

Abstract
Alzheimer disease (AD) is a devastating neurodegenerative disease affecting more than five million Americans. In this study, we have used updated genetic linkage data from chromosome 10 in combination with expression data from serial analysis of gene expression to choose a new set of thirteen candidate genes for genetic analysis in late onset Alzheimer disease (LOAD). Results in this study identify the KIAA1462 locus as a candidate locus for LOAD in APOE4 carriers. Two genes exist at this locus, KIAA1462, a gene associated with coronary artery disease, and “rokimi”, encoding an untranslated spliced RNA The genetic architecture at this locus suggests that the gene product important in this association is either “rokimi”, or a different isoform of KIAA1462 than the isoform that is important in cardiovascular disease. Expression data suggests that isoform f of KIAA1462 is a more attractive candidate for association with LOAD in APOE4 carriers than “rokimi” which had no detectable expression in brain.
Introduction
Alzheimer disease (AD) is a devastating neurodegenerative disease affecting more than five million Americans [1]–[3]. Symptoms begin late in life and progress over several years ultimately leaving the individual uncommunicative and bedridden. The cause of AD is complex, but the heritable component has been estimated to be as high as 80% [4]. Recent advances have been made in understanding the genetic component of AD, so that much of what we understand about the mechanism of AD we owe to genetics [5]. Investigations of early onset AD identified mutations in APP [6], PSEN1 [7], and PSEN2 [8], [9] as causative. APOE was identified in early candidate gene studies as associated with late onset AD (LOAD) [10], and remains the most replicated association in the 21 genome wide association studies (GWAS) that have been performed to date [5]. In fact, the association of APOE with LOAD still explains more of the population attributable risk than all current non-APOE GWAS findings together [5], underscoring the genetic complexity in this disease. Over 40 different loci have been highlighted in GWAS as LOAD susceptibility loci; only a handful of those have been confirmed by follow-up [5]. Thus, much of the heritability in LOAD remains unexplained.
The association between coronary vascular disease (CVD) and LOAD remains unclear. It has been theorized that atherosclerosis resulting in compromised blood flow to the brain and subsequent oxidative stress and inflammation could contribute to the risk for LOAD [11]. APOE has also been linked to CVD, although this association is controversial [12], [13]. It appears, however, that the contribution of APOE to AD pathology is not through enhanced CVD, but through more direct effects on amyloid beta processing and neurotoxicity [14].
In this study, we have used updated genetic linkage data from chromosome 10 in combination with expression data from serial analysis of gene expression to choose a new set of thirteen candidate genes for genetic analysis in LOAD. Chromosome 10 has long been of interest in LOAD genetics based on linkage studies [15]–[22]. Results in this study identify the KIAA1462 locus as a candidate locus for LOAD in APOE4 carriers. The likelihood that this gene is a candidate gene for LOAD in APOE carriers is discussed.
Materials and Methods
Study Populations
All individuals included in this study were Caucasian late-onset AD (LOAD) participants (minimum age at onset (AAO) = 60 years) and related unaffected relatives. LOAD was diagnosed according to the NINCDSADRDA criteria [23]. All unaffected individuals had results within the normal range in the Mini-Mental State Exam (MMSE) or Modified Mini-Mental State Exam (3 MS). Families were chosen by the presence of two or more affected individuals. Samples from all affected and at least one unaffected first degree relative were collected, resulting in an increased number of affected over unaffected individuals in this study. The overall data set of 441 families contains 1001 affected and 352 unaffected individuals (see Table 1 for details). The number of affected women is also more than the number of affected men, reflecting the increased incidence of LOAD in females [24] and the general tendency for women to participate in research at a higher rate then men. Samples were ascertained by the following centers: the National Cell Repository for Alzheimer’s Disease at Indiana University (NCRAD); the Collaborative Alzheimer Project (CAP), including the University of Miami, Vanderbilt University, the University of California at Los Angeles; and the National Institute of Mental Health repository (NIMH). Written consent was obtained from all participants in agreement with protocols approved by the institutional review board for the CAP participants at the University of Miami, Vanderbilt University, and the University of California at Los Angeles. Following informed consent, blood samples were collected from each individual and genomic DNA was extracted using the Puregene system (Gentra Systems, Minneapolis, MN). Extracted DNA was obtained from the NCRAD and NIMH repositories.
