–; Sostdc1–/– were generated by mating Sost–/– and Sostdc1–/– mice; E9.5 to E17.5 embryos were collected at various embryonic stages and geno-typed by PCR. E0.5 of gestation was considered to be noon on the day a copulatory plug was observed. Embryos earlier than E12.0 were stage-confirmed by somite counting for all subsequent analyses. All animal experiments were carried out in PHS-assured facilities in accordance with guidelines set by the Animal Care and Use Committee at University of California-Berkeley and Lawrence Livermore National Laboratory.
Identification of orthology and paralogy relationships
Human, rat, mouse, cow, chicken, and zebrafish orthologs of SOSTDC1 and SOST were identified from the Homologene database (Sayers et al., 2012) Release 66. HomoloGene homology searches rely on both proteins and their corresponding DNA sequences alignments, as well as synteny information, when applicable, and have been shown to perform well in phylogenetic and functional analyses where high specificity is required (Altenhoff and Dessimoz, 2009). In the case of frog, which is not included in the Homologene database, we used tBLASTn and BLASTp with the human protein sequences of SOSTDC1 and SOST to search the nucleotide and protein databases in NCBI, respectively; we only considered sequences represented in the current RefSeq (Pruitt et al., 2012).
Whole-mount in situ hybridization
Whole-mount in situ hybridizations were carried out using standard procedures (Collette et al., 2010). Briefly, digoxigenin-labeled antisense RNA probes were generated to the desired RNA sequence and hybridized to whole-mount embryos. Expression was visualized by binding BM Purple (Roche) to an alkaline-phosphatase conjugated anti-Digoxigenin antibody (Roche). Antisense RNA probes for Grem1 (MluI-SacII fragment of NM_011824), Fgf8 (PstI 3′cDNA and UTR fragment of NM_010205); (Crossley and Martin, 1995), Shh (MscI-NarI fragment of NM_009170); (Echelard et al., 1993) were generated as described (Hogan et al., 1994) with the following modification: proteinase K digestion was omitted for ectodermal or AER probes. Gli1 (NM_010296.2) probes were generated from gel-purified PCR fragments (Gli1 5′-TCCTCCTCTCATTCCACAGG-3′; 5′-TCCAGCTGAGTGT TGTCCAG-3′). A minimum of 4 embryos were used per genotype, per experiment.
LacZ stains
Embryos were dissected into ice-cold 1 × phosphate-buffered saline (PBS), pH 7.3 and fixed in 2% paraformaldehyde, 0.2% glutaraldehyde in 1 × PBS, 2 mM MgCl2 at 4°C for 30 min to 1 h, followed by extensive rinsing in 1 × PBS, 2 mM MgCl2. Embryos were stained overnight at 4°C in X-gal stain: 1 mg/ml X-gal, MgCl2, 5 mM EGTA, 0.02% Nonidet P-40, 5 mM potassium ferro-cyanide, 5 mM potassium ferricyanide, in 1 × PBS, pH 7.3. Neonates and adults (6 months of age) were skinned, eviscerated, and fixed as whole animals in 4% Paraformaldehyde in 1× PBS, 2 mM MgCl2 for 1 h at 4 °C followed by extensive rinsing and staining overnight (neonates) or 48 h (adults) at 4°C in LacZ staining solution, as for embryos. Prior to staining, adult bones were decalcified in 0.5 M EDTA, pH 7.3, by the weight loss-weight-gain method of decalcification endpoint determination. After staining, embryos were post-fixed in 4% paraformaldehyde in 1 × PBS, pH 7.3 at 4°C, and then cleared in glycerol for photography. For sectioning, neonate and adult tissues were post-fixed for 72 h in 4% paraformaldehyde, dehydrated and embedded into paraffin wax. Section were cut at 6 mm, baked at 42°C overnight, counterstained with Nuclear Fast Red and mounted with Permount for imaging.
Skeletal preparations
Skeletal preparations were carried out on neonate and adult mice (6 months of age) using Alcian Blue 8GX for cartilage and Alizarin Red S for bone as previously described (Collette et al., 2010); E12.5–E14.5 mouse embryos were stained with Alcian Blue 8GX for cartilage only (0.05% in 4% glacial acetic acid).
Lysotracker apoptosis stain
Embryos were dissected at E12.5 and E13.5 in Hank's balanced saline solution (HBSS) and placed in lysotracker staining solution (2.5 ml/ml in HBSS) for 30 min at 37°C. Embryos were washed with 1 × PBS (pH 7.3) 2 × and fixed overnight in 4% paraformaldehyde at 4°C and dehydrated in methanol and cleared in benzyl alcohol:benzyl benzonate (1:1) for photography.
Immunofluorescent antibody stain
Embryos were dissected at E12.5 into ice-cold PBS and fixed for 24 h in 4% paraformaldehyde at 4°C. Embryos were washed, dehydrated, and embedded into paraffin for sectioning. Slides were dewaxed and epitopes requiring antigen retrieval were incubated in Uni-Trieve (Innovex) for 30 min at 65°C unless otherwise indicated. Slides were blocked with 5% BSA/0.01% Triton X-100 (Sigma) or Rodent Block (Innovex, for mouse/rat monoclonal antibodies only), incubated in a humid chamber with primary antibody overnight at room temperature [1:200, anti-Gli3 (abcam), 1:200, anti-activated beta-catenin clone 8E7 (Millipore)], washed, and incubated for 2 h with Alexa-fluor-labeled secondary antibody (1:1000, Invitrogen/Molecular Probes), washed, and mounted using Prolong Gold/Prolog Gold with DAPI (Invitrogen/Molecular Probes) for imaging. Images were acquired using single-channel fluorescent filters on a Leica DM5000 compound microscope using a Qimaging color CCD camera and ImagePro software. Goat polyclonal anti-Sclerostin antibody (1:200, R&D Systems, cat# AF1589) and anti-goat Alexa-Fluor 488 secondary antibody (1:1000, Molecular Probes, cat# A21467) were used to determine Sost localization on bone paraffin sections as previously described (Collette et al., 2012).
Microarray analysis
Microarray data analysis was performed using R programming platform and Bioconductor (Gentleman et al., 2004). Bioconductor package ‘affy’ (Gautier et al., 2004) was used for data quality assessment. Data preprocessing and normalization were performed using Robust Multi-chip Average (RMA) protocol (Irizarry et al., 2003). Differentially expressed genes were identified using the empirical Bayes method implemented in Linear Models for Micro-Array (LIMMA) (Smyth, 2004) package. Probes were mapped to genes using Affymetrix Mouse Genome 430 2.0 Array annotation data from Bioconductor annotation package ‘mouse4302.db’. Fold change values were calculated as the ratio between the averages of normalized intensities of the two groups, Sost;–/– Sostdc1 –/– and wildtype. Fold change values for differentially expressed genes are reported in a log2 scale. Genes with fold change of 2 (log2 FC=1) or greater and P-value less than 0.05 were considered differentially expressed. Pathway enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Dennis et al., 2003; Huang da et al., 2009) and Kyoto Encyclopedia of Genes and Genomes (KEGG) option (Kanehisa et al., 2010). Pathways with EASE score, a modified Fisher Exact p-value less than 0.1 were considered as enriched. Microarray data is publicly available at NCBI (GS E44325).
Results
Sost and Sostdc1 arose as duplication/divergence events
The human genomic region containing SOSTDC1 protein on chr 7 is syntenic with the region containing SOST on chr 17 (Fig. 1). Both SOSTDC1 and SOST genes are extremely well conserved in the descendants of the ancestral Euteleostomi. Earlier chordates show some evidence of a SOSTDC1 or SOST ortholog, suggesting that the duplication event took place at least 500 million years ago and that the genes belong to an ancient gene family. Other genes that are neighbors of SOSTDC1 also have paralogs in the neighborhood of its paralog SOST, and this is true in all analyzed genomes. Thus, ETV1 and MEOX2 are paralogs of ETV4 and MEOX1, respectively. This situation is consistent with a large-scale, possibly genomic duplication event, such as those that took place in the vertebrate ancestor (Dehal and Boore, 2005). The duplication was followed by divergence, resulting in the present-day human SOSTDC1 and SOST sharing only 40–42% of their amino acids, with similarity spanning over 84–92% of the protein lengths. It is likely that additional paralogous genes in these syntenic regions have diverged beyond our ability to detect homology. Individually, each of the corresponding syntenic regions, including other genes and their relative order, is remarkably well conserved in Euteleostomi, suggesting fixation of the duplicated genes after a period of rapid divergence. Only in one examined lineage, the amphibians represented by X. tropicalis, we find Sost to be absent, suggesting that the duplicated region has been lost in this lineage. Finally, the noncoding regions in the vicinity of SOSTDC1 and SOST are not uniformly conserved; while SOSTDC1 is surrounded by a high density of evolutionary conserved noncoding sequences in mammals, only a few of these elements are shared among amniotes, suggesting neo- or sub-functionalization of the gene; in contrast some noncoding elements in the vicinity of SOST are conserved not only among amniotes, but, to a less extent, in tetrapods and fish (Loots and Ovcharenko, 2007).
