

Abstract
Histone lysine methylation has emerged as a critical player in the regulation of gene expression, cell cycle, genome stability, and nuclear architecture. Over the past decade, a tremendous amount of progress has led to the characterization of methyl modifications and the lysine methyltransferases (KMTs) and lysine demethylases (KDMs) that regulate them. Here, we review the discovery and characterization of the KMTs and KDMs and the methyl modifications they regulate. We discuss the localization of the KMTs and KDMs as well as the distribution of lysine methylation throughout the genome. We highlight how these data have shaped our view of lysine methylation as a key determinant of complex chromatin states. Finally, we discuss the regulation of KMTs and KDMs by proteasomal degradation, posttranscriptional mechanisms, and metabolic status. We propose key questions for the field and highlight areas that we predict will yield exciting discoveries in the years to come.
Introduction
Chromatin is a highly ordered structure that contains DNA, histones, and other chromosomal proteins. The basic building block of chromatin is the nucleosome, which is comprised of two copies of each histone: H2A, H2B, H3, and H4. The N-terminal tails of the histones are subject to posttranslational modifications (PTMs), which influence numerous biological processes including transcription, replication, and chromosome maintenance. Methylation of lysine residues within histones is tightly regulated by methyltransferases (KMTs) and demethylases (KDMs) to maintain cell fate and genomic stability. Over the past decade, a number of major discoveries and technological advances have increased our understanding of histone lysine methylation. These advances have emphasized the biological importance of lysine modifying enzymes, prompting discussion of links between these enzymes and important cellular functions. This review discusses the history of lysine methylation; summarizes our current understanding of lysine methylation dynamics, regulation and function; and highlights the bright and exciting prospects for future research in this field.
In the 1960s, RNA synthesis was demonstrated to be modulated by methylation at the ε-amino group of lysine in histone proteins (Allfrey and Mirsky, 1964; Murray, 1964) (Figure 1A). However, it was not until the year 2000 when a landmark discovery from Thomas Jenuwein and colleagues resulted in the identification of the first histone KMT, human and mouse SUV39H1 (KMT1A). This enzyme was demonstrated to be conserved from yeast to human (Rea et al., 2000). After the discovery of KMT1A, dozens of KMTs were identified through homology searches with the enzymatic SET domain (Table 1) (Dillon et al., 2005). The SET domain is a 130 amino acid catalytic domain initially found to be conserved in Su(var)3-9, E(z) (enhancer of zeste) and trithorax (Jenuwein, 2006). SET domain-containing enzymes are currently the larger of the two classes of KMTs. The second class of KMTs is represented solely by KMT4 (also known as Dot1p in yeast and Dot1L in human), which does not have a SET domain (Okada et al., 2005; van Leeuwen et al., 2002). Even though the catalytic domains of these two enzyme classes are distinct, both use S-adenosyl-L-methionine (SAM) as the methyl group donor (Figure 1B) (Dillon et al., 2005; Nguyen and Zhang, 2011).
Figure 1. History, Mechanism, and Specificity of KMTs and KDMs.

(A) Timeline chronicling important milestones in KMT and KDM research.
(B) Schematic depicting generalized reaction mechanisms of KMTs and KDMs.
(C) Schematic depicting substrate specificity of KMTs and KDMs.
Table 1.
List of Human KMTs and KDMs with Current Nomenclature, Official Gene Symbol, Previous Aliases, and Gene IDs
| Nomenclature | Official Symbol/Alias (H) | Gene ID (H) | |
|---|---|---|---|
| KMTs | KMT1A | SUV39H1 | 6839 |
| KMT1B | SUV39H2 | 79723 | |
| KMT1C | G9a/EHMT2 | 10919 | |
| KMT1D | GLP/EHMT1 | 79813 | |
| KMT1E | SETDB1 | 9869 | |
| KMT1F | SETDB2 | 83852 | |
| KMT2A | MLL | 4297 | |
| KMT2B | MLL2 | 8085 | |
| KMT2C | MLL3 | 58508 | |
| KMT2D | MLL4 | 9757 | |
| KMT2E | MLL5 | 55904 | |
| KMT2F | SETD1A | 9739 | |
| KMT2G | SETD1B | 23067 | |
| KMT2H | ASH1L | 55870 | |
| KMT3A | SETD2 | 29072 | |
| KMT3B | NSD1 | 64324 | |
| KMT3C | SMYD2 | 56950 | |
| KMT3D | SMYD1 | 150572 | |
| KMT3E | SMYD3 | 64754 | |
| KMT3F | WHSC1L1/NSD3 | 54904 | |
| KMT3G | WHSC1/NSD2 | 7468 | |
| KMT4 | DOT1L | 84444 | |
| KMT5A | SET8/PR-SET7/SETD8 | 387893 | |
| KMT5B | SUV420H1 | 51111 | |
| KMT5C | SUV420H2 | 84787 | |
| KMT6 | EZH2 | 2146 | |
| KMT7 | SET7/SET9/SETD7 | 80854 | |
| KMT8A | PRDM2/RIZ1 | 7799 | |
| KMT8B | PRDM9 | 56979 | |
| KMT8C | PRDM6 | 93166 | |
| KMT8D | PRDM8 | 56978 | |
| KMT8E | PRDM3 | 2122 | |
| KMT8F | PRDM16 | 63976 | |
| KDMs | KDM1A | KDM1A/LSD1 | 23028 |
| KDM1B | KDM1B/LSD2 | 221656 | |
| KDM2A | KDM2A/FBXL11/JHDM1A | 22992 | |
| KDM2B | KDM2B/FBXL10/JHDM1B | 84678 | |
| KDM3A | KDM3A/JHDM2A | 55818 | |
| KDM3B | KDM3B/JHDM2B | 51780 | |
| KDM3C | JMJD1C | 221037 | |
| KDM4A | KDM4A/JMJD2A/JHDM3A | 9682 | |
| KDM4B | KDM4B/JMJD2B | 23030 | |
| KDM4C | KDM4C/JMJD2C/GASC1 | 23081 | |
| KDM4D | KDM4D/JMJD2D | 55693 | |
| KDM5A | KDM5A/JARID1A | 5927 | |
| KDM5B | KDM5B/JARID1B/PLU1 | 10765 | |
| KDM5C | KDM5C/JARID1C/SMCX | 8242 | |
| KDM5D | KDM5D/JARID1D | 8284 | |
| KDM6A | KDM6A/UTX | 7403 | |
| KDM6B | KDM6B/JMJD3 | 23135 | |
| KDM7A | JHDM1D/KIAA1718 | 80853 | |
| KDM7B | PHF8 | 23133 | |
| KDM7C | PHF2 | 5253 | |
| KDM8 | JMJD5 | 79831 |
KMTs observe a high degree of enzymatic specificity for the lysine within the substrate and for the degree of methylation (Figure 1C). For example, KMT1A/B trimethylates histone 3 lysine 9 (H3K9me3) from a monomethylated state (H3K9me1) (Peters et al., 2001, 2003), while the H3K9 methyltransferase KMT1C (also known as. G9a) methylates to a dimethylated state (H3K9me2), with a preference for mono- to dimethylation (Tachibana et al., 2002). In addition, purified KMT2A (also known as MLL1) catalyzes the methylation of H3K4 to H3K4me2 (Milne et al., 2002; Nakamura et al., 2002); however, when associated with its endogenous interacting proteins, KMT2A can trimethylate H3K4 (Dou et al., 2006; Schneider et al., 2005). Therefore, KMTs can be highly specific, but their interacting partners can alter their target lysine or degree of activity.
Following a three-decade debate regarding the presence of lysine demethylases, Yang Shi and colleagues identified the first histone KDM, LSD1/KDM1A (Figure 1A and Table 1), as part of the C-terminal binding protein 1 (CtBP1) corepressor complex (Shi et al., 2003, 2004). KDM1A was found to be associated with other similar corepressor complexes, suggesting that this protein was a candidate repressor (Hakimi et al., 2002). KDM1A contains a flavin adenine dinucleotide (FAD)-dependent amine oxidase domain that demethylates H3K4me2 and H3K4me1 and modulates gene expression (Figures 1B and 1C) (Shi et al., 2004). Subsequent to the discovery of KDM1A, an additional class of KDMs was discovered. This enzyme class utilizes the JmjC domain (Figures 1A and 1C) to catalyze demethylation through the oxidation of methyl groups. JmjC proteins rely on α-ketoglutarate, molecular oxygen, and Fe(II) as cofactors for demethylation (Figure 1B) (Shi and Whetstine, 2007). In some cases, the JmjN domain is observed with the JmjC domain and is essential for enzymatic activity (Chen et al., 2006). The first documented JmjC demethylases were the KDM2A/B proteins, JHDM1A and JHDM1B (Figure 1A) (Tsukada et al., 2006). These enzymes, as well as the H3K9 di-demethylases KDM3A-KDM3C (also known as JMJD1A-JMJD1C), were identified by assaying chromatography fractions for the ability to release formaldehyde from methylated histones (Tsukada et al., 2006; Yamane et al., 2006). These observations reiterated that formaldehyde is a common byproduct of demethylase reactions, which was initially observed with KDM1A (Shi et al., 2004). The KDM2 and KDM3 families were unable to demethylate trimethylated lysines; however, this lack of activity was remedied by the discovery of the first tri-demethylase family, KDM4A-KDM4D (also known as JMJD2A-JMJD2D) (Figure 1A) (Cloos et al., 2006; Fodor et al., 2006; Klose et al., 2006; Trojer et al., 2009; Whetstine et al., 2006). KDM4A-KDM4D remove H3K9me3/H3K9me2, H3K36me3/H3K36me2, and H1.4K26me3/H1.4K26me2, but are unable to remove H3K9me1 or H3K36me1, emphasizing the specificity for both the site and degree of methylation. Subsequent to these discoveries, a multitude of groups proceeded to identify additional amine oxidase- and JmjC-containing KDMs (Figure 1C and Table 1). However, no enzyme that is capable of demethylating H4K20me3 or H3K79me1/H3K79me2/H3K79me3 has been discovered. Recently, LOXL2 has been demonstrated to remove methyl groups by deaminating lysine (Herranz et al., 2012). Therefore, we hypothesize that LOXL2 or other LOX family members may catalyze demethylation of H4K20me3 or H3K79, which is an exciting area for future research (Black and Whetstine, 2012b).