Table 1. AD family data details.
| ALL* | NCRAD* | NIMH* | CAP* | |
| Number of Families | 441 | 85 | 289 | 67 |
| Number of Individuals | 1354 | 250 | 877 | 227 |
| Number of Affected Ind. | 1001 | 189 | 644 | 168 |
| Sex | ||||
| Female | 708 | 133 | 477 | 98 |
| Male | 293 | 56 | 167 | 70 |
| Number of Unaffected Ind. | 352 | 61 | 232 | 59 |
| Sex | ||||
| Female | 208 | 32 | 144 | 32 |
| Male | 144 | 29 | 88 | 27 |
| Number of Unknown Ind. | 1 | 0 | 1 | |
| Sex | ||||
| Female | 0 | 0 | 0 | 0 |
| Male | 1 | 0 | 1 | 0 |
| Number of Families with: | ||||
| 0 affected | 1 | 0 | 1 | 0 |
| 1 affected | 7 | 0 | 7 | 0 |
| 2 affected | 336 | 71 | 219 | 46 |
| 3 affected | 73 | 11 | 50 | 12 |
| 4 affected | 20 | 2 | 11 | 7 |
| 5 affected | 2 | 0 | 1 | 1 |
| 6 affected | 1 | 1 | 0 | 0 |
| 7 affected | 1 | 0 | 0 | 1 |
| 8 affected | 0 | 0 | 0 | 0 |
| Number of Families | 441 | 85 | 289 | 67 |
ALL column lists the combined totals for all ascertainment sites. Samples were ascertained by the following centers: the NCRAD repository at Indiana University (NCRAD); the Collaborative Alzheimer Project (CAP), including the University of Miami and Vanderbilt University and University of California at Los Angeles; and the National Institute of Mental Health repository (NIMH). The
Gene Selection
The Serial Analysis of Gene Expression (SAGE) method was used to compare the gene expression levels in the brain tissue from LOAD patients and controls as described elsewhere [25], [26]. Candidate genes for analysis in this study were chosen by the convergence of their differential expression data in LOAD brain compared to control brain, and their position under the LOAD linkage peaks as shown in previous linkage studies.
SNP Selection and Genotyping
Tagging SNPs were selected using LDSelect with an r 2 threshold of 0.8 in the CEU subject data of HapMap Release 21 of phase II of the National Center for Biotechnology Information build 35 assembly. CEU subjects were Utah residents of northern and western European ancestry from the Centre d’Etude du Polymorphisme Humain. The minor allele frequency threshold was 0.05, as exceeded by any Caucasian study population of dbSNP, including CEU.
A total of 384 SNPs on chr10 were genotyped with the use of the midthroughput Sequenom genotyping platform, based on a single-base primer extension reaction coupled with mass spectrometry. The assays were designed using Sequenom SpectroDESIGNER software. Genomic DNA (5 ng) was amplified following the manufacturer recommendations (Sequenom). Single primer extension over the SNP was carried out in a final concentration of 1.25 µM of the extension primer. The extension step followed the manufacturer procedure. The reaction was then desalted by addition of 6 mg of resin followed by 15 min mixing and centrifugation (3000 rpm) to settle the contents of the plate. The extension product was then spotted onto a 384 well spectroCHIP before being flown in the MALDI-TOF mass spectrometer. Data was collected, real time, using SpectroTYPER Analyzer, SpectraAQUIRE and SpectroCALLER (Sequenom). DNA samples from cases and controls were randomly sorted, and duplicate samples were implemented across plates for genotyping quality control.
Statistical Methods
Association analysis in family data set
The allelic association analyses were conducted using the association in the presence of linkage (APL) program [27] and the pedigree disequilibrium test (PDT) [28]. These methods provide valid and robust tests for allelic association in trios and extended families. The Genotype-PDT (GenoPDT) tested genotypic association to the risk of LOAD [29]. Genotype efficiency, Hardy-Weinberg Equilibrium and linkage disequilibrium were checked using Haploview [30]. Linkage analysis was conducted on families using two-point heterogeneity LOD scores (HLOD) calculated using FASTLINK and HOMOG [31]. Both recessive and dominant models with disease allele frequencies of 0.01 and 0.001, respectively, were analyzed. This approach is robust for detecting linkage signals when the underlying model is unknown or complex [32].