Sost and Sostdc1 have non-overlapping expression patterns during limb development
LacZ was used to determine Sost and Sostdc1 expression in Sost-LacZ knock-in mice (referred to as SostLacZ for expression analysis, Sost–/– for phenotype analysis) and Sostdc1-LacZ knock-in (referred to as Sostdc1LacZ for expression analysis, Sostdc1–/– for phenotype analysis). SostLacZ was observed as early as E9.5 in the distal limb bud (Fig. 2A-A″). This expression is restricted to the ectoderm and is excluded from the apical ectodermal ridge (AER) and mesenchyme at all time points examined (Fig. 2A-F and b-d). Sostdc1LacZ emerges ectodermally at E10.5 in a small field on the posterior side of the limb near the zone of polarizing activity (ZPA) marked by sonic hedgehog (Shh) (Fig. 2h, corresponding Shh expression indicated in Fig. 6A). By E11.5 its expression is strongly present primarily in the mesenchyme of the proximal limb (Fig. 2I and i). By E12.5 Sostdc1 expression activated in the cartilage template outlining the ribs, vertebrae and digits (Fig. 2J and j). As limb development progresses, SostLacZ remains confined to the ectoderm, and by E14.5 its expression becomes fainter and restricted to the digits. In contrast, Sostdc1LacZ expands its expression domain, surrounding the condensing cartilage anlagen, and intensifying in the proximal limb. By E14.5 Sostdc1 is highly expressed in the limb in regions that include cartilage templates of the digits, mesenchyme and primary hair germs that are ectodermal derived, but is omitted from the most distal tips of the digits.
Fig. 2.

Sost and Sostdc1 expression during limb development visualized by LacZ expression. Sost (A–F) and Sostdc1 (G–L) expressions were examined in a time-course panel of E9.5–E14.5 heterozygous embryos referred to as SostLacZ and Sostdc1LacZ. At E9.5 in SostLacZ embryos, a dorsal view of the whole embryo (A′) and of the forelimb (A″) shows expression in the emerging limb bud while no limb expression is detected in Sostdc1LacZ (G). For E10.5 to E12.5 embryos (B–D and H–J), AER views (B′–D′), dorsal limb views (B″–D″ and H′–J′) and transverse section views (b–d; h–j; and i′, j′) are provided. For E13.5 and E14.5 embryos (E–F; K–L), dorsal limb views (E′–F′; K′–L′) are provided.
Fig. 6.

Altered SHH and FGF signaling causes polydactyly and syndactyly. Shh domain was expanded along the anterior–posterior and proximal–distal axis in Sost–/– and Sost–/–; Sostdc1–/– relative to WT and Sostdc1–/– limb buds at E10.5 (A, arrows, bracket) and E1 1.5 (D). Downstream of Shh, Gli1 expression was dramatically expanded in Sost–/–; Sostdc1–/– E11.5 limb buds relative to all other genotypes (E, brackets). Grem1 expression was absent in Shh positive region, and in Sostdc1–/– and Sost–/–; Sostdc1–/– limbs the Grem1 domain was reduced on the posterior side (B, brackets). Fgf8 AER expression domain was expanded and disorganized in Sost–/– and Sost–/–; Sostdc1–/– limbs, and on the anterior side of the Sost–/–; Sostdc1–/– limbs Fgf8 expression was reduced in all time points examined (B and F–G, arrows). A reduction in interdigital apoptosis was also detected on the anterior side of Sost–/–; Sostdc1–/– limb buds at E12.5 (I, arrow).
At E10.5–11.5 both Sost and Sostdc1 are expressed in the head and mark parts of the nervous system of the developing embryos, but Sost expression has some unique features. As early as E9.5 Sost marks a very thin layer of cells that line the edge of the neural folds (Fig. 2B, C and Sup. Fig. 1A). As the neural folds close, Sost expression becomes restricted to the base of the cerebellum (Fig. 2D-E), consistent with our previously results in the adult cerebellum (Collette et al., 2012). Along the trunk, at E11.5 Sost emerges symmetrically at the base of the spinal cord, at the level of the forelimbs (Fig. 2C; Sup. Fig. 1B and D) in a cluster of cells that likely mark the lateral motor neurons migrating into the limb. A zoomed in view allows the visualization of projections into the limb bud that resemble previously described projections of motor axons (Dasen et al., 2008) (Sup. Fig. 1D); by E13.5 Sost marks a network of axons that innervate the dorsal limb flank mesenchyme (Fig. 2E′; Sup. Fig. 1E), and appears near the base of the hindlimbs by E13.5 (Sup. Fig. 1C). Sostdc1 outlines the branchial arches as early as E10.5 (Fig. 2H); it marks the otic vesicle (Fig. 2H) and by E14.5 it is highly expressed in the ear, the developing skin and hair follicles (Fig. 2L). The E14.5 Sostdc1 ectodermal expression (Fig. 2L) is similar to the previously reported beta-catenin LacZ reporter strains (Zhang et al., 2012; Narhi et al., 2008).
Sost and Sostdc1 are expressed in adjacent tissues in the neonatal skeleton
Starting at E16.5, Sost expression becomes restricted primarily to the skeleton, while Sostdc1 expression spreads over the ectoderm, marks the hair follicles (Laurikkala et al., 2003; Narhi et al., 2008), tooth germs (Laurikkala et al., 2003) and many soft tissues throughout the late embryo. In neonates Sost marks the axial and appendicular skeleton (Fig. 3A–B), while Sostdc1 is more broadly expressed in the limb and other soft tissues adjacent to bone (Fig. 3A′–B′). In the neonatal bone, we find Sost expression primarily in a location consistent with osteocytes (Fig. 3C and c); no Sostdc1 expression is detected in this cell type; however Sostdc1 is highly expressed in the adjacent periosteum (Fig. 3C′ and c′), connective tissue, muscle, and periarticular chondrocytes of the epiphysis (Fig. 3D′), while Sost expression is missing in these tissue types (Fig. 3D). In the skull and jaw Sost marks putative osteoblasts and osteocytes cells in wholemount calvaria (Fig. 3E and e) while Sostdc1 is found in the membrane covering the calvaria (Fig. 3E′ and e′) and in the connective tissue surrounding the mandible (Fig. 3F′). Significant Sostdc1 expression was also found in the peripheral nervous system and in intervertebral disks (Fig. 3B′). Other sites of Sost neonatal expression included specific regions of the cardiovascular system (Sup. Fig. 2B, b, C and c). Sostdc1 was also found in the kidney and in the urogenital system in neonates.
Fig. 3.
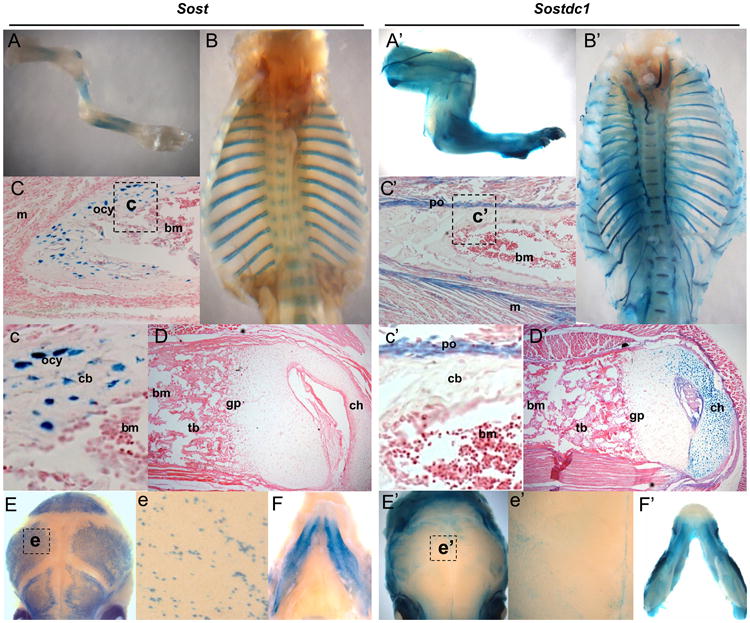
Sost and Sostdc1 expression in the neonatal skeleton. Sost expression marked cells in the appendicular (A) and axial (B, E) skeleton, while Sostdc1 was more broadly expressed in the limbs (A′) and rib cage (B′) to encompass connective tissue, muscle, cartilage and neurons. Sectioned long bones revealed Sost expression primarily in the osteocytes of cortical bone (C and c), but no obvious Sost expression was detected in the articular cartilage (D). Sostdc1 however was not detected in the mineralized bone; it was expressed in the periosteum (C and c′), the immediately adjacent muscles (C′) and the periarticular chondrocytes in the condyle (D′). Both Sost and Sostdc1 were also detected in the skull (E and E′) and mandible (F and F′); Sost expression was localized to osteoblasts and osteocytes in wholemount calvaria (e), while Sostdc1 was present in the connective tissue over the calvarial bones (e′); and m muscle; ocy osteocytes; bm bone marrow; po periosteum; cb cortical bone; gp growth plate; and ch chondrocytes.