Given the varying specificities of each enzyme, a major question facing both methyltransferase and demethylase biology is to elucidate the cellular and biological processes that these enzymes modulate and how they prevent or contribute to disease. In this review, we discuss the mechanisms of KMT and KDM targeting, specificity, and regulation. We also discuss the impact of these methylation events on chromatin states, transcription, and the cell cycle.
KMT and KDM Specificity
KMTs and KDMs have a high degree of specificity for particular lysine residues and the degree of methylation. Insights into this specificity have resulted from the evaluation of numerous KMT and KDM crystal structures (Table 2). The first crystal structures were resolved for two members of the SUV39 KMT family: decrease in DNA methylation 5 (DIM-5) from Neurospora crassa (Zhang et al., 2002) and CLR4 from S. pombe (Min et al., 2002). In contrast to other SAM-dependent methyltransferases, the structure of DIM-5 demonstrated that the SET domain within KMTs positions the SAM away from the peptide backbone and allows access to the lysine side chain through a narrow channel termed the “methylation pore.” The methylation pore is lined with conserved hydroxyl and carbonyl side chains that are important for transferring the methyl group to the lysine. By positioning the SAM away from the substrate peptide, the methylation pore allows for highly processive methyl addition, which is a characteristic of SET domain-containing KMTs. The first insight into the structural determinants of KMT specificity came from the KMT7 (Set7/Set9) crystal. Mutation of the side chains within the methylation pore convert KMT7 from an H3K4 monomethylase to an H3K4 trimethylase (Tyr245) or to an H3K9 dimethylase (Tyr305) (Dillon et al., 2005). The importance of this channel is evolutionarily conserved as mutation of Phe281 to Tyr281 in DIM-5 prevents it from performing trimethylation of H3K9. This became known as the F/Y switch, which is important for determining the specificity and extent of lysine methylation (Dillon et al., 2005). A number of additional methyltransferase crystals and cocrystals have since been resolved, including the Dot1p and KMT4 enzymes, which confirmed the presence of SAM as a cofactor for both classes of KMTs (Min et al., 2002, 2003).
Table 2.
List of Crystal Structures for Mammalian KMT and KDM Enzymes
| Enzyme | First Catalytic Domain | Crystal with Substrate | |
|---|---|---|---|
| KMTs | KMT1B | 2R3A | – |
| KMT1C | 2O8J | – | |
| KMT1D | 2IGQ | 3SW9 DNMT3AK44me0 and SFG | |
| 3SWC DNMT3AK44me2 and SAH | |||
| 3HNA H3K9me1 and SAH | |||
| 2RFI H3K9me2 and SAH | |||
| KMT2A | 2W5Y | 2W5Z histone peptide and SAH | |
| KMT2H | 3OPE | – | |
| KMT3A | 3H6L | – | |
| KMT3B | 3OOI | – | |
| KMT3C | 3QWV | 3S7D p53me and SAH | |
| 3S7F p53 and SAM | |||
| 3TG5 p53 and SAH | |||
| KMT3E | 3MEK | – | |
| KMT4 | 1NW3 | – | |
| KMT5A | – | 1ZKK H4 peptide (16–24) and SAH | |
| 3F9Y Y334F H4K20me1 and SAH | |||
| 3F9X Y334F H4K20me2 and SAH | |||
| KMT5B | 3S8P | – | |
| KMT5C | 3RQ4 | – | |
| KMT7 | 1MUF | 3OS5 Dnmt1K142me1 and SAH | |
| 2F69 TAF10 and SAH | |||
| 3M55 TAF10K189me1 and SAH | |||
| 3M56 TAF10K189me2 and SAH | |||
| 3M5A TAF10K189me3 and SAH | |||
| 1XQH p53 and SAH | |||
| KMT8A | 2JV0 | – | |
| KDMs | JMJD6 | 3K2O | – |
| KDM1A | 2H94 | 2V1D H3, COREST and FAD | |
| 2UXN H3, COREST and FDA | |||
| KDM1B | 3KV5 | – | |
| KDM2A | 2YU2 | – | |
| KDM4A | 2GP3 | 2OT7 H3K9me1 OGA | |
| 2OX0 H3K9me2 OGA | |||
| 2OQ6 H3K9me3 OGA | |||
| 2Q8C H3K9me3 AKG | |||
| 2PXJ H3K36me1 OGA | |||
| 2Q8D H3K36me2 SIN | |||
| 2OS2 H3K36me3 OGA | |||
| 2YPB H3K36me3 2HG | |||
| 2YBS H3K36me3 S2G | |||
| 3U4S H3 O8P | |||
| 2P5B H3k36me3 OGA | |||
| KDM4C | 2XML | – | |
| KDM4D | 3DXU | – | |
| KDM6A | 3AVS | 3AVR H3K27me3 and OGA | |
| KDM6B | 2XXZ | – | |
| KDM7B | 2WWU | 3KV4 H3 and OGA | |
| KDM8 | 3UYJ | – |
OGA, N-oxalylglycine; SIN, succinic acid; AKG, 2-oxoglutaric acid; SAH, S-adenosyl-L-homocysteine; SAM, S-adenosylmethionine; SFG, sine-fungin; FAD, flavin-adenine; FDA, dihydroflavine-adenine dinucleotide.
The understanding of KDM biology has also been greatly enhanced through structural analyses, which have identified key interacting partners and amino acid residues that regulate activity and specificity. For example, cocrystals of KDM1A and interacting partner Co-Rest demonstrated that the interaction with Co-Rest allowed the nucleosome to be opened up so that KDM1A could access the target (Yang et al., 2006). Structures of the JmjC catalytic domain revealed that it consists of a β-barrel structure that coordinates Fe(II) and α-ketoglutarate (Clissold and Ponting, 2001). In the case of KDM4A, amino acids within a specific β sheet in the β-barrel structure were predicted to impact the degree of demethylation. These residues differ between KDM4A and KDM4D. Upon mutating KDM4A residues Ser288 and Thr289 to match the corresponding amino acids in KDM4D (Ala291 and Ile292, respectively), the degree of demethylation by KDM4A matched that of KDM4D (Chen et al., 2006). Studies of the KDM4A crystal in complex with a monomethylated histone tail peptide also demonstrated that KDM4A does not recognize the monomethyl state because the methyl group is oriented away from the iron atom (Wilson, 2007). In addition, crystallographic studies demonstrated that both amino acids in KDM4A and the H3 tail were important for lysine specificity (Wilson, 2007 and references therein). For example, when isoleucine at position 71 of KDM4A is mutated to leucine, H3K9me3 demethylase activity is retained but H3K36me3 activity is greatly reduced in vitro (Hillringhaus et al., 2011). The histone tail also provides specificity through the amino acid sequence surrounding H3K9 or H3K36. A diglycine motif present either before (H3K36) or after (H3K9) the target lysine allows the peptide flexibility to enter the catalytic pocket for demethylation. Mutating the glycines, or substituting a proline residue to mimic the residues surrounding H3K27, significantly reduces demethylation activity (Wilson, 2007). Intriguingly, H3G34 in the diglycine motif adjacent to H3K36 has recently been demonstrated to have somatic mutations in some gliomas and glioblastomas (Khuong-Quang et al., 2012; Schwartzentruber et al., 2012; Wu et al., 2012). This suggests that cancers can acquire mutations in histone tails that may inactivate specific targets of KMTs and KDMs.
The RCSB protein data bank has many additional KMTs and KDMs crystallized alone or in complex with their substrates or inhibitors (Table 2; http://www.rcsb.org). Taken together, the abovementioned studies have highlighted the exquisite substrate specificity of KMTs and KDMs. Below, we discuss how lysine specificity and degree of methylation impact the determination of chromatin states, regulation of transcription, and progression of the cell cycle.
Methylation as Key Determinants in Chromatin States
The development of highly specific antibodies prompted the study of how specific lysines and the degree of methylation impact chromatin biology (Bannister and Kouzarides, 2004). These critical tools and the availability of large-scale, genome-wide data sets from individual laboratories as well as the ENCODE and modENCODE projects have enabled computational comparisons of the patterns of histone modifications and chromatin associated proteins. The distribution of methyl modifications, KMTs, and KDMs at both active and inactive open reading frames (ORFs) has now been elucidated (Figure 2). These findings have provided additional support to the notion that specific lysine residues, their degree of methylation, and their position within the genome have important roles and could have specific consequences. Genome-wide studies have led to the definition of “chromatin states,” which can be distinguished by histone methylation patterns. For example, different computational approaches have led to the determination of anywhere from four states in Arabidopsis (Roudier et al., 2011) to 5–9 states in Drosophila (Kharchenko et al., 2011; Riddle et al., 2011) to up to 51 states in human cells (Ernst and Kellis, 2010). The different numbers of defined states were derived principally from the subclassification of promoter states, transcribed states, active intergenic regions, repressed intergenic regions, and repetitive elements. These categories can also be subdivided. For example, in human cells 11 different promoter states can be defined by different patterns of H3K4me1/H3K4me2/H3K4me3 methylation, H3K79 methylation, H4K20me1, and promoter acetylation.
Figure 2. Distribution of Histone Methylation, KMTs, and KDMs from Genome-wide Profiling Studies.

The distribution of methyl modifications is shown relative to chromosomal location, as well as in relationship to active and inactive genes. Green represents euchromatic regions, while red coloring represents heterochromatic regions. Distributions of modifications are represented by bars, and gradients were derived from metagene analysis published as part of the genome-wide data sets from the work of numerous labs. We have included plots from metagene analyses that included both a transcription start site (TSS) and a transcription termination site (TTS). Enzymes marked with an * indicate the distribution depicted from metagene analyses that did not include both a TSS and TTS or from distributions published as heatmaps centered on the TSS. In the active gene model, the green E represents expressed exons, while the red E represents a nonexpressed exon. I denotes introns. Distributions uncovered in specific species are indicated by the following: Y, Saccharomyces cerevisiae; D, Drosophila. The lack of a denotation indicates conservation across multiple species. BV denotes bivalent genes, NBV denotes nonbivalent genes, HOX denotes patterns at Hox genes, and ZNF denotes zinc finger genes. This figure is the compiled work of numerous laboratories. We apologize for being unable to reference everyone’s contributions due to space limitations.