Reverse Transcriptase PCR and Real Time Quantitative PCR
Frozen superior frontal cortex samples were obtained from the Harvard Brain Tissue Resource Center from four control and four LOAD brains. RNA was isolated from these samples using TRIzol reagent (Invitrogen) and converted to first strand cDNA using the SuperScript® III First-Strand Synthesis System for RT-PCR (Invitrogen). Oligo(dT), random hexamers, and a gene specific primer were all used separately to create first strand synthesis for the rokimi putative transcript, while oligo(dT) was used as a first strand synthesis primer for the KIAA1462 mRNA quantitation. TaqMan probes specific for the junction between exons 2 and 3 (Hs1584907_m1), between exons 3 and 4 (Hs1584907_m1), and the 3′UTR (6835–6843 of NM_020848) were used for quantitative real time PCR. Relative levels of the KIAA1462 transcript were measured with quantitative real time PCR on an ABI 7900HT Fast Real Time PCR System using Taqman Gene Expression Assays. Absolute quantitation was performed using standard curves based on cDNAs cloned into the pCR4-TOPO vector using the TOPO-TA cloning kit (Invitrogen). Endogenous control assays to housekeeping gene GAPDH or ACTB were run in triplicate. The presence or absence of the rokimi transcript in brain was determined by the presence/absence of an RT-PCR product of the correct size. Two sets of primers were used, primer set 1 (5′-ctcctgcccttctcccatc-3′ roki1.for and 5′-ggcacgatcttggctcat-3′ roki1.rev) primers set 2 (5′-CACTCCTAGGCGGGGCTCCT-3′ roki2.for and 5′-TGCGTACCTCACCGAGGTTTC-3′ roki2.rev).
Results
Genomic Convergence Identified Genes and Genotyped SNPs
To identify new candidate genes for Alzheimer disease, linkage data from chromosome 10 was combined with expression data from SAGE analysis. Genes that showed significant differential expression in AD brain in at least two SAGE comparisons [33] and were under previously identified linkage peaks [16], [18], [34]–[36] were chosen for further study. Thirteen genes were selected for detailed genetic analysis, and all of these genes have evidence for expression in brain in the Unigene database. Tagging SNPs were chosen from both intronic and exonic regions of the genes, except for PRKG1 where size necessitated limiting SNPs to the exonic regions. The gene description, size, and number of genotyped SNPs are summarized in Table 2.
Table 2. Candidate Genes on Chromoesome 10 Identified by Genomic Convergence.
| Gene Name | UniGene | Expression Ratio* | start position | Size (kb) | # of SNPs genotyped |
| DNMBP | Hs.500771 | −14.4 | 101625324 | 134342 | 12 |
| TMEM10 | Hs.12449 | −6.3 | 98092968 | 16081 | 13 |
| SORBS1 | Hs.38621 | 1.5 | 97061520 | 249605 | 102 |
| HELLS | Hs.546260 | 1.7 | 96295563 | 56282 | 6 |
| LOC387700 | Hs.530338 | 1.6 | 91180035 | 105256 | 32 |
| IFIT3 | Hs.549041 | 1.7 | 91082265 | 8023 | 3 |
| FAS | Hs.244139 | 1.6 | 90740267 | 25254 | 30 |
| C10orf58 | Hs.500333 | 1.7 | 82167953 | 6188 | 7 |
| PRKG1 | Hs.2689 | 1.7 | 52504298 | 1220982 | 92 |
| C10orf72 | Hs.522928 | 2 | 49893191 | 3018 | 21 |
| CUL2 | Hs.82919 | 2.2 | 35338813 | 80487 | 5 |
| EPC1 | Hs.167805 | 1.7 | 32610792 | 65360 | 14 |
| KIAA1462 | Hs.533953 | 3.1 | 30344113 | 13294 | 47 |
[33]. Expression ratio is from SAGE data
Association Analysis and Real Time Quantitative PCR of KIAA1462
The allelic association of 384 genotyped SNPs in thirteen genes was analyzed in the overall data set of 441 families containing 1001 affected and 352 unaffected individuals (Table 1). The Genotype-PDT (GenoPDT) results are presented in Figure 1 and the linkage analysis results using two-point heterogeneity LOD scores (HLOD) are presented in Figure 2. Fourteen SNPs in six genes (KIAA1462; MIM# 6, CUL2; MIM#603135, PRKG1; MIM# 176894, FAS; MIM#134637, SLC16A12; MIM#611910, and SORBS1; MIM#605264) showed association with AD at nominal p<0.05. Two of those SNPS, rs2488024 in KIAA1462 and rs10823056 in PRKG1, also had calculated two point LOD scores greater than 1.0.