Sostdc1 and Sost have broad tissue distribution in the adult
In adult tissues, Sost and Sostdc1 expression domains comprehensively encompass nearly every organ system and tissue in the body. Sost is robustly expressed in the skeleton, primarily in osteocytes, but low levels of Sost expression were also detected in osteoblasts and osteoclasts (Sup. Fig. 3A and C). This expression is consistent with its previously described roles in bone formation (Collette et al., 2012; Li et al., 2008), B-cell maintenance in the bone marrow niche (Cain et al., 2012), as well as recent reports that Sost is expressed in osteoclasts of aged mice (Ota et al., 2013). Other sites of Sost expression included the epididymis and vas deferens of the testis (Sup. Fig. 4A and a), the pyloric sphincter (Sup. Fig. 4B), parts of the cerebellum (Sup. Fig. 4C and c) and the kidney (Sup. Fig. 4E and e). Contrary to previous reports, Sost expression was not detected in the liver or cartilage, suggesting some differences between human and mouse endogenous Sost expression (Geetha-Loganathan et al., 2010). However, Sost cartilage expression has recently been linked to osteoarthritis, and there is a possibility that Sost expression turns on in the articular cartilage in response to joint trauma (Chan et al., 2011).
Sost expression was also detected in a highly restricted cluster of cells in the heart (Sup. Fig. 2D, d, and e), and in the ascending aorta branches (carotid arteries) of both the neonatal and adult heart (Sup. Fig. 2B, C, b, c, and E). The cardiovascular neonatal expression we observed is consistent with previous reports where Sost expression was detected in the smooth muscle cells of the ascending aorta, aortic arch, brachiocephalic artery, common carotids, and pulmonary trunk (van Bezooijen et al., 2007a). In contrast, Sostdc1 expression was present in the cardiac plexus that innervates the heart (Sup. Fig. 2F and f).
Sostdc1 expression, however, has not been fully characterized. Sostdc1 is expressed in the skin and hair follicles (Fig. 4A and a–a′), in the brain (Fig. 4B and B′), the stomach and intestines (Fig. 4C, C′, D and D′), pancreas (Fig. 4E and E′), kidney (Fig. 4F and F′), nerves (Fig. 4G and I), lungs (Fig. 4H), smooth and skeletal muscles (Fig. 4J and K), vasculature (Fig. 4L), the urogenital system, teeth, connective tissue and periosteum. Previously published reports have described several phenotypes associated with these expression domains including roles in tooth development (Kassai et al., 2005), hair follicle development (Narhi et al., 2008, 2012), urogenital system development (Maeda et al., 2007), kidney development and toxicity (Tanaka et al., 2008), and more recently pancreas metabolism (Henley et al., 2012). While Sostdc1 expression has not been described in the context of muscle tissue, the robust intermittent expression pattern is consistent with a described role for WNT signaling in the identity of muscle fiber types (Tee et al., 2009; von Maltzahn et al., 2012).
Fig. 4.
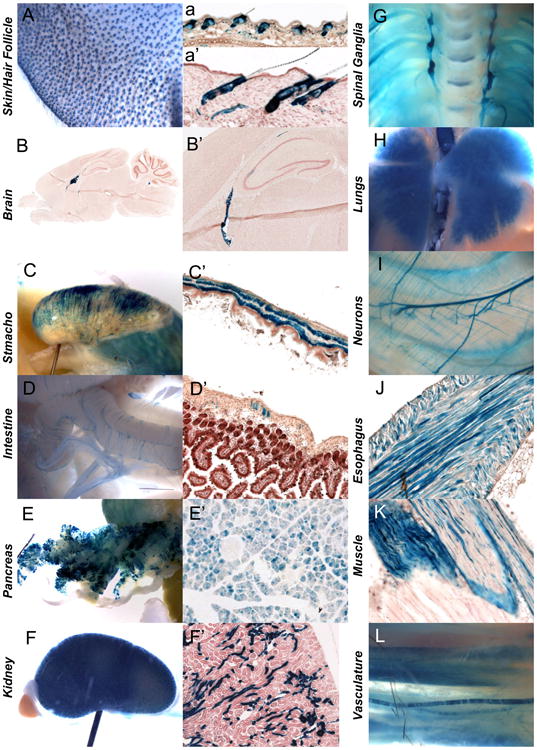
Sostdc1 expression in adult tissues. Sostdc1 expression was examined in wholemount and sectioned LacZ stained tissues, and was detected in the skin and hair follicles (A, a, and a′). A highly specialized region in the brain was positive for Sostdc1 (B and B′). Smooth muscles of the stomach (C and C′), intestine (D and D′) and esophagus (J), and skeletal muscle (K) expressed Sostdc1. Sostdc1 was also robustly expressed in the pancreas (E and E′) and kidney (F and F′) and in the nervous system Sostdc1 stained spinal ganglia (G) and the lungs (H). Neurons (I) and vasculature was also positive for Sostdc1 (L).
Preaxial polydactyly in Sost–/–; Sostdc1–/– mice
Sostdc1 has not been previously associated with functions during skeletal development and Sostdc1–/– mice do not exhibit any obvious limb patterning defects. In contrast, Sost has been described in limb development in the context of Sclerosteosis. Sclerosteosis patients show variably penetrant limb developmental anomalies in the autopod, with a range of phenotypes that include soft and/or bony tissue syndactyly of anterior digits, nail dysplasia and radial deviation of digits (Hamersma et al., 2003; Itin et al., 2001). We also recently described SOST gain-of-function mice where overexpression of SOST caused severe limb patterning defects (Collette et al., 2010). Upon closer examination of Sost–/– mice we found 4% of neonates to display all hand defects previously described for Sclerosteosis patients (Fig. 5B-B2; Table 1). In addition, both Sost–/– and Sostdc1–/– mice had varying degrees of ventral pigmentation and ectopic hair growth on the autopod (Fig. 5b′; Table 1). When Sost–/– mice were mated to Sostdc1–/– to generate double knockout mice, ∼50% of the double knockout embryos displayed hand defects. Also, a new autopod defect emerged consisting of preaxial polydactyly primarily of digit 1 and occasionally of digit 2 (Fig. 5C–C″ and C4–C4′). The accompanying syndactyly of anterior digits seen in Sclerosteosis occurred in 6% of the embryos, but was not statistically different from the single mutant (Table 1). The preaxial polydactyly was visualized as early as E11.5 of development, in the form of ectopic tissue thickening in the anterior limb, during digit specification and templating of the autopod and is evident as polydactyly by as early as E12.5 (Fig. 5C1, arrow). We observed varying degrees of duplication, from incomplete soft-tissue duplication, to duplication of multiple projections with or without bone, at the site of the expected first digit, and protruding from the ventral autopod, the majority of defective Sost–/–; Sostdc1–/– adults had a rudimentary duplicated thumb (Fig. 5C4′ and C4″) or an occasional branching off digit 2 (Fig. 5C4).
Fig. 5.