Initial studies on the biological roles of histone methylation have focused heavily on their roles and links to the regulation of gene expression. This subject has been well reviewed (Kouzarides, 2007; Li et al., 2007; Shilatifard, 2006; Smith and Shilatifard, 2010); therefore, we will highlight key paradigms in transcriptional regulation in the context of the genomic data in the subsequent sections. Below, we review the role of methylation in defining: enhancers, promoters, and gene bodies; exons, introns, and splicing; and heterochromatin and repetitive regions. We also discuss the function of large methylation domains.
Promoters and Gene Bodies
One of the major demarcations of a promoter or transcription start site (TSS) is the presence of H3K4me3 (Barski et al., 2007; Mikkelsen et al., 2007; Santos-Rosa et al., 2002). H3K4me3 is highly enriched at the TSS and can be found at both inactive and active promoters; thus it demarks not only a transcribed/active gene, but also genes that may become active. This is the case for genes that are silent in G0 but active in G1 (Smith et al., 2009). The enrichment of H3K4me3 near the TSS may play a functional role in initiation of transcription. For example, TAF3 binds H3K4me3 through its PHD finger and could help facilitate recruitment of RNA polymerase II (Vermeulen et al., 2007).
In addition to H3K4me3, the sequence immediately flanking the TSS is also enriched for H3K4me1/2 (Barski et al., 2007; Ernst and Kellis, 2010; Ernst et al., 2011; Filion et al., 2010; Gerstein et al., 2010; Kharchenko et al., 2011; Mikkelsen et al., 2007; Riddle et al., 2011; Roy et al., 2010) (Figure 2). While the exact role remains unclear at the TSS, H3K4me1 is important at distal gene regulatory elements termed enhancers. Computational analysis of H3K4me1 localization, as well as p300 binding and H3K27acetylation, accurately predict location of enhancer sequences (Hon et al., 2009b; Rada-Iglesias et al., 2011).
Similar to enhancers, analysis of methyl modifications at promoters allows them to be categorized based on gene activity. For example, active promoter classes are associated with gene bodies enriched in H4K20me1, H2BK5me1, and H3K36me3 (Hon et al., 2009a) while inactive promoters are often marked by H3K27me3 or H3K9me3. A special subset of inactive promoters, the bivalent genes, are marked by methylation for both H3K4me3 and H3K27me3 (Figure 2) (Bernstein et al., 2006). It is unclear if these distributions of modifications are enriched on the same nucleosome or different nucleosomes at the same promoter, or if they represent the effects of population averaging. Most recently, Voigt and colleagues demonstrated that bivalent nucleosomes exist with H3K4me3 and H3K27me3 present on different H3 tails within the same nucleosome (Voigt et al., 2012). These data suggest that intranucleosomal tail-to-tail crosstalk may be an important regulatory mechanism.
The histones in the coding regions of actively transcribed genes are decorated with multiple modified lysines, of which several are conserved in yeast, worm, fly, and mammalian systems (Figure 2). While the biological significance of these precisely distributed modifications remains unclear, the conservation of the modifications implies important regulatory or functional roles. For example, H3K79me1/H3K79me2/H3K79me3 has been observed to slightly overlap with H3K4me1/H3K4me2/H3K4me3 and continue throughout the body of the gene. Initial genome-wide studies revealed that all three H3K79 methylation marks are enriched closest to the TSS and gradually decrease throughout the gene body (Barski et al., 2007). However, recent studies with more specific antibodies have demonstrated that H3K79me2 and H3K79me3 may have different meta gene profiles (Figure 2) (Ernst et al., 2011; Liu et al., 2011; Roy et al., 2010). This is particularly true in yeast, where the H3K79me2 and H3K79me3 signals do not overlap and H3K79me2 peaks slightly further into the gene body (Schulze et al., 2009). Adjacent to the H3K79me1/H3K79me2/H3K79me3, there is a modest enrichment for methylation of H2BK5me1, H3K9me1, H3K27me1, and H4K20me1. These modifications cover the gene body, but decrease prior to the transcriptional stop site. In contrast, H3K36me3, which has the strongest correlation with level of expression, is the highest at the 3′ end of active genes and increases gradually as H3K79 methylation decreases. This general pattern is highly conserved across species (Barski et al., 2007; Ernst and Kellis, 2010; Ernst et al., 2011; Filion et al., 2010; Gerstein et al., 2010; Kharchenko et al., 2011; Mikkelsen et al., 2007; Riddle et al., 2011; Roy et al., 2010). Unlike H3K36me3, H3K27me3 in gene bodies correlates with transcriptional repression through the inhibition of transcriptional elongation (Chen et al., 2012). This inhibition can be relieved through demethylation by KDM6B (Figure 2). These results suggest that antagonism between H3K27me3 and H3K36me3 could be an important regulator of transcriptional elongation.
Interestingly, chromatin state analyses in Drosophila and C. elegans have demonstrated that active genes in heterochromatin, pericentromeric chromatin, and euchromatin have distinct methylation patterns. For instance, worm and fly genes that are expressed in heterochromatic regions have higher levels of H3K9me2/H3K9me3 in their coding regions, while euchromatic genes lack this modification (Kharchenko et al., 2011; Liu et al., 2011; Riddle et al., 2011). These studies also suggest that gene length can influence methylation patterns. In C. elegans, long genes are distinguished by H3K36me1 enrichment (Liu et al., 2011), while in yeast, long genes are enriched in H3K36me3 (Li et al., 2007). H3K36me3 recruits the RPD3S complex, which promotes deacetylation to prevent spurious transcription (Carrozza et al., 2005; Keogh et al., 2005; Li et al., 2007). H3K36me3 likely plays additional important roles as the C. elegans H3K36me3 KMT, Mes-4, is required for proper inheritance of gene expression in primordial germ cells (Furuhashi et al., 2010). These data suggest that H3K36me3 could not only suppress spurious transcription, but also act as an epigenetic memory for proper transcription.
Exons, Introns, and Splicing
In-depth analysis of genome-wide data sets has demonstrated that introns and exons are demarcated by specific methylation patterns. For example, exons that are expressed within an ORF are enriched in H3K4me3, H3K36me3, H2BK5me1, H4K20me1, and H3K79me1 (reviewed in Hnilicová and Stanĕk, 2011) (Figure 2). The marking of exons suggests a functional role for these modifications, especially when considering that they are not evenly distributed across gene bodies. For instance, H3K4me3 is enriched in the two exons closest to the promoter, while H3K79me1 is enriched in exons toward the 3′ end of genes (Dhami et al., 2010). The demarcation of expressed exons by H3K36me3 is conserved in worm and fly, but it remains unclear if expressed exons in these organisms are also preferentially marked by H2BK5me1, H4K20me1, and H3K79me1 (Kharchenko et al., 2011; Kolasinska-Zwierz et al., 2009; Liu et al., 2011; Riddle et al., 2011). The reason these modifications are enriched in expressed exons remains elusive. However, it is possible that they may serve to link splicing and/or elongation to transcription.
Introns are also enriched for H3K4me1 and H3K36me1 (Dhami et al., 2010; Spies et al., 2009). The identification of a specific methylation associated with introns suggests that this observation is not merely a function of increased nucleosome occupancy, but could reflect a functional significance in the marking of exons and introns. Furthermore, this suggests there may be methylation patterns specific for alternative exons not included in the transcript or even in introns adjacent to nonexpressed exons.
At the level of specific genes, data are beginning to support the notion that methyl marks can influence splicing. Mistelli and colleagues demonstrated that modulation of H3K36me3 and H3K4me3 regulated inclusion or exclusion of alternatively spliced exons at FGFR2 through the modulation of polypyrimidine binding protein recruitment (Luco et al., 2010). Sims and colleagues also demonstrated that H3K4me3 can influence splicing through recruitment of CHD1 (Sims et al., 2007). In addition, when H3K9me3 is enriched within the body of specific genes, there appears to be a role in regulating alternative splicing upon induction (Saint-André et al., 2011). These results suggest that the histone modifications themselves may be important for directly or indirectly recruiting splicing factors and thus determining transcript composition.
Heterochromatin and Repetitive Regions
The yeast, worm, fly, and mammalian genomes contain numerous repetitive genomic elements, including satellite sequences, rRNA clusters, pericentromeric chromatin, and the repetitive arms of the C. elegans chromosomes. Regardless of the organism, these regions are characterized by heavy enrichment of H3K9me2/H3K9me3 and H4K20me3 (Figure 2) (Barski et al., 2007; Ernst and Kellis, 2010; Ernst et al., 2011; Filion et al., 2010; Gerstein et al., 2010; Kharchenko et al., 2011; Liu et al., 2011; Mikkelsen et al., 2007; Riddle et al., 2011; Roy et al., 2010). It is unclear what biological function these regions serve and why they are decorated with these specific methylation signatures. However, it is apparent that proper regulation of these regions is crucial for maintaining genomic stability. For instance, in Drosophila, loss of H3K9 methylation results in increased DNA damage in heterochromatin and mitotic defects (Peng and Karpen, 2009). In mice, loss of KMT1A/KMT1B (Suv39H1/2) resulted in loss of H3K9me2/H3K9me3, disruption of heterochromatin, and an increase in telomere length compared to wild-type littermates (Benetti et al., 2007; García-Cao et al., 2004; Peters et al., 2001). A similar lengthening of telomeres is observed in MEFs deficient for KMT5B/KMT5C (Suv420H1/2), which correlates with the loss of H4K20me3 from telomeres. Furthermore, loss of either KMT1A/KMT1B or KMT5B/KMT5C results in increased telomere recombination frequency (Benetti et al., 2007).