Figure 1. Allelic association test of candidate gene SNPs in the overall data set.

KIAA1462 and SORBS1 were associated with LOAD at a p value<.01.
Figure 2. Two point heterogeneity LOD scores of candidate gene SNPs in the overall data set.

The Association of KIAA1462 Locus SNPs and LOAD is Restricted to APOE Carriers
GWAS studies have identified KIAA1462 as a novel locus for coronary artery disease [37], [38]. The association between non-stroke cardiovascular disease (CVD) and Alzheimer’s disease has been debated, but a recent study reports that CVD increases risk of AD only in carriers of the APOE4 allele (Hazard Ratio 2.39, 95% confidence interval) [39]. From these studies, a model is suggested in which variation in KIAA1462 increases the risk of coronary artery disease, which in turn increases the risk of LOAD only in APOE4 carriers. To test this model, the genetic analysis of the association between SNPs in KIAA1462 and LOAD was performed again after stratifying the families by APOE status. The APOE status of a family was defined as positive if more than 50% of the family members had the APOE4 allele. By this definition, 279 APOE positive families and 162 APOE negative families were analyzed. When the analysis is stratified by APOE status, the association between KIAA1462 and LOAD is significant almost exclusively in the APOE positive group (Table 3).
Table 3. Linkage of SNPs on KIAA1462/rokimi region of chr10 and LOAD unstratified (all families) and stratified by APOE status.
| All families | APOE positive families | APOE negative families | ||||||||||
| HETLOD | HETLOD | PDT | MLOD | MLOD | MLOD | MLOD | ||||||
| RS# | dom | REC 2pt | SUM | FBAT | dom | rec | PDTsum | FBAT | dom | rec | PDTsum | FBAT |
| RS2256283 | 0.33 | 0.10 | 1.000 | 0.997 | 0.24 | 0.05 | 0.739 | 0.879 | 0.10 | 0.05 | 0.587 | 0.850 |
| RS3006544 | 0.00 | 0.00 | 0.665 | 0.968 | 0.00 | 0.00 | 0.962 | 0.351 | 0.00 | 0.02 | 0.549 | 0.255 |
| RS12764479 | 0.00 | 0.00 | 0.677 | 0.878 | 0.00 | 0.00 | 0.232 | 0.359 | 0.08 | 0.14 | 0.091 | 0.073 |
| RS7917431 | 0.24 | 0.25 | 0.483 | 0.528 | 0.02 | 0.03 | 0.652 | 0.453 | 0.41 | 0.45 | 0.587 | 0.891 |
| RS1774240 | 0.90 | 0.76 | 0.213 | 0.207 | 0.43 | 0.28 | 0.214 | 0.341 | 0.68 | 0.57 | 0.685 | 0.390 |
| RS1774243 | 0.64 | 0.84 | 0.212 | 0.445 | 0.76 | 1.02 | 0.475 | 0.854 | 0.02 | 0.04 | 0.283 | 0.266 |
| RS1418278 | 1.73 | 1.66 | 0.515 | 0.360 | 1.11 | 0.89 | 0.291 | 0.160 | 0.69 | 0.79 | 0.775 | 0.870 |
| RS1063205 | 0.70 | 0.66 | 0.294 | 0.193 | 0.28 | 0.34 | 0.351 | 0.261 | 0.48 | 0.32 | 0.602 | 0.394 |
| RS3739998 | 0.86 | 0.70 | 0.308 | 0.265 | 0.69 | 0.56 | 0.390 | 0.494 | 0.20 | 0.15 | 0.580 | 0.295 |