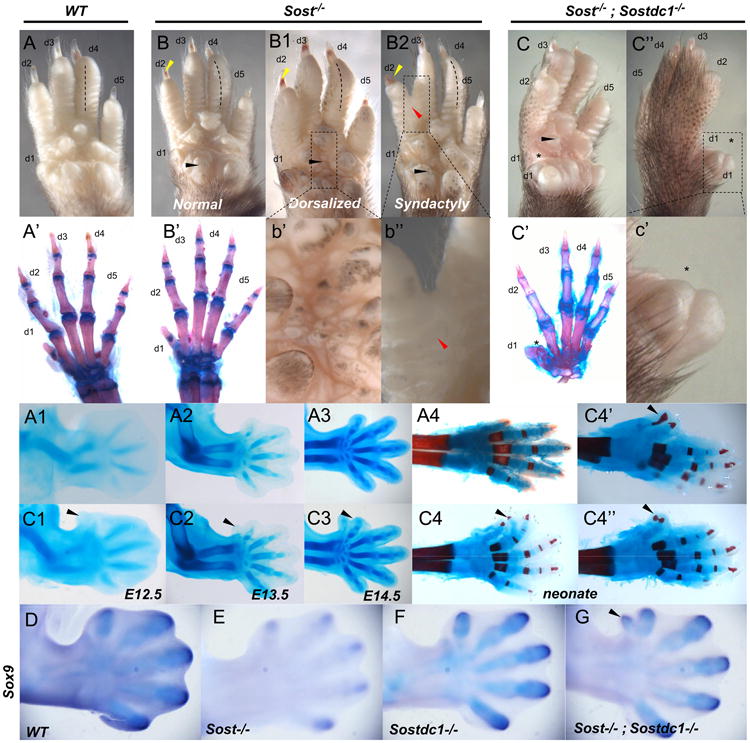
Limb defects in Sost–/– and Sost–/–; Sostdc1–/– mice. Compared to adult WT autopods (A), Sost–/– autopods (B, B1, and B2, insets at b′ and b″) displayed pigmentation on the ventral side (B1 and b′), digit 2–3 syndactyly (B2 and b″; red arrow), nail dysplasia (B, B1 and B2; yellow arrows) and radial deviation of digits, primarily observed for digit 4 (B, B1 and B2, dotted lines). Ventral pigmentation was also observed in Sost–/–; Sostdc1–/– autopods. Unlike WT and Sost–/– autopods that had normal digit patterning (A′–B′), Sost–/–; Sostdc1–/– digit 1 was thicker (C, C″ and c′; asterisk) and skeletal preparation indicated the presence of extra bones (C′) in digit 1. A time course skeletal preparation examination revealed that an ectopic digit 1 was distinguishable as a tissue projection as early as E12.5 (A1-3 vs. C1-3); and the neonate Sost–/–; Sostdc1–/– limbs displayed a range of extra digits (C4 and C4″) associated with ectopic projections primarily from digit 1 (C4′ and C4″) and in rare occasions from digit 2 (C4; black arrow). Sox9 in situ hybridization on E13 embryos revealed an ectopic digit 1 field in Sost–/–; Sostdc1–/– autopods. d digit.
Table 1.
Autopod phenotypic analysis of Sost and Sostdc1 single and double knockout mice.
| Genotype | Normal | Syndactyly | Ventral pigment | Digit duplication |
|---|---|---|---|---|
| Sost–/– | 95 | 4 | 1 | 0 |
| Sostdc1–/– | 83 | 0 | 17 | 0 |
| Sost–/–; Sostdc1–/– | 53 | 6 | 10a | 36b |
1 neonate had an extra dermal pad.
1 neonate had syndactyly; 4 had ventral pigmentation.
Sox9 is expressed in committed chondroprogenitor cells and differentiated chondrocytes and has been previously shown to function as an essential regulator of chondrogenesis. When Sox9 was expressed ectopically in Sox9-transgenic mice, the cell density of the anterior limb bud mesenchyme at the site of Sox9 transgene expression increased around E13.5, and a nubbin emerged ∼E14.5 highly similar to the ectodermal protrusion observed in Sost–/–; Sostdc1–/– autopods (Fig. 5C3) (Akiyama et al., 2007). The Sox9 transgenics showed increased proliferation at Sox9 ectopic sites and the subsequent differentiation of Sox9 positive cells into chondrocytes (Akiyama et al., 2007). To determine if Sox9 is involved in the Sost–/–; Sostdc1–/– autopod defect, we examined Sox9 expression in E13.5 embryos. Expression of Sox9 appeared reduced in Sost–/– and enhanced in Sostdc1–/– embryos, although these patterns are not associated with cartilage templating defects in the single mutants. In Sost–/–; Sostdc1–/– autopods, ectopic expression of Sox9 was observed in the anterior region of digit 1 (Fig. 5G) which corresponded to the site of digit 1 duplication.
Changes in Gli1/Gli3 expression promote ectopic Sox9 and polydactyly in Sost–/–; Sostdc1–/–
Digit 1 formation has been described as Shh-independent since Shh–/– mice are missing all but digit 1, however altered morphogen diffusion or ectopic Shh affects anterior digits and has been shown to cause digit duplication. A number of mouse mutants with preaxial polydactyly exhibit ectopic Shh expression in the anterior mesenchyme of the limb bud during development; these include Extra toes (Xt), Strong's luxoid (lst), luxate, X-linked polydactyly, Rim4, Hemimelic extra toes, as well as Msx1–/–;Msx2–/– mutants (Bensoussan-Trigano et al., 2011; Buscher and Ruther, 1998; Chan et al., 1995; Masuya et al., 1995; Sharpe et al., 1999). In particular paired-type homeodomain transcription factor Alx4 is expressed in the mesenchyme of the anterior limb, and when mutated causes preaxial polydactyly slightly more severe than the polydactyly observed in Sost–/–; Sostdc1–/– mice. Since Sostdc1 expression domain slightly overlaps the ZPA at E10.5 (Fig. 2H′) and Alx4 as well as Msx1–/– ;Msx2–/– mice display ectopic Shh in the anterior digit 1 field of the autopod, we first compared Shh expression in single and double mutant embryos to WT embryos.
Consistent with loss of Shh expression in SOSTtg limbs (Collette et al., 2010), the Shh field was expanded both anteriorly and distally in Sost–/– and Sost–/–; Sostdc1–/– embryos as early as E10.5 of development; in double mutants, this was before the preaxial polydactyly was visually detected (Fig. 6A). Shh expression in E11.5 Sost–/–; Sostdc1–/– limbs was estimated to be 3.66-fold above WT levels as determined by microarray expression analysis (Table 2). However, unlike other mutants with digit 1 polydactyly, we detected no ectopic Shh expression in the anterior region of the E10.5 or E11.5 autopod (Fig. 6A and D) in all Sostdc1–/–; Sostdc1–/– embryos examined (N=28). Consistent with Shh expansion, Grem1 domain was reduced posteriorly since Shh positive cells repress Grem1 expression (Fig. 6B), and was also missing in the anterior mesenchyme where ectopic digits form in double mutants (Fig. 7K′ and K″), but this reduction did not translate into a significant quantitative change in Grem1 expression by microarray analysis in the E11.5 limb (Log FC: 0.81, FC: 1.75, p-value: 0.37133).
Table 2.
Differentially expressed genes in the Sost; Sostdc1 KO E11.5 forelimb.