Interestingly, heterochromatin integrity is also regulated by the H3K9me1 KMTs PRDM3 and PRDM16 (Pinheiro et al., 2012). PRDM3 and PRDM16 methylate H3K9me1 on free histones in the cytoplasm, allowing incorporation of premonomethylated histones into heterochromatin. Consistent with this, PRDM3 and PRDM16 are required for efficient H3K9me3 in mouse cells. Monomethylation by PRDM3 and PRDM16 is essential for heterochromatin formation and proper nuclear lamina formation. These results suggest that proper regulation of repetitive sequences is key to maintaining genome stability, which may be a reflection of altered heterochromatin structure and/or ncRNAs in these regions.
Genome-wide data analysis has helped define borders between repetitive heterochromatin and euchromatin in C. elegans and Drosophila. In Drosophila, these borders are defined by a sharp decrease in H3K9me2 (Roy et al., 2010) while in C. elegans the boundaries between chromosome arms and euchromatin are typified by a more gradual decrease in H3K9me2/H3K9me3 and an increase in H3K4me3 and H3K79 methylation (Liu et al., 2011). Interestingly, these defined borders appear to be consistent across tissues and over developmental time. The specific methylation states, combinations of modifications, and impact of misregulating methylation state at these regions strongly suggest that the repetitive regions play an important role within the cells that may be independent of development or differentiation.
Large Methylation Domains
In mammalian systems, H3K9me2, H3K9me3, and H3K27me3 can often be found in large broad domains (Hawkins et al., 2010; Pauler et al., 2009; Wen et al., 2009). H3K9me2 domains, or LOCKs (Large Organized Chromatin K modifications), are conserved between mouse and human and arise and spread upon differentiation (Wen et al., 2009). The majority of H3K9me2 in these domains is dependent on the H3K9me2 KMT G9a/KMT1C. These domains have been hypothesized to lock certain chromatin domains and prevent expression in differentiated cell types. In agreement with this hypothesis, many of the genes that are contained in these domains are developmentally regulated. Another interesting observation from these studies is that LOCKs coincide with regions that are enriched in Lamin B1. These data suggest that H3K9me2 could also be playing a role in docking LOCKs at the nuclear periphery. Consistent with this idea, lamin-associated domains (LADs) change during differentiation and have a high correlation with LOCKs across cell types (Guelen et al., 2008; Wen et al., 2009). This correlation appears to be conserved in C. elegans because binding of LEM-2 (C. elegans Lamin homolog) is highly correlated with H3K9me3, H3K9me2, repetitive regions, and inactive genes (Liu et al., 2011). These data suggest that H3K9me2 domains may be important determinants of higher-order chromosome structure and nuclear architecture.
Hawkins and colleagues observed that H3K9me3, like H3K9me2, can also exist in broad chromatin domains (Hawkins et al., 2010). The authors identified numerous H3K9me3 domains in human embryonic stem cells (ESCs) and in differentiated human fibroblasts. The H3K9me3 domains are not particularly enriched in developmental genes, but are enriched for gene families with high sequence homology (e.g., zinc fingers and olfactory receptors). It is unclear why these gene families are enriched; however, many have highly similar domains and nucleic acid sequences. Therefore, one possible explanation for the enriched H3K9me3 is that this modification state could mark repetitive gene regions to recruit proteins that prevent inappropriate recombination (Blahnik et al., 2011; García-Cao et al., 2004).
Collectively, these results suggest that one role for large domains of H3K9 methylation is to maintain association of inactive genomic regions with the nuclear lamina. Consistent with this model, loss of the H3K9me3 KMT in C. elegans results in detachment of an integrated complex array from the nuclear lamina and loss of lamin from regions normally associated with the nuclear lamina (Towbin et al., 2012). This is consistent with the role of PRDM3 and PRDM16 in maintaining the integrity of heterochromatin and the nuclear lamina as mentioned above. Thus, H3K9 methylation, through these large tracts, may serve as a key determinant in the three-dimensional nuclear architecture and chromatin arrangement within the nucleus. Consistent with this notion, disruption of the methylation or lamina function has dire consequences for cellular function, which was observed with the aging disorder Hutchinson-Gilford Progeria Syndrome (reviewed in Black and Whetstine, 2011).
H3K27me3 also exists in large domains originally described in mouse embryonic fibroblasts as broad local enrichments (BLOCs), which are enriched in repressed differentiation genes (Pauler et al., 2009). BLOCs are also enriched in repressed genes in human fibroblasts and ESCs (Hawkins et al., 2010). Interestingly, the H3K27me3 domains in human ESCs were more focal and also contained developmental genes and neural specific promoters, which the authors speculate expand during differentiation to silence genes that are no longer needed. In support of this model, the authors demonstrated that induced pluripotent stem cells reprogrammed from fibroblasts have an H3K27me3 pattern that is more similar to ESCs.
It remains unclear how these large tracks of histone methylation are established, maintained, and regulated. Even more perplexing is what the biological function of these domains may be. We would postulate that BLOCs and LOCKs may also serve to demarcate specific chromatin domains for replication or for progression through other phases of cell cycle. This large domain would help ensure that the structure is inherited and would also safeguard against losing the methylation at regions due to nucleosome turnover or replicative dilution (Deal et al., 2010). Therefore, it becomes critical to increase our understanding of which factors are important for modulating these domains and how KMTs and KDMs are recruited to establish and regulate these domains.
Targeting of KMTs and KDMs
The unique distribution of methylation marks described above demands that KMTs and KDMs arrive at the proper time and locale. Cells have evolved a multitude of mechanisms to recruit KMTs and KDMs, including domains that recognize chromatin states as well as direct interactions with transcription factors, DNA, RNA polymerase II (polII), and noncoding RNAs. Many of these strategies have recently been extensively reviewed (Smith and Shilatifard, 2010). We will focus on emerging paradigms and how these are supported by the large genomics data sets.
Pioneering studies in yeast demonstrated that enzymes involved in lysine methylation can directly interact with RNA polymerase or can be recruited to areas of polII enrichment through association with the polymerase associated factor (Paf) complex (reviewed in Smith and Shilatifard, 2010). For instance, set2 directly binds phosphorylated polII carboxy-terminal domain (CTD) in yeast and is required for H3K36 trimethylation in the coding region of active genes. Similarly, the evolutionarily conserved H3K4 KMT-containing COMPASS complex interacts with the RNA polymerase II-associated Paf complex (reviewed in Eissenberg and Shilatifard, 2010).
Another important targeting paradigm emerged with the discovery that the catalytically inactive JmjC domain-containing protein JARID2 recruits the PRC2 complex to polycomb targets in mammalian cells. In Drosophila, PRC2 is recruited to sequence-specific DNA elements, or polycomb responsive elements (PREs) (Müller and Kassis, 2006). Despite substantial efforts, the identification of PREs or sequence-specific targeting transcription factors in mammalian systems remained elusive; therefore, another targeting mechanism is likely present. Recent evidence suggests that JARID2, a component of PRC2 in ESCs, is involved in modulating PRC2 methylation activity and is required for PRC2 recruitment to target genes (Li et al., 2010; Pasini et al., 2010; Peng et al., 2009; Shen et al., 2009). Interestingly, Li et al. demonstrated that the ARID domain within JARID2 bound CG-rich sequences, raising the possibility that this serves as a recruitment mechanism for PRC2 (Li et al., 2010). CG-rich DNA and CpG islands likely function to recruit other KMTs and KDMs. For example, the zinc finger CxxC domain of KDM2A has increased affinity for nonmethylated CpG islands (Blackledge et al., 2010).
Demethylases and methyltransferases can also be recruited to target regions through interactions with noncoding RNAs. Silencing of the X chromosome in mammals requires the archetypal long noncoding (lncRNA), Xist (Payer and Lee, 2008). Xist derives at least part of its function from the ability to recruit PRC2 through interactions with KMT6 (EZH2) and Suz12, which results in H3K27me3 and transcriptional silencing (Zhao et al., 2008). Other lncRNAs, including both Air and Kcnq1ot1, are able to repress nearby target genes in cis through recruitment of KMT1C and KMT6, respectively (Nagano et al., 2008; Umlauf et al., 2004; Wagschal et al., 2008). LncRNAs can also function to repress genes in trans, which is best exemplified by recruitment of PRC2 and KDM1A by HOTAIR (Tsai et al., 2010). Recent work also indicates that lncRNAs are not exclusively involved in repression of target genes. The HOTTIP lncRNA is expressed from the HoxA locus and recruits MLL through interaction with WDR5, which results in H3K4me3 promoter methylation and activation of the HoxA cluster (Wang et al., 2011). In upcoming years, there will be a need to resolve the intricate interplay between KMTs, KDMs, and noncoding RNAs. These relationships will likely impact transcription, cell cycle, 3D genome organization, development, and disease.
KMTs and KDMs Modulate Cell Cycle
The recent influx of genomic data has provided an excellent snapshot of chromatin domains and states. However, these snapshots most likely do not adequately represent the truly dynamic nature of histone modifications in living cells. One major driving force for modification dynamics is progression through cell cycle. In order to divide, each cell must open, replicate, and then condense its chromatin to allow division, while preserving the inherited information. Thus, control of cell cycle-dependent gene expression and chromatin structure represent important regulatory targets of KMTs and KDMs.
Control of Cell Cycle through Regulation of Gene Expression
KMTs and KDMs can impact cell cycle by directly modulating expression of important cell-cycle genes. In yeast H3K79me2, but not H3K79me3, marks cell cycle-regulated genes for expression in G1/S (Schulze et al., 2009). H3K79me2 levels are low in G1-arrested cells but increase throughout S phase to a peak in G2/M. Yeast lacking the H3K79 methyltransferase Dot1p accumulate in G1, suggesting H3K79me2 is important for S phase entry. It is not clear if the cell-cycle defects are only due to misregulation of specific cell-cycle genes; however, it has been postulated that H3K79me2 could serve as an epigenetic memory for genes transcribed in the previous cell cycle (Schulze et al., 2009). Consistent with this idea, genes that are transcribed in G1 are marked with H3K4me3 during G0 (Smith et al., 2009). Taken together, these data suggest that modifications could serve as memory modules to mark passage through cell cycle.