| RS2185724 | 0.11 | 0.10 | 0.572 | 0.416 | 0.04 | 0.08 | 0.513 | 0.464 | 0.07 | 0.02 | 0.904 | 0.733 |
| RS9337951 | 1.00 | 0.69 | 0.362 | 0.375 | 0.64 | 0.36 | 0.317 | 0.390 | 0.36 | 0.38 | 0.874 | 0.543 |
| RS2487928 | 1.24 | 0.95 | 0.077 | 0.072 | 1.02 | 0.77 | 0.151 | 0.152 | 0.26 | 0.21 | 0.299 | 0.154 |
| RS1342150 | 0.48 | 0.29 | 0.077 | 0.046 | 0.17 | 0.06 | 0.236 | 0.317 | 0.37 | 0.30 | 0.186 | 0.032 |
| RS2478833 | 0.90 | 0.68 | 0.020 | 0.012 | 0.26 | 0.10 | 0.100 | 0.021 | 0.74 | 1.07 | 0.094 | 0.156 |
| RS2505087 | 1.42 | 1.30 | 0.063 | 0.027 | 1.39 | 1.01 | 0.155 | 0.037 | 0.15 | 0.43 | 0.209 | 0.180 |
| RS1122730 | 1.96 | 1.07 | 0.235 | 0.235 | 1.67 | 0.76 | 0.581 | 0.430 | 0.37 | 0.32 | 0.203 | 0.257 |
| RS2255515 | 0.18 | 0.06 | 0.109 | 0.119 | 0.19 | 0.02 | 0.283 | 0.224 | 0.02 | 0.05 | 0.128 | 0.248 |
| RS6481654 | 0.18 | 0.16 | 0.050 | 0.235 | 0.38 | 0.32 | 0.113 | 0.231 | 0.00 | 0.00 | 0.245 | 0.446 |
| RS4749527 | 0.37 | 0.30 | 0.286 | 0.395 | 1.31 | 1.13 | 0.298 | 0.292 | 0.00 | 0.00 | 0.752 | 0.783 |
| RS2505127 | 0.30 | 0.15 | 0.021 | 0.056 | 0.15 | 0.00 | 0.064 | 0.087 | 0.24 | 0.44 | 0.131 | 0.281 |
| RS3847404 | 0.25 | 0.13 | 0.088 | 0.064 | 0.57 | 0.35 | 0.201 | 0.095 | 0.00 | 0.00 | 0.234 | 0.237 |
| RS2505126 | 0.94 | 0.60 | 0.005 | 0.013 | 1.07 | 0.68 | 0.017 | 0.015 | 0.03 | 0.01 | 0.129 | 0.173 |
| RS12220246 | 0.00 | 2.91 | 0.445 | 0.032 | 0.09 | 2.02 | 0.589 | 0.033 | 0.00 | 0.93 | 0.582 | 0.030 |
| RS2488024 | 1.00 | 0.65 | 0.000 | 0.002 | 0.77 | 0.48 | 0.004 | 0.009 | 0.23 | 0.18 | 0.030 | 0.041 |
| RS11007904 | 0.72 | 0.56 | 0.096 | 0.167 | 1.27 | 1.16 | 0.098 | 0.087 | 0.00 | 0.00 | 0.630 | 0.750 |
| RS1887317 | 1.15 | 1.18 | 0.100 | 0.265 | 0.58 | 0.51 | 0.065 | 0.146 | 0.62 | 0.73 | 0.889 | 0.921 |
| RS10826765 | 0.22 | 0.14 | 0.029 | 0.056 | 0.46 | 0.36 | 0.146 | 0.100 | 0.00 | 0.00 | 0.079 | 0.212 |
The Association of the KIAA1462 Locus with LOAD in a Publicly Available Genome-wide Dataset
The region of interest was examined for association with LOAD using the dataset assembled by the ADGC (stage 1, 8,309 individuals with LOAD (cases) and 7,366 cognitively normal elders (CNEs) as controls) from eight cohorts and a ninth newly assembled cohort from the 29 National Institute on Aging (NIA)-funded Alzheimer Disease Centers (ADCs) [40]. No significant association with LOAD was seen at this locus in this dataset. Although these data might suggest that KIAA1462 is not associated with LOAD, it is also possible that an association would only be detected after stratification by APOE status or presence of cardiovascular disease in this dataset. Thus a large scale GWAS study cannot exclude that this gene is still important in a subset of LOAD.