| Up | LogFC | FC | p-value | Down | LogFC | FC | p-value | |
|---|---|---|---|---|---|---|---|---|
| WNT signaling | Tcf12 | 2.52 | 5.74 | 0.00020 | Sfrp1 | −3.06 | 8.31 | 0.00024 |
| Id2 | 2.48 | 5.61 | 0.00008 | Dvl2 | −2.93 | 7.61 | 0.00102 | |
| Fn1 | 2.41 | 5.34 | 0.00671 | Fzd1 | −2.92 | 7.57 | 0.00038 | |
| Gsk3b | 2.08 | 4.22 | 0.00039 | Ror1 | −2.66 | 6.32 | 0.00230 | |
| Bcl9 | 1.93 | 3.82 | 0.00042 | Ctnnb1 | −2.40 | 5.27 | 0.00015 | |
| Ccnd2 | 1.85 | 3.61 | 0.00512 | Snai1 | −1.97 | 3.91 | 0.00004 | |
| Nlk | 1.81 | 3.51 | 0.00055 | Apc | −1.91 | 3.75 | 0.00036 | |
| Lef1 | 1.72 | 3.29 | 0.01267 | Frzb | −1.84 | 3.57 | 0.00422 | |
| Wnt5a | 1.52 | 2.86 | 0.01145 | Fzd6 | −1.83 | 3.55 | 0.00022 | |
| Tcf4 | 1.35 | 2.56 | 0.00264 | Csnk1d | −1.77 | 3.40 | 0.00048 | |
| RhoA | 1.33 | 2.51 | 0.00155 | Axin2 | −1.65 | 3.15 | 0.00170 | |
| Wnt6 | 1.16 | 2.23 | 0.00010 | Cdc42 | −1.62 | 3.07 | 0.00010 | |
| Csnk1a1 | 1.06 | 2.08 | 0.00400 | Fzd7 | −1.48 | 2.79 | 0.00348 | |
| Tsc1 | 1.06 | 2.06 | 0.00187 | Rac1 | −1.46 | 2.75 | 0.00004 | |
| Axin1 | −1.44 | 2.72 | 0.00025 | |||||
| Dkk3 | −1.12 | 2.16 | 0.00094 | |||||
| BMP signaling | Twsg1 | −3.94 | 15.33 | 0.00171 | ||||
| Smad3 | −3.66 | 12.63 | 0.00022 | |||||
| Nog | −2.47 | 5.56 | 0.00002 | |||||
| Bmp1 | −2.30 | 4.94 | 0.00017 | |||||
| Bmpr1b | −2.05 | 4.14 | 0.00388 | |||||
| Bmp7 | −1.40 | 2.64 | 0.00101 | |||||
| Bmp2k | −1.12 | 2.17 | 0.00715 | |||||
| Bmpr2 | −1.07 | 2.09 | 0.00832 | |||||
| FGF signaling | Fgf8 | 1.28 | 2.43 | 0.00453 | Fgfr2 | −2.52 | 5.72 | 0.00007 |
| Fgfr1 | −2.31 | 4.95 | 0.00001 | |||||
| Fgfr3 | −1.51 | 2.84 | 0.00469 | |||||
| Homeobox transcription factors | Hoxa10 | 2.06 | 4.16 | 0.00675 | Hoxd13 | −3.60 | 12.12 | 0.00006 |
| Hoxd4 | 1.93 | 3.80 | 0.00014 | Tbx15 | −3.06 | 8.38 | 0.00061 | |
| Hoxc8 | 1.06 | 2.08 | 0.03535 | Sox4 | −2.96 | 7.80 | 0.00230 | |
| Prrx1 | −2.64 | 6.23 | 0.00080 | |||||
| Hoxc5 | −2.39 | 5.25 | 0.00016 | |||||
| Hoxd12 | −2.11 | 4.33 | 0.00027 | |||||
| Sox6 | −1.95 | 3.85 | 0.00548 | |||||
| Sox5 | −1.50 | 2.83 | 0.00700 | |||||
| Hoxd11 | −1.36 | 2.56 | 0.00054 | |||||
| Pitx2 | −1.34 | 2.53 | 0.028202 | |||||
| Hoxc6 | −1.27 | 2.42 | 0.00275 | |||||
| Sox7 | −1.05 | 2.06 | 0.00024 | |||||
| SHH signaling | Shh | 1.83 | 3.65 | 0.03943 | Gli3 | −1.88 | 3.68 | 0.00352 |
| Ptch1 | 1.78 | 3.44 | 0.00635 | |||||
| Gli1 | 1.20 | 2.30 | 0.005846 | |||||
| TGFβ signaling | Tgfbr1 | −4.75 | 27.03 | 0.00161 | ||||
| Smad3 | −3.66 | 12.63 | 0.00022 | |||||
| Tgfbr2 | −2.13 | 4.37 | 0.00108 | |||||
| Smurf2 | −1.67 | 3.18 | 0.0001 | |||||
| Tgfbr3 | −1.51 | 2.84 | 0.00085 | |||||
| Acvr1 | −1.48 | 2.79 | 0.003522 | |||||
| Smurf1 | −1.41 | 2.66 | 0.01048 | |||||
| Tgfb1 | −1.02 | 2.02 | 0.00096 |
Fig. 7.

Gli3, Grem1, HoxD13, Bmp4 and Bmp7 expression is affected in Sost–/–; Sostdc1–/– E11.5 limbs. Consistent with a reduction in mRNA expression of Gli3, Gli3 activator protein expression was dramatically reduced in the ectoderm of Sost–/–; Sostdc1–/– E11.5 limbs (A and D). Higher magnification images of the anterior region of the limb showed a dramatic reduction in Gli3 both in the ectoderm (marked by dashed lines) and the underlying mesenchyme (B and E). Similarly, the pre-chondrocytes in the cartilage condensation stained positive for Gli3 in the WT limbs, but had little expression in the double knockouts (F). Grem1 expression was reduced in the anterior mesenchyme in double knockout (K′–K″) relative to WT limbs (G′–G″). Asterisks indicate region of lost anterior expression. HoxD13 was ectopically up-regulated in the anterior mesenchyme in the regions corresponding to digit 1 (L, L′, and L″; green arrows) and on the ventral side of the autopod in an ectodermal nubbin (L′ green arrow). Both Bmp4 and Bmp7 expression was absent from the AER (M and N; red asterisks). Views are indicated at the bottom of the figure.
SHH signaling utilizes Gli transcription factors to mediate anterior-posterior limb patterning, and these proteins can function as either activators or repressors of transcription. Gli3, which functions primarily as a repressor, but the full-length protein can also serve as an activator, has been suggested to be the main effector of SHH signaling. In the absence of Gli3 repression, anterior digits are duplicated and take on anterior digit character that is dependent on the timing of Gli3 inactivation, such that inactivation at E10.5 causes digit 1 duplication highly similar to the phenotype observed in Sost–/–; Sostdc1–/– (Bowers et al., 2012). Gli1, a downstream transcriptional activator of SHH, while not deemed essential for limb development in single KOs, does contribute to the formation of a posterior tissue nubbin in Gli1–/–; Gli2–/– autopods (Park et al., 2000), and has been shown to activate Sox9 expression via a Gli1-dependent transcriptional regulatory element (Bien-Willner et al., 2007). Consistent with these previous observations, we find Gli1 to be up-regulated in Sost–/–; Sostdc1–/– at E11.5, through both a dramatic anterior expansion (Fig. 6E) as well as a 2.3-fold change in transcript levels. Quantitatively Gli3 was reduced by 3.68-fold in Sost–/–; Sostdc1–/– in E11.5 limbs (Table 2). Immunofluorescent stains for activated Gli3 confirmed a dramatic reduction of Gli3 activator in the ectoderm, mesenchyme and in the cartilage anlangen in the E12.5 Sost–/–; Sostdc1–/– limbs (Fig. 7D–F). In addition we observed ectopic anterior Hoxd13 expression (Fig. 7L′–L″) in E11.5 Sost–/–; Sostdc1–/– limbs consistent with results described for a hypermorphic activator allele of Gli3 that resulted in preaxial polydactyly (Wang et al., 2007). Comprehensively, these findings suggest that the preaxial polydactyly in Sost–/–; Sostdc1–/– limbs is the result of altered SHH signaling that induces ectopic Sox9 expression via Gli3 derepression to promote tissue nubbins or extra rudimentary copies of anterior digits.
Altered FGF and BMP signaling cause syndactyly in Sost–/–; Sostdc1–/–
Sostdc1 has been previously characterized as both a WNT- and BMP-antagonist (Henley et al., 2012; Murashima-Suginami et al., 2008; Tanaka et al., 2008), and ectodermal derived BMPs and Fgf8 have been shown to control interdigital apoptosis (Hernandez-Martinez et al., 2009), a mechanism involved in the establishment of both polydactyly and syndactyly. Consistent with previous findings that Fgf8 expression promotes cell survival and growth in the distal limb mesenchyme and that Fgf8 repression triggers interdigital apoptosis associated with syndactyly we found Sost–/–; Sostdc1–/– embryos to display both an increase in Fgf8 expression characterized by disorganized expansion of the AER domain, as well as a disruption of the AER continuity characterized by speckled down-regulation of Fgf8 primarily in the anterior region of the limb (Fig. 6C, F, and G). Quantitatively Fgf8 was found to be 2.43-fold above WT levels in Sost–/–; Sostdc1–/– E11.5 forelimbs, while Fgf receptors 1 through 3 were down-regulated (Table 2). Since ∼6% of Sost–/–; Sostdc1–/– embryos display syndactyly of anterior digits, we examined whether regions of Fgf8 down-regulation corresponded to a decrease in interdigital apoptosis, and found a reduction in apoptosis in the 1-2 interdigital field at E12.5 (Fig. 6I).
Since Bmp2/4 restrict Shh expression and antagonize Fgf signaling in the early limb, and Bmp7 induces cell death in the distal mesenchyme and inhibits Fgf8 expression in the ectoderm at later developmental times, we examined whether members of the Bmp family are transcriptionally affected in Sost–/–; Sostdc1–/– embryos. In situ hybridization for Bmp4 and Bmp7 showed a complete absence of expression in the AER of Sost–/–; Sostdc1–/– embryos (Fig. 7M–N). Additionally, most Bmp-related transcripts were down-regulated in Sost–/–; Sostdc1–/– E11.5 forelimbs suggesting an overall reduction in BMP-signaling in the limb (Table 2) which may account for both Shh up-regulation and Fgf8 ectodermal/mesenchymal expansion.