In mammalian cells, KMT6 is a key regulator of genes that are critical for regulating cell-cycle progression (e.g., Cyclins A2, D1, and E1) (Bracken et al., 2003). Similarly, the KDM7B (PHF8) demethylase is regulated over cell cycle with peak levels in G1/S, which corresponds to the role of KDM7B in regulating E2F1 targets and G1/S transition (Figure 3) (Liu et al., 2010). These examples demonstrate how methyltransferases and demethylases can regulate genes important for cell-cycle progression.
Figure 3. Histone Methylation as Well as KMTs and KDMs Are Dynamically Regulated during the Cell Cycle.
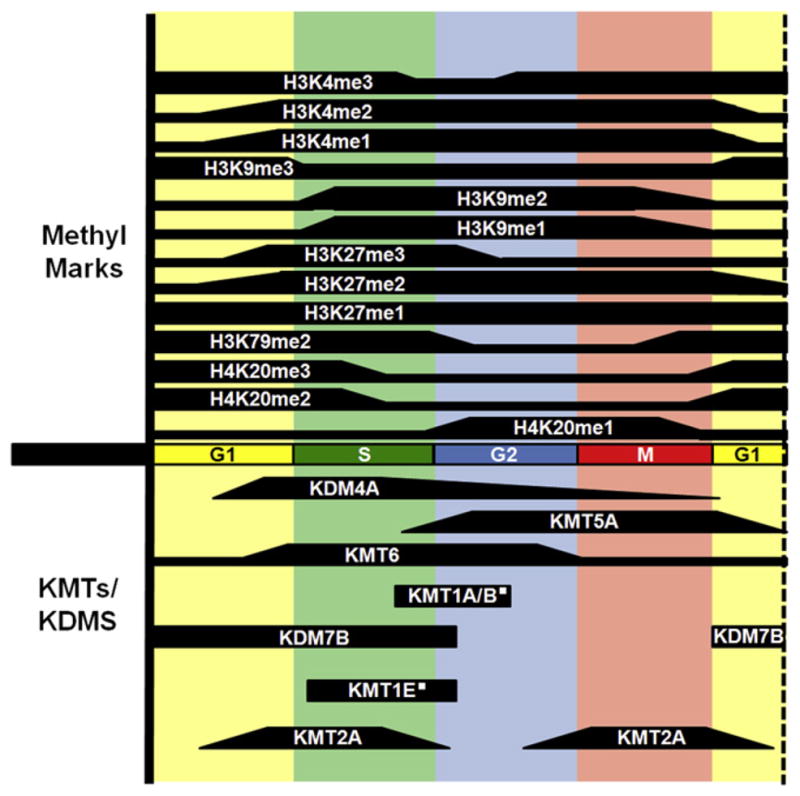
Solid shapes denote published information for the levels or functions of the indicated enzymes and modifications during that phase of cell cycle. Histone methylation positions and degree are indicated above the cell-cycle bar, and KMTs and KDMs are indicated below the cell-cycle bar. The white square denotes that the indicated enzyme is important during this time in cell cycle but that expression levels over cell cycle have not been reported. This figure is compiled from work generated by numerous groups. We apologize for being unable to reference everyone’s contributions due to space limitations.
Control of Cell Cycle through Chromatin Structure
KMTs and KDMs are not restricted to the direct regulation of cyclin genes, E2F targets, or other important cell-cycle genes. They could regulate chromatin structure and, in turn, cell-cycle progression. This could provide a “chromatin checkpoint,” an additional regulatory mechanism to ensure proper chromatin state during cell division. Recent genome-wide approaches have demonstrated a strong correlation between chromatin accessibility and replication timing, which agrees with a structural role for histone modifications and chromatin structure in replication (reviewed in Black and Whetstine, 2011). In mammals, transcriptionally active regions and a more open chromatin structure correlate with early replicating regions while H3K9me3- and H4K20me3-enriched regions replicate later in S phase. Consistent with these data, KMTs and KDMs regulate cell cycle by coordinating replication timing and progression of S phase by altering the chromatin structure (Figure 3). This has been demonstrated in S. pombe, where the Clr4 KMT regulates transcription of repetitive DNA surrounding the centromere. The transcription and repression of these repeats is crucial for proper centromere function, and loss of Clr4 results in altered S phase progression (Chen et al., 2008; Grewal and Jia, 2007; Li et al., 2011b). Furthermore, the elongating DNA polymerase epsilon subunit is required for small RNA generation and H3K9 methylation, suggesting that the region could be replicated prior to the formation of heterochromatin (Li et al., 2011b).
In mammalian systems, the H3K9 KMTs—KMT1E (SETDB1), KMT1A/KMT1B, and KMT1C—associate with PCNA and replication forks (reviewed in Groth et al., 2007), but their direct role in the regulation of replication still needs to be elucidated. It is likely, however, that methylation serves an important function, as overexpression of the H3K9me3 demethylase KDM4A resulted in faster progression through S phase that correlated with more open chromatin, an increase in replication forks, and altered replication timing at heterochromatin regions (Black et al., 2010). Similarly, loss of KDM4A in MDA-MB-231 breast cancer cells resulted in a G1/S arrest and decreased proliferation rates (Li et al., 2011a). Consistent with these results, H3K9me3 levels are reduced in S phase, while H3K9me1/2 levels increase (Figure 3) (O’Sullivan et al., 2010). These results suggest that H3K9me3 domains may serve as a conserved determinant of replication timing.
H4K20 methylation is also regulated during cell cycle. H4K20me1 accumulates during late S phase and into G2/M where it is converted into H4K20me3 by KMT5B/KMT5C (Figure 3) (reviewed in Beck et al., 2012). Consistent with these dynamic patterns observed during cell cycle, the expression of the H4K20me1 methyltransferase KMT5A (PR-SET7/SET8) is tightly regulated during cell cycle and is required for proper chromatin condensation and G2/M progression. Interestingly, KMT5A remains associated with chromatin through mitosis and is segregated to daughter cells, which suggests that this enzyme could be an important inherited factor that could mark replication origins (Rice et al., 2002). In agreement with this idea, loss of KMT5A results in decreased association of CDC6 and MCM proteins with chromatin and reduced origin firing (Tardat et al., 2010).
Similar to KMT5A, PRC2 appears to associate with chromatin through mitosis (Aoto et al., 2008). Consistent with this observation, H3K27me3 domains arise during the G1 phase in regions that were occupied by PRC2 prior to cell division (Figure 3) (Aoto et al., 2008). Establishment of these domains is crucial for the G1/S transition, and loss of the PRC2 component Suz12 results in decreased S phase progression. Similar to PRC2 and H3K27me3, the H3K4me3 methyltransferase MLL also persists on chromatin through mitosis (Blobel et al., 2009). Not all genes are marked by MLL; however, genes occupied by MLL tend to reactivate with faster kinetics in the following interphase. This faster reactivation is dependent on the continual association of MLL through mitosis. These data strongly support the notion that important gene regulatory information needs to persist through mitosis, and certain chromatin modifiers are required for this process.
We are just beginning to understand the regulation and distribution of histone methylation as well as KMTs and KDMs during cell cycle (Figure 3), and it is clear that expression of many KMTs and KDMs is exquisitely controlled during cell cycle. As we move forward, it will be important to remember that the majority of epigenomic and gene expression analyses are conducted on populations of asynchronous cells and thus potentially mask the important contributions that modifying enzymes or their associated modifications have on the cell cycle. The dynamic regulation of multiple KMTs/KDMs during S and G2/M suggests that activating or inactivating key KMTs or KDMs may result in substantial changes to replication, chromosome segregation, and chromosome stability (Figure 3). The direct roles of these enzymes in cell-cycle progression or regulation could be a major factor influencing cancer development and/or progression. It remains to be determined how many other chromatin modulators directly impact cell-cycle progression and genome integrity.
We also need to consider the notion that KMTs and KDMs are impacting specific proteins during cell cycle, which could be influencing cell division directly. This raises two questions: what nonhistone substrates are targets of KMTs and KDMs, and what are the functional outcomes of the regulation of nonhistone targets? The best-characterized nonhistone substrate, p53, is an important cell-cycle regulator, and its activity can be improved or repressed by methylation (reviewed in Huang and Berger, 2008). Nonhistone targets are not limited to p53 and in some cases may be the preferred substrates for the KMT. This may be the case for KMT7, which is capable of methylating numerous transcription factors and coactivators (Del Rizzo and Trievel, 2011). Thus, KMTs and KDMs may impact gene expression not only by altering chromatin environment, but through regulation of the transcriptional machinery. In support of this hypothesis, Sims and colleagues demonstrated that a single arginine in a noncanonical repeat in the RPB1 C-terminal domain is methylated (Sims et al., 2011). This raises the possibility that one of the eight lysine residues in the noncanonical polII CTD repeats may also be regulated by methylation. As in the case of automethylation of KMT1C, KMTs and KDMs themselves may represent important nonhistone targets (Chin et al., 2007; Sampath et al., 2007). Therefore, we suspect nonhistone targets will be important KMT and KDM substrates and may directly impact cell cycle.
Posttranscriptional Regulation of KMTs and KDMs
KMTs and KDMs play intricate roles in transcription, replication, and cell division; therefore, it is not surprising that cells have developed posttranscriptional methods of regulating their levels, activity, and localization. While our understanding of the regulation of these enzyme families is still in its nascent stage (Figure 4), it is clear they can be regulated by a diverse array of posttranscriptional control mechanisms. In this section of the review, we summarize some of the recent work describing the modification of KMTs and KDMs by ubiquitination, phosphorylation, miRNA, and metabolites.
Figure 4. KMTs and KDMs Are Dynamically Regulated through Multiple Mechanisms.