Analysis of the Genomic Structure at the KIAA1462 Locus Identifies another Putative Gene
While three SNPS in the KIAA1462 locus showed nominal association to AD in a previous study, none of these associations survived multiple testing correction [41]. More recently, other SNPs in the locus were discovered in two GWAS to be strongly associated with coronary artery disease (CAD) (p = 8.78×10−6) [42], [37]. When Haploview [30] was used to analyze linkage disequilibrium, the groups of SNPs associated with CAD and LOAD appear to cluster in two different LD blocks, suggesting that they are inherited separately (Figure 3). In addition, when the SNPs from the two studies were overlaid on the genomic structure of the locus using the UCSC genome browser (February 2009 human reference sequence (GRCh37)) [43], the SNPs associated with CAD are more distal than the SNPs associated with LOAD (Figure 4). The CAD SNPs are clustered over the protein coding exons of KIAA1462, while the LOAD SNPs are clustered proximal to the protein coding exons.
Figure 3. Linkage disequilibrium plot of SNPs associated with LOAD and CAD in KIAA1462 region.

Figure 4. CAD and LOAD associated SNPs mapped to intron/exon map of expressed genes.
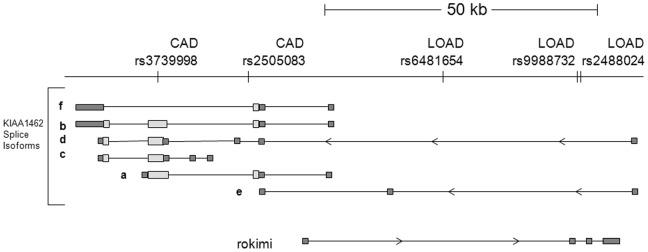
SNPs associated with CAD and LOAD are shown on the top line. Isoforms of KIAA1462 and “rokimi” are shown in relation underneath. Direction of transcription is symbolized by arrows. Exons are shown as boxes, and introns as lines. Putative protein coding regions of exons are shown as lighter colored boxes, and noncoding regions as darker boxes.
KIAA1462 has seven predicted splice isoforms, four of which have protein coding exons (Aceview) (Figure 4). The isoform b structure, which contains all three coding exons, is conserved in mouse, and the predicted protein is expressed in mouse [42]. The SNPs associated with CAD are more distal than the SNPs associated with LOAD, which are clustered upstream of the predicted start of transcription of KIAA1462 isoforms a, b, c, and f, and in the introns of isoforms d and e. Another gene, designated “rokimi” in Aceview, was recently identified as a noncoding spliced RNA completely internal to the KIAA1462 gene, but on the opposite strand of DNA. The sequence of the “rokimi” gene is defined by two GenBank accessions from one cDNA clone from synovial membrane tissue from a patient with rheumatoid arthritis. The SNPs associated with LOAD are also within introns of the “rokimi” gene.
Analysis of Expression Levels of KIAA1462 and “rokimi”
The location of the SNPs associated with LOAD suggests that they might alter the transcription of KIAA1462 or “rokimi”. The expression of KIAA1462 was investigated in brain of LOAD and control individuals using real time quantitative PCR. KIAA1462 mRNA was measured in brain samples taken from 14 control and 14 LOAD individuals (Table 4) using TaqMan based real time quantitative PCR. Three TaqMan probes were used individually, one specific for the junction between exons 2 and 3 of isoform b, one specific for the junction between exons 3 and 4 of isoform b, and one in the 3′ untranslated region of isoform b. These probes were designed to measure expression of isoforms a, b, c, and d. KIAA1462 was not significantly differentially expressed in AD brains as compared to controls (data not shown) using any of these probes. In addition, KIAA1462 was not differentially expressed in the brain based on any APOE genotype (data not shown).