WNT signaling is both up- and down-regulated in Sost–/–; Sostdc1–/– limbs
Sost and Sostdc1 have been previously described as antagonists of both WNT and BMP signaling (Collette et al., 2010; Holdsworth et al., 2012; Krause et al., 2010; Tanaka et al., 2010; Winkler et al., 2003). Our previous work of examining limb defects in transgenic mice overexpressing SOST showed that the BatGal transgene, a reporter of canonical WNT signaling was down-regulated in the limb mesenchyme, in response to elevated levels of SOST in the limb ectoderm (Collette et al., 2010). This data suggested that SOST functions as a WNT antagonist in the limb, in gain-of-function transgenic mice. Based on these previous findings, we anticipated Sost–/–; Sostdc1–/– limb buds to display elevated WNT and/or possibly BMP signaling.
To determine what signaling pathways are altered due to lack of Sost and Sostdc1 in the limb, we compared gene expression between Sost–/–; Sostdc1–/– and wildtype E11.5 forelimbs using Affymetrix gene expression arrays (Mouse Genome 430 2.0 Array). We found 1218 and 1701 transcripts to be more than 2-fold up- or down-regulated in Sost–/–; Sostdc1–/– forelimbs (p ≤ 0.05), respectively. Consistent with the molecular marker analysis and WNT signaling function, pathway analysis identified WNT and SHH signaling among the top most significantly enriched in up-regulated genes; while all significantly altered transcripts associated with the BMP and TGFb signaling pathways were down-regulated in Sost–/–; Sostdc1–/– limbs (Table 2). Interestingly, the WNT signaling was also identified among the top enriched in down-regulated genes, with 16 transcripts dramatically reduced in Sost–/–; Sostdc1–/– limbs (Table 2).
To further determine what changes in WNT signaling occurred as a relationship of the signal transduction from receptor to transcriptional targets, we mapped each transcriptionally altered transcript on the WNT signaling map in Fig. 8. We depicted known inhibitory relationships among molecules (either at the transcript or protein level) by red lines and positive relationships by blue lines. We also marked the genes with significant transcriptional changes identified in Sost–/–; Sostdc1–/– E11.5 limb buds with a red star for down-regulated genes or a green star for up-regulated genes. Contrary to our hypothesis that both Sost- and Sostdc1 interfere with canonical WNT signaling, we found β-catenin (CTNNB1; Fig. 8; yellow box) transcript levels reduced by 5.27-fold. In addition we found two inhibitors of canonical WNT signaling: GSK3B and NLK to be significantly up-regulated, 4.22-and 3.51-fold, respectively; and hence to further contribute to blunting β-catenin activator function, in the Sost–/–; Sostdc1–/– limbs. Immunofluorescent stains of E12.5 sectioned limbs showed a marked increase in both ectodermal and mesenchymal β-catenin activity in Sost–/– limbs, while Sostdc1–/– limbs had a slight reduction in mesenchyme. Consistent with the microarray expression data, the double knockouts exhibited a dramatic reduction in both ectodermal and mesenchymal activated β-catenin (Fig. 9). In addition, two known non-canonical WNT ligands, WNT5A and WNT6 were significantly up-regulated 2.86- and 2.23-folds, respectively, and so were several transcription factors known to activate downstream WNT targets, including TCF4, TCF12, Lef1, and BCL9, along with several known WNT target genes, CCND, ID2 and FN1 (Table 2). While the microarray data conclusively high-lighted WNT signaling as the only pathway significantly up-regulated, it also suggested that the phenotypes are driven by a β-catenin independent mechanism, and likely facilitated by the overabundance of the two non-canonical WNT ligands.
Fig. 8.

Transcriptional changes in the WNT signaling pathway of E11.5 Sost–/–; Sostdc1–/– limbs. Genes found to be transcriptionally up- or down- regulated by more than 2-fold in Sost–/–;Sostdc1–/– limb buds are marked by a green or red star, respectively. Red arrows mark inhibitory relationships, and blue arrows mark other relationships such as transcriptional up-regulation or protein stabilization. β-catenin (CTNNB1) was found to be down-regulated (yellow box).
Fig. 9.

Activated β-catenin is dramatically reduced in Sost–/–; Sostdc1–/– limbs. Lack of Sost up-regulates WNT signaling as evidenced by increased staining for activated β-catenin in the ectoderm (marked by dashed lines) and underlining mesenchyme (B). Sost–/– ectoderm also appears thicker than all other genotypes; lack of Sostdc1 has little effect on ectodermal β-catenin (C), but causes a slight reduction in the mesenchyme; removing both Sost and Sostdc1 dramatically reduces both ectodermal and mesenchymal activated β-catenin protein (D).
Discussion
The WNT signaling pathway is involved in a broad range of developmental and physiological processes ranging from cell proliferation, cell fate, body axis determination, tissue morphogenesis, and tissue homeostasis. Thus, its dysregulation has been linked to multiple congenital and degenerative diseases, as well as cancer. Sost and Sostdc1 have been previously described as WNT antagonists, and therefore loss and/or gain-of-function mutations in these molecules are likely to interfere with aspects of WNT signaling pathway involved in critical developmental and metabolic processes. Here we showed that both Sost and Sostdc1 have a broad tissue distribution in both the developing embryo and the adult mouse, broadening our current understanding of their expression pattern and therefore highlighting new potential functional sites where these two molecules could interfere with WNT and/or other signaling pathways. Their adjacent expression domains in the developing limb show epithelial-mesenchymal interactions that overlap to influence anterior digit patterning, especially in that several genes do not appear to be differentially regulated by in situ hybridization in single mutants, but show altered expression only in Sost–/–; Sostdc1–/– double mutants. Second, we establish that the combined lack of Sost and Sostdc1 causes preaxial polydactyly through modulating SHH signaling, through Gli3 transcriptional repression, up-regulation of Gli1 and subsequent ectopic activation of Sox9 in the digit 1 field. Ectopic Sox9 expression in Sost–/–; Sostdc1–/– mice is likely a consequence of misregulated limb patterning genes upstream of Sox9, since we show that patterning genes such as Grem1, Fgf8 and Shh are misregulated in the developing limb of Sost–/–; Sostdc1–/– mice but they remain unaffected in Sox9 gain-of-function mutant (Akiyama et al., 2007). The phenotype we describe herein is highly similar to the recently described conditional inactivation of Gli3 in the developing autopod using a Cre deletor under the control of Hoxa13 locus (Lopez-Rios et al., 2012). In this study, Lopez-Rios et al. were able to show that Gli3 acts in the anterior mesenchyme to restrict and terminate Grem1 expression in the anterior autopod in a spatiotemporally controlled manner, to promote BMP-dependent exit of progenitors from the proliferation phase to the chondrogenic differentiation stage. This is consistent with our data that show increased Gli3 activation in the anterior limb restricts Grem1 expression despite the lack of ectopic Shh. In the absence of Gli3 repressor, chondrogenic differentiation is delayed, resulting in an accumulation and subsequent increase in the pool of chondrogenic progenitor cells, which ultimately create new digit fields in the anterior region of the autopod (Lopez-Rios et al., 2012).
Lopez-Rios et al. show that the timing of Gli3 inactivation determines the severity of the polydactyly phenotype, which in turn is directly related to the duration of the proliferative expansion of the progenitor cells phase, such that conditional inactivation of Gli3 using Hoxa13-Cre results in the dissipation of the Gli3 transcripts by E11.75, and subsequent duplication of digit 1 only (Lopez-Rios et al., 2012). This timing coincides with the emergence of Sostdc1 in the limb, since Sostdc1 expression initiates at E10.5 on the ventral side of the autopod and subsequently expands to the proximal region of the E11.5 autopod. The cumulative absence of Sost and Sostdc1 from the developing limb represses Gli3 transcript levels by 3.68-fold which is sufficient to generate a phenotype highly similar to the removal of Gli3 allele at ∼E11.5. As loss of Gli3 transcription results primarily in the loss of Gli3 repressor, our data shows that increased Gli3 activator without increased Gli3 transcription induces a similar mild preaxial polydactyly phenotype, similar to a study that demonstrated a hypermorphic allele of Gli3 increased the activator form of the protein and resulted in mild preaxial polydactyly (Wang et al., 2007).