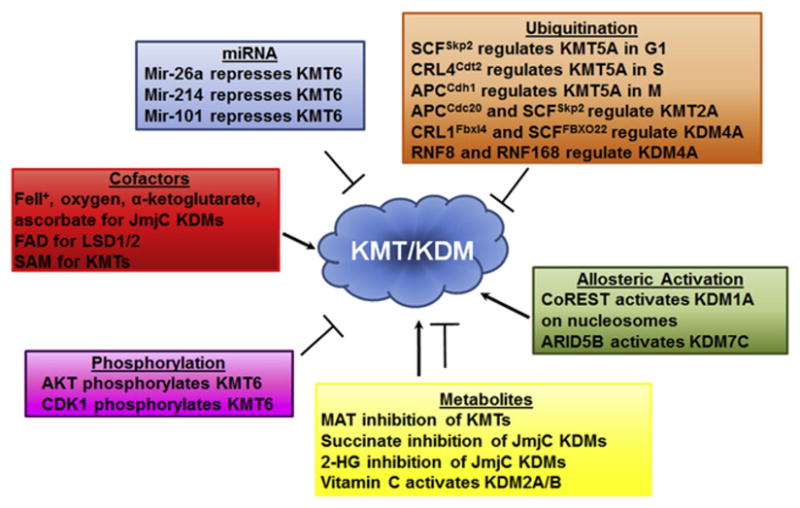
KMTs and KDMs are subject to inhibition or activation through the regulatory mechanisms that are indicated.
Regulation by Ubiquitination
Protein polyubiquitination often leads to degradation by the proteasome. Recently, this particular mode of regulation has emerged as an instrumental mechanism for regulating KMTs and KDMs; it is likely, however, that we have only just scratched the surface regarding how proteasome-mediated degradation impacts KMT and KDM biology.
The role of ubiquitin-mediated regulation of KMTs is best exemplified by the studies on KMT5A. KMT5A levels are low during S phase and peak in expression during G2/M (Figure 3). At least three ubiquitination complexes regulate KMT5A during different phases of cell cycle (Figure 4). In G1, KMT5A is degraded following ubiquitination by the SCFSkp2 complex. This degradation is necessary for proper entry into S phase (Yin et al., 2008). During S phase, KMT5A interacts with the DNA replication protein PCNA through a direct interaction via KMT5A’s PIP box. This interaction is required for ubiquitination and degradation of KMT5A by the CRL4Cdt2 complex (Beck et al., 2012 and references therein). Stabilization of KMT5A during S phase by mutations in the PIP box leads to premature chromatin compaction, a failure to enter mitosis, and an increase in apoptosis. Finally, during early anaphase KMT5A is evicted from mitotic chromosomes and ubiquitinated by APCCdh1. Together, these data highlight the intricate network of enzymes used to modulate levels of KMT5A during cell cycle. However, KMT5A is not the only KMT that is modulated by ubiquitination. KMT2A is also specifically ubiquitinated and degraded during cell cycle (Figures 3 and 4). KMT2A exhibits a biphasic expression pattern during cell cycle with enrichment in both G1 and G2/M (Liu et al., 2007). KMT2A is degraded upon S phase entry by the SCFSkp2 complex and by APCCdc20 in late S phase. This degradation is necessary to ensure proper G1/S and M phase progression.
Ubiquitination has also emerged as an important regulatory mechanism for histone demethylases. For example, KDM4A was shown to be targeted by two SCF complexes, one containing Cullin1 and the F-box protein FbxL4 and a second containing FBXO22: the former is necessary for proteasomal degradation and cell-cycle progression (Van Rechem et al., 2011) and the later impacts transcription (Tan et al., 2011). Finally, KDM4A is also regulated by ubiquitination in response to DNA damage by the RNF8 and RNF168 complexes (Mallette et al., 2012). These data suggest that the ubiquitin regulatory complexes could modulate the demethylase during specific nuclear events or at specific times during cell cycle.
Yeast Jhd2 has also been shown to be targeted for ubiquitin-dependent degradation by the E3 ligase Not4 (Mersman et al., 2009). This is conserved in mammals as the human Jhd2 homolog, KDM5C (JARID1C/SMCX), is also targeted for degradation (Mersman et al., 2009). Interestingly, the stability of Jhd2 is partially mediated by the catalytic domain. Mutation of the β strands of the catalytic domain elicits the proteasome-mediated protein degradation response (Huang et al., 2010). In a catalytically active enzyme, this ubiquitination site is unavailable, which suggests that this degradation mechanism might be triggered by reaction byproducts or natural inhibitors. Interestingly, the SCFFBXO22 complex interacts with the catalytic domain of KDM4A, and the stability of the yeast KDM4 homolog, Gis1, is dependent on the catalytic domain (Quan et al., 2011; Tan et al., 2011). These data suggest a conserved role for the JmjC domain in regulating KDMs, especially inactivated enzymes.
Regulation by Phosphorylation
To date, only a limited number of KMTs and KDMs have been demonstrated to be phosphorylated; however, these events are functionally important for KMT and KDM regulation. For example, AKT-dependent phosphorylation of Ser21 within KMT6 exemplifies this regulation because the affinity for H3 is abrogated, which leads to reduced H3K27me3 (Figure 4) (Cha et al., 2005). Expression of the S21D phosphomimetic form of KMT6 enhanced cell growth and promoted tumor development in xenograft models even though enzymatic activity was decreased. In contrast, expression of the nonphosphorylatable S21A KMT6 completely blocked tumor development. These data suggest that misregulation of KMT6-specific targets or that methylation of nonhistone targets could be important for the oncogenic phenotypes. This mechanism could be particularly important in tumors with altered AKT signaling (Cha et al., 2005).
Phosphorylation also impacts KDM regulation. For example, KDM7C (PHF2) is phosphorylated at Ser1056 following activation of PKA by Forskolin. The phosphorylated KDM7C forms a complex with ARID5B and demethylates Lys336 within ARID5B. The KDM7C-ARID5B complex is then recruited to target promoters where it facilitates gene activation via demethylation of H3K9me2 (Figure 4) (Baba et al., 2011). Given the need to tightly regulate KDMs and KMTs, which is emphasized by ubiquitin-mediated regulation and phosphorylation, it seems likely that there are more regulatory phosphorylation events for both KMTs and KDMs yet to be identified.
Regulation by miRNA
Recent work has highlighted the importance of miRNAs in regulating KMTs and KDMs. By regulating these enzymes, miRNAs can have a broad impact on genome organization and cellular function. For example, KMT6 is regulated by several miRNAs, which results in altered PRC2 function and impacts differentiation, development, and cancer evasion. The regulation of KMT6 by mir-214, mir-26a, and mir-let-7b is important in myogenesis, senescence, lymphoma, and cancer (Juan et al., 2009; Kottakis et al., 2011; Tzatsos et al., 2011). The miRNAs themselves can also be regulated by KMT6 and KDM2B, which suggests that both positive and negative feedback loops between KMTs, KDMs, and miRNAs are important in the regulation of development and disease prevention (Juan et al., 2009; Kottakis et al., 2011; Tzatsos et al., 2011). It remains unclear the extent to which other methyltransferases and demethylases are modulated by miRNAs. Analysis of Target Scan suggests that many KMTs and KDMs have multiple miRNA seed sequences within their UTRs, making this an important regulatory area for future work. However, at this early stage, these data provide an indication that chromatin modifying enzymes and miRNA networks will fine-tune one another.
Regulation by Metabolites and Cofactors
KMTs and KDMs rely on cofactors, many of which are key intermediates from metabolic processes (Figures 1B and 4). These cofactors include oxygen, FAD, α-ketoglutarate, and S-adenosyl methionine (SAM) (Shi and Whetstine, 2007; Shilatifard, 2006). Therefore, these enzymes are potentially sensors of metabolic states and may be directly regulated by metabolic processes. FAD and α-ketoglutarate are predominantly produced in the mitochondria, while SAM is synthesized cytoplasmically (Huang, 2002). These cofactors are believed to passively diffuse through the cell. However, it remains unclear what the nuclear availability of these cofactors are or if they can be produced within the nucleus by alternative, undefined pathways. It is possible that the three-dimensional nuclear architecture could create sequestered pockets of available or excluded cofactors to regulate the activity of KMTs and KDMs.
Catabolites, oncometabolites, and metabolic derivatives can inhibit KMTs and KDMs (Figure 4). S-adenosylhomocysteine (SAH) is a byproduct of methylation reactions using SAM and a key intermediate in the production of cysteine. SAH can act as a competitive inhibitor for methyltransferases (Figure 1B). The levels of SAM and SAH are primarily balanced by the SAH hydrolase that converts SAH into adenosine and homocysteine (Huang, 2002). Thus SAH hydrolase activity, production of cysteine, or the act of methylation could provide a natural feedback loop to regulate histone methylation. The JmjC-containing demethylases are also inhibited by catabolites. Succinate is a catabolite produced from α-ketoglutarate that can act as a competitive inhibitor for α-ketoglutarate and inhibit this class of KDMs (Figure 1B) (Smith et al., 2007). These data suggest that the TCA cycle and the availability of specific catabolites could control KDM activity. Consistent with this idea, mutations in succinate dehydrogenase (SDH) cause overproduction of succinate and familial paraganglioma (Koivunen et al., 2012; Smith et al., 2007). It is intriguing to speculate that this cancer could be the result of inappropriate inhibition of KDMs, which if true would argue for the identification of KDM agonists or conditions that could be used to stabilize the levels and activity of these enzymes for therapeutic purposes (Black and Whetstine, 2012a).
A recently discovered oncometabolite, 2-hydroxyglutarate (2-HG), inhibits Fe(II) and α-ketoglutarate using enzymes (e.g., KDM4 family) (Ward and Thompson, 2012; Xu et al., 2011). 2-HG is generated when the isocitrate dehydrogenase genes (IDH1 or IDH2) are mutated, which has been observed in numerous cancers. These data would suggest that loss of function of KDMs could be linked to tumorigenesis, especially if the inhibited demethylase is a tumor suppressor. Another important aspect of 2-HG is that this molecule is enantiomeric, which raises the possibility that the two enantiomers could have differential effects on KDM4 enzymes (Ward and Thompson, 2012). Recent data suggest this is a possibility, which needs to be evaluated further in the context of histone methylation (Koivunen et al., 2012).
There is the potential for cancer cells to regulate KMTs and KDMs through changes in the metabolic programs as well as through mutations in metabolic genes. Given their key regulatory role in access to the genome for transcription and replication, there is an important need to consider KMTs and KDMs in the overall molecular picture of all diseases. We predict that this will be an exciting area for future exploration.