Table 4. Origin of Brain Samples for RNA Purification.
| Sample | Gender | BRAAK Stage | Age of Onset | Age at Exam | ApoE Allele 1 | ApoE Allele 2 | Origin |
| C01 | M | 68 | 3 | 3 | HBTRC | ||
| C02 | M | 66 | 3 | 4 | HBTRC | ||
| C03 | M | 67 | 2 | 3 | HBTRC | ||
| C04 | M | 76 | 3 | 3 | HBTRC | ||
| C05 | M | 88 | 3 | 3 | HBTRC | ||
| C06 | F | 95 | 3 | 3 | HBTRC | ||
| C07 | M | 78 | 3 | 3 | HBTRC | ||
| C08 | M | 78 | 3 | 3 | HBTRC | ||
| C09 | M | 67 | 3 | 3 | HBTRC | ||
| C10 | F | 78 | 3 | 3 | HBTRC | ||
| C11 | F | 84 | Xu, et.al. 2007 | ||||
| C12 | M | 75 | Xu, et.al. 2007 | ||||
| C13 | M | 74 | Xu, et.al. 2007 | ||||
| C14 | F | 79 | Xu, et.al. 2007 | ||||
| AD1 | M | IV | 69 | 77 | 4 | 4 | HBTRC |
| AD2 | M | III | 76 | 84 | 3 | 3 | HBTRC |
| AD3 | M | V | 73 | 81 | 4 | 4 | HBTRC |
| AD4 | F | V | 68 | 76 | 4 | 4 | HBTRC |
| AD5 | M | IV | 69 | 75 | 4 | 4 | HBTRC |
| AD6 | M | VI | 71 | 75 | 3 | 4 | HBTRC |
| AD7 | F | VI | 75 | 81 | 3 | 4 | HBTRC |
| AD8 | F | V | 75 | 86 | 3 | 3 | HBTRC |
| AD9 | F | VI | 81 | 89 | 3 | 3 | HBTRC |
| AD10 | F | V | 85 | 91 | 3 | 3 | HBTRC |
| AD11 | M | VI | 70 | Xu, et.al. 2007 | |||
| AD12 | F | VI | 79 | Xu, et.al. 2007 | |||
| AD13 | M | VI | 77 | Xu, et.al. 2007 | |||
| AD14 | F | VI | 87 | Xu, et.al. 2007 |
The presence of expression of the rokimi gene was examined using RT-PCR. RNA from brain and HeLa cell lines was used to perform RT-PCR with two sets of rokimi primers. Because rokimi appears to be a noncoding RNA and it was unclear if the message would be polyadenylated or not, in addition to oligo(dT) primers for first strand synthesis, random hexamers and a gene specific primer were also used. None of these experimental conditions yielded a PCR product, suggesting that rokimi is not expressed in brain tissue or HeLa cells. We have also examined the KIAA1462/rokimi locus for QTLs using both the eQTL Browser [44] and Aperture, an online tool designed by the William Bush laboratory (http://gwar.mc.vanderbilt.edu/aperture) to provide LocusZoom-style plots for over 28 million eQTL associations. No significant eQTLs were found at this locus.
Discussion
Ours is not the first genomic convergence study to investigate KIAA1462 in relation to AD and find similar results. Chapuis et al. selected genes from areas of genetic interest in LOAD, analyzed their expression in LOAD brain compared to controls via custom expression microarrays, and found that KIAA1462 was upregulated approximately 2.5 fold in AD brain [41]. Follow-up examination of eighteen SNPs in KIAA1462 showed that three were associated with AD, including rs2488024, the most strongly associated SNP in our study, and five were associated with age on onset of AD. The significance of the association of these SNPs did not, however, survive multiple correction testing [41].
Polymorphisms in KIAA1462 have also been associated with other diseases and phenotypes. KIAA1462 was one of six loci associated with variation in human recombination rates [45]. Specifically rs2505089 in KIAA1462 was found by GWAS to be associated with maternal recombination rate with a combined p value = 4.42×10−7. In addition, recent reports link KIAA1462 to CAD with genome wide significance scores [37], [38]. The first study showed association of rs3739998, a nonsynonymous SNP in KIAA1462, with CAD in the German MI Family Study with a combined p value = 1.27×10−11 [38]. The second study, a meta-analysis of four combined GWAS with subjects from both European and South Asian background, revealed five major loci associated with CAD, including KIAA1462 (p value = 3.87×10(−8)) [37]. The function of KIAA1462 is unknown, but the protein encoded by isoform b is localized to endothelial cell-cell junctions, and this localization is VE-cadherin dependent [42].