The SHH/GREM1/AER-FGF feedback loop has been studied extensively and significant evidence exists that indicate that BMP activity is at low levels during the proliferative expansion of digit progenitors, but at higher levels during chondrogenic differentiation (Bandyopadhyay et al., 2006; Lopez-Rios et al., 2012), and that SHH modulates these downstream effects. In the present study, we show that WNT signaling events upstream of SHH can produce alterations in Gli3 expression which ultimately result in the same chondrogenic differentiation defects that cause preaxial polydactyly, positioning components of WNT signaling as novel candidates for congenital malformations observed in patients with preaxial polydactyly. Finally, extensive gene network and pathway analysis revealed that the preaxial polydactyly phenotype observed in Sost–/–; Sostdc1–/– limbs, while consistent with a lack of WNT inhibition molecular output, it is b-catenin independent, and likely to be mediated by two non-canonical WNT ligands: Wnt5A and Wnt6. In particular, since Wnt6 has been previously described as an ectodermally derived negative regulator of chondrogenesis (Geetha-Loganathan et al., 2010), the link between Sost, Sostdc1, and Wnt6, chondrogenic differentiation and proliferation should be further explored.
Supplementary Material
Acknowledgments
We would like to thank the National Institutes of Health (NIH) Knock-Out Mouse Program (KOMP) and Regeneron for providing the Sost and Sostdc1 knockout mice. NMC, CY, DM and GGL were supported by NIH Grant HD47853 and DK075730. This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. LT was supported by the Intramural Research Program of the NIH, NLM.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.ydbio.2013.08.015.
References
- Ahn Y, Sanderson BW, Klein OD, Krumlauf R. Inhibition of Wnt signaling by Wise (Sostdc1) and negative feedback from Shh controls tooth number and patterning. Development. 2010;137:3221–3231. doi: 10.1242/dev.054668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Stadler HS, Martin JF, Ishii TM, Beachy PA, Nakamura T, de Crombrugghe B. Misexpression of Sox9 in mouse limb bud mesenchyme induces polydactyly and rescues hypodactyly mice. Matrix Biol: J Int Soc Matrix Biol. 2007;26:224–233. doi: 10.1016/j.matbio.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol. 2009;5:e1000262. doi: 10.1371/journal.pcbi.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensoussan-Trigano V, Lallemand Y, Saint Cloment C, Robert B. Msx1 and Msx2 in limb mesenchyme modulate digit number and identity. Dev Dyn. 2011;240:1190–1202. doi: 10.1002/dvdy.22619. [DOI] [PubMed] [Google Scholar]
- Bien-Willner GA, Stankiewicz P, Lupski JR. SOX9cre1, a cis-acting regulatory element located 1.1 Mb upstream of SOX9, mediates its enhancement through the SHH pathway. Hum Mol Genet. 2007;16:1143–1156. doi: 10.1093/hmg/ddm061. [DOI] [PubMed] [Google Scholar]
- Blish KR, Clausen KA, Hawkins GA, Garvin AJ, Willingham MC, Turner JC, Torti FM, Torti SV. Loss of heterozygosity and SOSTDC1 in adult and pediatric renal tumors. J Exp Clin Cancer Res: CR. 2010;29:147. doi: 10.1186/1756-9966-29-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers M, Eng L, Lao Z, Turnbull RK, Bao X, Riedel E, Mackem S, Joyner AL. Limb anterior–posterior polarity integrates activator and repressor functions of GLI2 as well as GLI3. Dev Biol. 2012;370:110–124. doi: 10.1016/j.ydbio.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscher D, Ruther U. Expression profile of Gli family members and Shh in normal and mutant mouse limb development. Dev Dyn. 1998;211:88–96. doi: 10.1002/(SICI)1097-0177(199801)211:1<88::AID-AJA8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Cain CJ, Rueda R, McLelland B, Collette NM, Loots GG, Manilay JO. Absence of sclerostin adversely affects B cell survival. J Bone Miner Res. 2012;27:1451–1461. doi: 10.1002/jbmr.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, Jackson MT, Cake MA, Read RA, Bateman JF, Sambrook PN, Little CB. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage. 2011;19:874–885. doi: 10.1016/j.joca.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Chan DC, Laufer E, Tabin C, Leder P. Polydactylous limbs in Strong's Luxoid mice result from ectopic polarizing activity. Development. 1995;121:1971–1978. doi: 10.1242/dev.121.7.1971. [DOI] [PubMed] [Google Scholar]
- Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, Lane NE, Harland RM, Loots GG. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci USA. 2012;109:14092–14097. doi: 10.1073/pnas.1207188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collette NM, Genetos DC, Murugesh D, Harland RM, Loots GG. Genetic evidence that SOST inhibits WNT signaling in the limb. Dev Biol. 2010;342:169–179. doi: 10.1016/j.ydbio.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121:439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- Dasen JS, De Camilli A, Wang B, Tucker PW, Jessell TM. Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell. 2008;134:304–316. doi: 10.1016/j.cell.2008.06.019. [DOI] [PubMed] [Google Scholar]
- Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3:e314. doi: 10.1371/journal.pbio.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Geetha-Loganathan P, Nimmagadda S, Christ B, Huang R, Scaal M. Ectodermal Wnt6 is an early negative regulator of limb chondrogenesis in the chicken embryo. BMC Dev Biol. 2010;10:32. doi: 10.1186/1471-213X-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet. 2003;63:192–197. doi: 10.1034/j.1399-0004.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- He JW, Yue H, Hu WW, Hu YQ, Zhang ZL. Contribution of the sclerostin domain-containing protein 1 (SOSTDC1) gene to normal variation of peak bone mineral density in Chinese women and men. J Bone Miner Metab. 2011;29:571–581. doi: 10.1007/s00774-010-0253-5. [DOI] [PubMed] [Google Scholar]
- Henley KD, Gooding KA, Economides AN, Gannon M. Inactivation of the dual Bmp/Wnt inhibitor Sostdc1 enhances pancreatic islet function. Am J Physiol Endocrinol Metab. 2012;303:E752–E761. doi: 10.1152/ajpendo.00531.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Martinez R, Castro-Obregon S, Covarrubias L. Progressive interdigital cell death: regulation by the antagonistic interaction between fibroblast growth factor 8 and retinoic acid. Development. 2009;136:3669–3678. doi: 10.1242/dev.041954. [DOI] [PubMed] [Google Scholar]
- Hoffmann FG, Storz JF, Gorr TA, Opazo JC. Lineage-specific patterns of functional diversification in the alpha- and beta-globin gene families of tetrapod vertebrates. Mol Biol Evol. 2010;27:1126–1138. doi: 10.1093/molbev/msp325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan BL, Beddington R, Costantini F, Lacey E. Manipulating the Mouse Embryo: A Laboratory Manual. 2nd. Cold Spring Harbor Press; New York: 1994. [Google Scholar]
- Holdsworth G, Slocombe P, Doyle C, Sweeney B, Veverka V, Le Riche K, Franklin RJ, Compson J, Brookings D, Turner J, Kennedy J, Garlish R, Shi J, Newnham L, McMillan D, Muzylak M, Carr MD, Henry AJ, Ceska T, Robinson MK. Characterization of the interaction of sclerostin with the low density lipoprotein receptor-related protein (LRP) family of Wnt co-receptors. J Biol Chem. 2012;287:26464–26477. doi: 10.1074/jbc.M112.350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Itin PH, Keseru B, Hauser V. Syndactyly/brachyphalangy and nail dysplasias as marker lesions for sclerosteosis. Dermatology. 2001;202:259–260. doi: 10.1159/000051649. [DOI] [PubMed] [Google Scholar]
- Jarvinen E, Tummers M, Thesleff I. The role of the dental lamina in mammalian tooth replacement. J Exp Zool Part B, Mol Dev Evol. 2009;312B:281–291. doi: 10.1002/jez.b.21275. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassai Y, Munne P, Hotta Y, Penttila E, Kavanagh K, Ohbayashi N, Takada S, Thesleff I, Jernvall J, Itoh N. Regulation of mammalian tooth cusp patterning by ectodin. Science. 2005;309:2067–2070. doi: 10.1126/science.1116848. [DOI] [PubMed] [Google Scholar]