The Future of Lysine Methylation: The Disease Connection
The last decade has led to the identification of numerous KMTs and KDMs and demonstrated that their activities are crucial for regulating gene expression, cell cycle, and differentiation. The chromatin community has determined that histone methylation patterns exist and can define genes, enhancer elements, and even large chromosomal domains. The field is just beginning to understand the regulation of these enzymes and the biological significance of histone methylation. It is becoming clear that numerous regulatory mechanisms impinge on KMTs and KDMs and that misregulation and genomic lesions of KMTs and KDMs may be critical determinants in cancer and neurological disease (Black and Whetstine, 2012a; Greer and Shi, 2012).
It is important to note that changes in lysine methylation state have not been shown to directly cause disease. However, amplification, deletion, misregulation, and mutation of numerous KMTs and KDMs have been linked to cancer, including KMT2A, KMT6, KMT1C, KDM4B, KDM4C, KDM1A, and KDM6A (Black and Whetstine, 2012a; Varier and Timmers, 2011). While the enzymes that regulate these modification states are clearly the target of genomic lesions, it remains to be proven whether the histones are the important disease targets. However, genetic evidence from medulloblastomas suggests that histone methylation could be an important target, as the H3K9 methyltransferase KMT1C and the H3K9 methylation binding proteins L3MBTL2 and L3MBTL3 are deleted in medulloblastomas (Northcott et al., 2009). Other medulloblastomas have amplification or increased expression for the KDM4B/KDM4C H3K9me3 demethylases. These results suggest that H3K9 hypomethylation or the inability to recognize H3K9 methylation are important in medulloblastoma progression and suggest that the histone mark may be a critical target. Furthermore, oncometabolites such as 2-HG generated from mutant IDH1/IDH2 also target H3K9 demethylases and likely other JmjC enzymes, implicating improper regulation of methylation in other cancers. Similarly, H3K27me3 may also be a critical contributor to oncogenesis as hypomethylation of H3K27me3 is observed in numerous cancers. Consistent with this idea, the H3K27 methyltransferase KMT6 and the H3K27 demethylases KDM6A/KDM6B are misregulated and mutated in numerous cancers (Greer and Shi, 2012; Varier and Timmers, 2011). Furthermore, somatic mutations in H3K27 have been observed in some gliomas, suggesting this lysine residue might be important in cancer (Khuong-Quang et al., 2012; Schwartzentruber et al., 2012; Wu et al., 2012). A major question facing KMT and KDM biology is how the enzymes and the methyl modifications are influencing cancer progression and clinical response. It is possible that multiple KMTs and KDMs cooperate to induce the phenotypic changes observed in cancers or that single enzymes are sufficient to promote or potentiate tumors as observed with KMT6 and MLL.
Regulation of lysine methylation is not just important in cancer, but has also emerged as a critical regulator of neurological function and disease. For example, H3K9 methylation imbalance has been linked to cognitive impairment and disorders. KMT1C and its homolog KMT1D (GLP [G9a-like-Protein]/EHMT1) are responsible for mono- and dimethylation of H3K9. Deletion of kmt1d (located in 9q34) or substitution within the protein have been linked to mental retardation, epileptic seizures, and autism (Greer and Shi, 2012). Furthermore, failure to properly interpret H3K9me3 may be linked to α-thalassemia/mental retardation, X-linked syndrome. Many of the mutations in the ATRX gene that lead to this syndrome are clustered in the cysteine-rich ADD domain, which is now known to bind H3K9me3 (Greer and Shi, 2012). In addition to mental retardation, autism spectrum disorders may also be impacted through misregulation of H3K9 methylation. Recent work has identified single-nucleotide polymorphisms in KDM4C and genomic deletions containing KDM4B in autistic patients (Kantojärvi et al., 2010; Pinto et al., 2010). However, the link between lysine methylation and cognitive disorders is not just through H3K9 methylation. Other demethylases, including KDM7B and KDM5C, are mutated in patients with cognitive or neurological impairments (Black and Whetstine, 2012a). Whether KMTs and KDMs regulate neuronal function through modulation of gene expression and/or regulation of chromatin architecture is an exciting area of future research.
Due to the recent links to disease, KMTs and KDMs have come under heavy investigation as potential drug targets for the treatment of both cancers and neurological disease. The strides made in understanding the structural and biochemical properties of the enzymes have enabled these advancements, but it will also be important to consider what happens to genomic architecture following treatment with inhibitors to KMTs and KDMs. The disruption of nuclear architecture and chromatin methylation is a hallmark of Hutchinson Gilford Progeria Syndrome and might be a potential consequence of altering activity of KMTs and KDMs (Black and Whetstine, 2011). These observations emphasize the need to balance methylation and genome organization so that disease is avoided, but also highlight the importance in understanding how to regulate enzymes and pathways so that when targeting them, other pathologies are not created.
The ability to translate the lessons learned from epigenomic profiling, structural studies, and regulatory mechanisms to treatment will be an important focus of the next wave of lysine methylation research. However, it will also be important to continue advancing our understanding of how KMTs and KDMs exert their function. The foundation established has poised the community to address several key questions: What are the functions of the chromatin states, and how do cells “read” them? Is chromatin compartmentalized in the nucleus in such a way that chromatin states are clustered together? Are KMTs and KDMs key regulators of this 3D organization through controlling LOCKs and BLOCs? Is histone methylation able to dictate inclusion and exclusion of exons and thus contribute to the array of different protein isoforms? What is the function of the intricate regulation of histone methylation and KMTs and KDMs during the cell cycle? Are KMTs and KDMs metabolic sensors, and can they coordinate responses to changes in metabolites? Are KMTs and KDMs more than just passenger enzymes in cancers? Finally, can the enzymes be successfully targeted to improve treatment of cancer and neurological diseases? While these questions highlight how far we have come in the last decade, they also emphasize that there is a tremendous amount of discovery left to understand the importance of lysine methylation, KMTs, and KDMs.
Acknowledgments
We would like to thank Nick Dyson and Brendon Ladd for helpful comments and suggestions on the manuscript. J.R.W. is supported by the Ellison Medical Foundation, CA059267 and R01GM097360. J.C.B. is a fellow of The Jane Coffin Childs Memorial Fund for Medical Research. This investigation has been aided by a grant from The Jane Coffin Childs Memorial Fund for Medical Research.
References
- Allfrey VG, Mirsky AE. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science. 1964;144:559. doi: 10.1126/science.144.3618.559. [DOI] [PubMed] [Google Scholar]
- Aoto T, Saitoh N, Sakamoto Y, Watanabe S, Nakao M. Polycomb group protein-associated chromatin is reproduced in post-mitotic G1 phase and is required for S phase progression. J Biol Chem. 2008;283:18905–18915. doi: 10.1074/jbc.M709322200. [DOI] [PubMed] [Google Scholar]
- Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y, Ni M, Meyer CA, Igarashi K, Kanno J, et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat Cell Biol. 2011;13:668–675. doi: 10.1038/ncb2228. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Histone methylation: recognizing the methyl mark. Methods Enzymol. 2004;376:269–288. doi: 10.1016/S0076-6879(03)76018-2. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Beck DB, Oda H, Shen SS, Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012;26:325–337. doi: 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetti R, Gonzalo S, Jaco I, Schotta G, Klatt P, Jenuwein T, Blasco MA. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol. 2007;178:925–936. doi: 10.1083/jcb.200703081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Black JC, Whetstine JR. Chromatin landscape: methylation beyond transcription. Epigenetics. 2011;6:9–15. doi: 10.4161/epi.6.1.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JC, Whetstine J. Tipping the Lysine Methylation Balance in Disease. Biopolymers. 2012a doi: 10.1002/bip.22136. in press Published online August 8 2012 http://dx.org/10.1002/bip.22136. [DOI] [PMC free article] [PubMed]
- Black JC, Whetstine JR. LOX out, histones: a new enzyme is nipping at your tails. Mol Cell. 2012b;46:243–244. doi: 10.1016/j.molcel.2012.04.023. [DOI] [PubMed] [Google Scholar]
- Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschöp K, Rinehart C, Quiton J, Walsh R, Smallwood A, et al. Conserved antagonism between JMJD2A/KDM4A and HP1γ during cell cycle progression. Mol Cell. 2010;40:736–748. doi: 10.1016/j.molcel.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ. CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell. 2010;38:179–190. doi: 10.1016/j.molcel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blahnik KR, Dou L, Echipare L, Iyengar S, O’Geen H, Sanchez E, Zhao Y, Marra MA, Hirst M, Costello JF, et al. Characterization of the contradictory chromatin signatures at the 3′ exons of zinc finger genes. PLoS ONE. 2011;6:e17121. doi: 10.1371/journal.pone.0017121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel GA, Kadauke S, Wang E, Lau AW, Zuber J, Chou MM, Vakoc CR. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell. 2009;36:970–983. doi: 10.1016/j.molcel.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- Chen S, Ma J, Wu F, Xiong LJ, Ma H, Xu W, Lv R, Li X, Villen J, Gygi SP, et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 2012;26:1364–1375. doi: 10.1101/gad.186056.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin HG, Estève PO, Pradhan M, Benner J, Patnaik D, Carey MF, Pradhan S. Automethylation of G9a and its implication in wider substrate specificity and HP1 binding. Nucleic Acids Res. 2007;35:7313–7323. doi: 10.1093/nar/gkm726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clissold PM, Ponting CP. JmjC: cupin metalloenzyme-like domains in jumonji, hairless and phospholipase A2beta. Trends Biochem Sci. 2001;26:7–9. doi: 10.1016/s0968-0004(00)01700-x. [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328:1161–1164. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rizzo PA, Trievel RC. Substrate and product specificities of SET domain methyltransferases. Epigenetics. 2011;6:1059–1067. doi: 10.4161/epi.6.9.16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami P, Saffrey P, Bruce AW, Dillon SC, Chiang K, Bonhoure N, Koch CM, Bye J, James K, Foad NS, et al. Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution. PLoS ONE. 2010;5:e12339. doi: 10.1371/journal.pone.0012339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339:240–249. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20:1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi H, Takasaki T, Rechtsteiner A, Li T, Kimura H, Checchi PM, Strome S, Kelly WG. Trans-generational epigenetic regulation of C. elegans primordial germ cells. Epigenetics Chromatin. 2010;3:15. doi: 10.1186/1756-8935-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Cao M, O’Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet. 2004;36:94–99. doi: 10.1038/ng1278. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci USA. 2002;99:7420–7425. doi: 10.1073/pnas.112008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Dave N, Millanes-Romero A, Morey L, Díaz VM, Lórenz-Fonfría V, Gutierrez-Gallego R, Jerónimo C, Di Croce L, García de Herreros A, Peiró S. Lysyl oxidase-like 2 deaminates lysine 4 in histone H3. Mol Cell. 2012;46:369–376. doi: 10.1016/j.molcel.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Hillringhaus L, Yue WW, Rose NR, Ng SS, Gileadi C, Loenarz C, Bello SH, Bray JE, Schofield CJ, Oppermann U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J Biol Chem. 2011;286:41616–41625. doi: 10.1074/jbc.M111.283689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnilicová J, Stanĕk D. Where splicing joins chromatin. Nucleus. 2011;2:182–188. doi: 10.4161/nucl.2.3.15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G, Wang W, Ren B. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput Biol. 2009a;5:e1000566. doi: 10.1371/journal.pcbi.1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Ren B. Predictive chromatin signatures in the mammalian genome. Hum Mol Genet. 2009b;18(R2):R195–R201. doi: 10.1093/hmg/ddp409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. Histone methyltransferases, diet nutrients and tumour suppressors. Nat Rev Cancer. 2002;2:469–476. doi: 10.1038/nrc819. [DOI] [PubMed] [Google Scholar]
- Huang J, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins. Curr Opin Genet Dev. 2008;18:152–158. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Huang F, Chandrasekharan MB, Chen YC, Bhaskara S, Hiebert SW, Sun ZW. The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J Biol Chem. 2010;285:24548–24561. doi: 10.1074/jbc.M110.117333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T. The epigenetic magic of histone lysine methylation. FEBS J. 2006;273:3121–3135. doi: 10.1111/j.1742-4658.2006.05343.x. [DOI] [PubMed] [Google Scholar]
- Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell. 2009;36:61–74. doi: 10.1016/j.molcel.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantojärvi K, Onkamo P, Vanhala R, Alen R, Hedman M, Sajantila A, Nieminenvon Wendt T, Järvelä I. Analysis of 9p24 and 11p12-13 regions in autism spectrum disorders: rs1340513 in the JMJD2C gene is associated with ASDs in Finnish sample. Psychiatr Genet. 2010;20:102–108. doi: 10.1097/YPG.0b013e32833a2080. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 2011;43:285–298. doi: 10.1016/j.molcel.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Li B, Gogol M, Carey M, Pattenden SG, Seidel C, Workman JL. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007;21:1422–1430. doi: 10.1101/gad.1539307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010;24:368–380. doi: 10.1101/gad.1886410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BX, Zhang MC, Luo CL, Yang P, Li H, Xu HM, Xu HF, Shen YW, Xue AM, Zhao ZQ. Effects of RNA interference-mediated gene silencing of JMJD2A on human breast cancer cell line MDA-MB-231 in vitro. J Exp Clin Cancer Res. 2011a;30:90. doi: 10.1186/1756-9966-30-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Martienssen R, Cande WZ. Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature. 2011b;475:244–248. doi: 10.1038/nature10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Cheng EH, Hsieh JJ. Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution: a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev. 2007;21:2385–2398. doi: 10.1101/gad.1574507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, Ohgi KA, Benner C, Garcia-Bassets I, Aggarwal AK, et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2011;21:227–236. doi: 10.1101/gr.115519.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersman DP, Du HN, Fingerman IM, South PF, Briggs SD. Polyubiquitination of the demethylase Jhd2 controls histone methylation and gene expression. Genes Dev. 2009;23:951–962. doi: 10.1101/gad.1769209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Min J, Zhang X, Cheng X, Grewal SI, Xu RM. Structure of the SET domain histone lysine methyltransferase Clr4. Nat Struct Biol. 2002;9:828–832. doi: 10.1038/nsb860. [DOI] [PubMed] [Google Scholar]
- Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Müller J, Kassis JA. Polycomb response elements and targeting of Polycomb group proteins in Drosophila. Curr Opin Genet Dev. 2006;16:476–484. doi: 10.1016/j.gde.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Murray K. The Occurrence of Epsilon-N-Methyl Lysine in Histones. Biochemistry. 1964;3:10–15. doi: 10.1021/bi00889a003. [DOI] [PubMed] [Google Scholar]
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–1720. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. 2010;464:306–310. doi: 10.1038/nature08788. [DOI] [PubMed] [Google Scholar]
- Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I, Sommer A, Aszodi A, Jenuwein T, Barlow DP. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res. 2009;19:221–233. doi: 10.1101/gr.080861.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–772. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- Peng JC, Karpen GH. Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS Genet. 2009;5:e1000435. doi: 10.1371/journal.pgen.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. 2009;139:1290–1302. doi: 10.1016/j.cell.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schöfer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M, et al. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell. 2012;150:948–960. doi: 10.1016/j.cell.2012.06.048. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan Z, Oliver SG, Zhang N. JmjN interacts with JmjC to ensure selective proteolysis of Gis1 by the proteasome. Microbiology. 2011;157:2694–2701. doi: 10.1099/mic.0.048199-0. [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Rice JC, Nishioka K, Sarma K, Steward R, Reinberg D, Allis CD. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 2002;16:2225–2230. doi: 10.1101/gad.1014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D, et al. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res. 2011;21:147–163. doi: 10.1101/gr.110098.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F, Ahmed I, Bérard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011;30:1928–1938. doi: 10.1038/emboj.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-André V, Batsché E, Rachez C, Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol. 2011;18:337–344. doi: 10.1038/nsmb.1995. [DOI] [PubMed] [Google Scholar]
- Sampath SC, Marazzi I, Yap KL, Sampath SC, Krutchinsky AN, Mecklenbräuker I, Viale A, Rudensky E, Zhou MM, Chait BT, Tarakhovsky A. Methylation of a histone mimic within the histone methyltransferase G9a regulates protein complex assembly. Mol Cell. 2007;27:596–608. doi: 10.1016/j.molcel.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are trimethylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell. 2005;19:849–856. doi: 10.1016/j.molcel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Schulze JM, Jackson J, Nakanishi S, Gardner JM, Hentrich T, Haug J, Johnston M, Jaspersen SL, Kobor MS, Shilatifard A. Linking cell cycle to histone modifications: SBF and H2B monoubiquitination machinery and cell-cycle regulation of H3K79 dimethylation. Mol Cell. 2009;35:626–641. doi: 10.1016/j.molcel.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, Yuan GC, Lee Y, Orkin SH. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell. 2009;139:1303–1314. doi: 10.1016/j.cell.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affar B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Rojas LA, Beck D, Bonasio R, Schüller R, Drury WJ, 3rd, Eick D, Reinberg D. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science. 2011;332:99–103. doi: 10.1126/science.1202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Shilatifard A. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol Cell. 2010;40:689–701. doi: 10.1016/j.molcel.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EH, Janknecht R, Maher LJ., 3rd Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16:3136–3148. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- Smith AE, Chronis C, Christodoulakis M, Orr SJ, Lea NC, Twine NA, Bhinge A, Mufti GJ, Thomas NS. Epigenetics of human T cells during the G0—>G1 transition. Genome Res. 2009;19:1325–1337. doi: 10.1101/gr.085530.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Nielsen CB, Padgett RA, Burge CB. Biased chromatin signatures around polyadenylation sites and exons. Mol Cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MK, Lim HJ, Harper JW. SCF(FBXO22) regulates histone H3 lysine 9 and 36 methylation levels by targeting histone demethylase KDM4A for ubiquitin-mediated proteasomal degradation. Mol Cell Biol. 2011;31:3687–3699. doi: 10.1128/MCB.05746-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 2010;12:1086–1093. doi: 10.1038/ncb2113. [DOI] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150:934–947. doi: 10.1016/j.cell.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-containing JMJD2/KDM4 Proteins. J Biol Chem. 2009;284:8395–8405. doi: 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Tzatsos A, Paskaleva P, Lymperi S, Contino G, Stoykova S, Chen Z, Wong KK, Bardeesy N. Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J Biol Chem. 2011;286:33061–33069. doi: 10.1074/jbc.M111.257667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet. 2004;36:1296–1300. doi: 10.1038/ng1467. [DOI] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- Van Rechem C, Black JC, Abbas T, Allen A, Rinehart CA, Yuan GC, Dutta A, Whetstine JR. The SKP1-Cul1-F-box and leucine-rich repeat protein 4 (SCF-FbxL4) ubiquitin ligase regulates lysine demethylase 4A (KDM4A)/Jumonji domain-containing 2A (JMJD2A) protein. J Biol Chem. 2011;286:30462–30470. doi: 10.1074/jbc.M111.273508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta. 2011;1815:75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Voigt P, Leroy G, Drury WJ, 3rd, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. Asymmetrically modified nucleosomes. Cell. 2012;151:181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagschal A, Sutherland HG, Woodfine K, Henckel A, Chebli K, Schulz R, Oakey RJ, Bickmore WA, Feil R. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol Cell Biol. 2008;28:1104–1113. doi: 10.1128/MCB.01111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Wilson JR. Targeting the JMJD2A histone lysine demethylase. Nat Struct Mol Biol. 2007;14:682–684. doi: 10.1038/nsmb0807-682. [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006;23:377–387. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Yin Y, Yu VC, Zhu G, Chang DC. SET8 plays a role in controlling G1/S transition by blocking lysine acetylation in histone through binding to H4 N-terminal tail. Cell Cycle. 2008;7:1423–1432. doi: 10.4161/cc.7.10.5867. [DOI] [PubMed] [Google Scholar]
- Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU, Cheng X. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002;111:117–127. doi: 10.1016/s0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]