- Krause C, Korchynskyi O, de Rooij K, Weidauer SE, de Gorter DJ, van Bezooijen RL, Hatsell S, Economides AN, Mueller TD, Lowik CW, ten Dijke P. Distinct modes of inhibition by sclerostin on bone morphogenetic protein and Wnt signaling pathways. J Biol Chem. 2010;285:41614–41626. doi: 10.1074/jbc.M110.153890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N. Sclerostin is a novel secreted osteoclast-derived bone morpho-genetic protein antagonist with unique ligand specificity. J Biol Chem. 2003;278:24113–24117. doi: 10.1074/jbc.M301716200. [DOI] [PubMed] [Google Scholar]
- Laurikkala J, Kassai Y, Pakkasjarvi L, Thesleff I, Itoh N. Identification of a secreted BMP antagonist, ectodin, integrating BMP, FGF, and SHH signals from the tooth enamel knot. Dev Biol. 2003;264:91–105. doi: 10.1016/j.ydbio.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- Lintern KB, Guidato S, Rowe A, Saldanha JW, Itasaki N. Characterization of wise protein and its molecular mechanism to interact with both Wnt and BMP signals. J Biol Chem. 2009;284:23159–23168. doi: 10.1074/jbc.M109.025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots G, Ovcharenko I. ECRbase: database of evolutionary conserved regions, promoters, and transcription factor binding sites in vertebrate genomes. Bioinformatics. 2007;23:122–124. doi: 10.1093/bioinformatics/btl546. [DOI] [PubMed] [Google Scholar]
- Lopez-Rios J, Speziale D, Robay D, Scotti M, Osterwalder M, Nusspaumer G, Galli A, Hollander GA, Kmita M, Zeller R. GLI3 constrains digit number by controlling both progenitor proliferation and BMP-dependent exit to chondrogenesis. Dev Cell. 2012;22:837–848. doi: 10.1016/j.devcel.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Lee DS, Yanagimoto Ueta Y, Suzuki H. Expression of uterine sensitization-associated gene-1 (USAG-1) in the mouse uterus during the periimplantation period. J Reprod Dev. 2007;53:931–936. doi: 10.1262/jrd.18154. [DOI] [PubMed] [Google Scholar]
- Masuya H, Sagai T, Wakana S, Moriwaki K, Shiroishi T. A duplicated zone of polarizing activity in polydactylous mouse mutants. Genes Dev. 1995;9:1645–1653. doi: 10.1101/gad.9.13.1645. [DOI] [PubMed] [Google Scholar]
- Moleirinho A, Carneiro J, Matthiesen R, Silva RM, Amorim A, Azevedo L. Gains, losses and changes of function after gene duplication: study of the metallothionein family. PLoS One. 2011;6:e18487. doi: 10.1371/journal.pone.0018487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munne PM, Tummers M, Jarvinen E, Thesleff I, Jernvall J. Tinkering with the inductive mesenchyme: Sostdc1 uncovers the role of dental mesenchyme in limiting tooth induction. Development. 2009;136:393–402. doi: 10.1242/dev.025064. [DOI] [PubMed] [Google Scholar]
- Murashima-Suginami A, Takahashi K, Sakata T, Tsukamoto H, Sugai M, Yanagita M, Shimizu A, Sakurai T, Slavkin HC, Bessho K. Enhanced BMP signaling results in supernumerary tooth formation in USAG-1 deficient mouse. Biochem Biophys Res Commun. 2008;369:1012–1016. doi: 10.1016/j.bbrc.2008.02.135. [DOI] [PubMed] [Google Scholar]
- Narhi K, Jarvinen E, Birchmeier W, Taketo MM, Mikkola ML, Thesleff I. Sustained epithelial beta-catenin activity induces precocious hair development but disrupts hair follicle down-growth and hair shaft formation. Development. 2008;135:1019–1028. doi: 10.1242/dev.016550. [DOI] [PubMed] [Google Scholar]
- Narhi K, Tummers M, Ahtiainen L, Itoh N, Thesleff I, Mikkola ML. Sostdc1 defines the size and number of skin appendage placodes. Dev Biol. 2012;364:149–161. doi: 10.1016/j.ydbio.2012.01.026. [DOI] [PubMed] [Google Scholar]
- Ota K, Quint P, Ruan M, Pederson L, Westendorf JJ, Khosla S, Oursler MJ. Sclerostin is expressed in osteoclasts from aged mice and reduces osteoclast-mediated stimulation of mineralization. J Cell Biochem. 2013;114:1901–1907. doi: 10.1002/jcb.24537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Krasnov S, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Karsch-Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012;40:D13–D25. doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–26775. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- Sharpe J, Lettice L, Hecksher-Sorensen J, Fox M, Hill R, Krumlauf R. Identification of sonic hedgehog as a candidate gene responsible for the polydactylous mouse mutant Sasquatch. Curr Biol. 1999;9:97–100. doi: 10.1016/s0960-9822(99)80022-0. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Yasuhara T. Sclerosteosis a case report. J Bone J Surg Am. 1975;57:273–277. [PubMed] [Google Scholar]
- Tanaka M, Asada M, Higashi AY, Nakamura J, Oguchi A, Tomita M, Yamada S, Asada N, Takase M, Okuda T, Kawachi H, Economides AN, Robertson E, Takahashi S, Sakurai T, Goldschmeding R, Muso E, Fukatsu A, Kita T, Yanagita M. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J Clin Invest. 2010;120:768–777. doi: 10.1172/JCI39569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Endo S, Okuda T, Economides AN, Valenzuela DM, Murphy AJ, Robertson E, Sakurai T, Fukatsu A, Yancopoulos GD, Kita T, Yanagita M. Expression of BMP-7 and USAG-1 (a BMP antagonist) in kidney development and injury. Kidney Int. 2008;73:181–191. doi: 10.1038/sj.ki.5002626. [DOI] [PubMed] [Google Scholar]
- Tee JM, van Rooijen C, Boonen R, Zivkovic D. Regulation of slow and fast muscle myofibrillogenesis by Wnt/beta-catenin and myostatin signaling. PLoS One. 2009;4:e5880. doi: 10.1371/journal.pone.0005880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P, Krause C, de Gorter DJ, Lowik CW, van Bezooijen RL. Osteocyte-derived sclerostin inhibits bone formation: its role in bone morphogenetic protein and Wnt signaling. J Bone J Surg Am. 2008;90(Suppl 1):31–35. doi: 10.2106/JBJS.G.01183. [DOI] [PubMed] [Google Scholar]
- Turk T, Leeuwis JW, Gray J, Torti SV, Lyons KM, Nguyen TQ, Goldschmeding R. BMP signaling and podocyte markers are decreased in human diabetic nephropathy in association with CTGF overexpression. J Histochem Cytochem: Off J Histochem Soc. 2009;57:623–631. doi: 10.1369/jhc.2009.953224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bezooijen RL, Deruiter MC, Vilain N, Monteiro RM, Visser A, van der Wee-Pals L, van Munsteren CJ, Hogendoorn PC, Aguet M, Mummery CL, Papapoulos SE, Ten Dijke P, Lowik CW. SOST expression is restricted to the great arteries during embryonic and neonatal cardiovascular development. Dev Dyn. 2007a;236:606–612. doi: 10.1002/dvdy.21054. [DOI] [PubMed] [Google Scholar]
- van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, Quax PH, Vrieling H, Papapoulos SE, ten Dijke P, Lowik CW. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007b;22:19–28. doi: 10.1359/jbmr.061002. [DOI] [PubMed] [Google Scholar]
- van Bezooijen RL, ten Dijke P, Papapoulos SE, Lowik CW. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 2005;16:319–327. doi: 10.1016/j.cytogfr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- von Maltzahn J, Renaud JM, Parise G, Rudnicki MA. Wnt7a treatment ameliorates muscular dystrophy. Proc Natl Acad Sci USA. 2012;109:20614–20619. doi: 10.1073/pnas.1215765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Pan Y, Wang B. A hypermorphic mouse Gli3 allele results in a polydactylous limb phenotype. Dev Dyn. 2007;236:769–776. doi: 10.1002/dvdy.21082. [DOI] [PubMed] [Google Scholar]
- Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. Embo J. 2003;22:6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagita M, Okuda T, Endo S, Tanaka M, Takahashi K, Sugiyama F, Kunita S, Takahashi S, Fukatsu A, Yanagisawa M, Kita T, Sakurai T. Uterine sensitization-associated gene-1 (USAG-1), a novel BMP antagonist expressed in the kidney, accelerates tubular injury. J Clin Invest. 2006;116:70–79. doi: 10.1172/JCI25445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Shi J, Huang Y, Lai L. Expression of canonical WNT/beta-CATENIN signaling components in the developing human lung. BMC Dev Biol. 2012;12:21. doi: 10.1186/1471-213X-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.