Our results support those from another study examining chromosome 10 variation in LOAD [41]. Taken together, these results bolster the idea that KIAA1462 might play some role in the development of LOAD, although the role would likely be quite small. We can not rule out, however, that this locus is not associated with LOAD, considering that the data associating the locus with late onset AD is modest. The role of KIAA1462 in the genetics of CAD, however, seems much clearer from recent GWAS [37], [38]. The association between non-stroke cardiovascular disease (CVD) and Alzheimer’s disease has been debated, but a recent study reports that CVD increases risk of AD only in carriers of the APOE4 allele (Hazard Ratio 2.39, 95% confidence interval) [39]. The present study shows that KIAA1462, which is associated with CVD, is associated with APOE positive LOAD. The discovery of a genetic modifier of CVD that is associated with the APOE4 class of LOAD might explain some of the conflicting results in the literature regarding CVD and LOAD. These results suggest a model in which underlying CAD increases the risk of LOAD only in APOE4 carriers.
Several different lines of evidence suggest that variation in two different gene products at this locus may be responsible for the association with CAD and LOAD respectively. The CAD associated SNP rs3739998, which is a nonsynonymous change in exon 3 of isoform b of KIAA1462 that changes a serine to a threonine, is associated with CAD with a p value of 1.27×10−11, but is not significantly associated with LOAD in our study. Secondly, the genomic arrangement of the SNPs associated with CAD and LOAD suggests that they are in two different linkage disequilibrium blocks. The more proximal nature of the LOAD associated SNPs suggest that the gene product important in this association is either “rokimi”, or a different isoform of KIAA1462 than the isoform that is important in CVD. Since expression of “rokimi” was not detected in brain, it is unlikely that “rokimi” is the gene of interest in LOAD. We also did not see any differential expression of KIAA1462 in LOAD brain, but this could be a result of the methods used. With the three different TaqMan probes used, we would have observed a difference in expression of isoforms a, b, c, and d. Isoform e was not recognized by these probes, and isoform f has alternative polyadenylation sites, therefore some forms of isoform f may not have been detected by these TaqMan assays. Isoform e is represented by one Genbank entry from skeletal muscle. Isoform f is represented by 175 Genbank entries from many tissues, including multiple entries from brain, making isoform f a more attractive candidate for association with LOAD.
Acknowledgments
We would like to thank all of the individuals who participated in this study. This work used the core resources of the GCRC and the CHGR at VUMC, and the MIHG at University of Miami. Brain tissues were provided by the Harvard Brain Tissue Resource Center.
Funding Statement
NIH grants AG019757, AG021547, AG20135, AG21886; Alzheimer Association (IIRG-00-2006); Vanderbilt GCRC (MO1 RR-00095); Harvard Brain Tissue Resource Center (R24MH068855). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Alzheimer’s Association (2012) Alzheimer’s disease facts and figures. www.alz.org, Accessed 22 October 2013.
- 2. Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60 (8): 1119–1122 10.1001/archneur.60.8.1119 [DOI] [PubMed] [Google Scholar]
- 3. Hebert LE, Beckett LA, Scherr PA, Evans DA (2001) Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis Assoc Disord 15 (4): 169–173. [DOI] [PubMed] [Google Scholar]
- 4. Bergem AL, Engedal K, Kringlen E (1997) The role of heredity in late-onset Alzheimer disease and vascular dementia. A twin study. Arch Gen Psychiatry 54 (3): 264–270. [DOI] [PubMed] [Google Scholar]
- 5. Bertram L, Tanzi RE (2012) The genetics of Alzheimer’s disease. Prog Mol Biol Transl Sci 107: 79–100 10.1016/B978-0-12-385883-2.00008-4 [DOI] [PubMed] [Google Scholar]
- 6. Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, et al. (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349 (6311): 704–706 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- 7. Rogaev EI, Sherrington R, Wu C, Levesque G, Liang Y, et al. (1997) Analysis of the 5′ sequence, genomic structure, and alternative splicing of the presenilin-1 gene (PSEN1) associated with early onset Alzheimer disease. Genomics 40 (3): 415–424 10.1006/geno.1996.4523 [DOI] [PubMed] [Google Scholar]
- 8. Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, et al. (1995) A familial Alzheimer’s disease locus on chromosome 1. Science 269 (5226): 970–973. [DOI] [PubMed] [Google Scholar]
- 9. Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, et al. (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376 (6543): 775–778 10.1038/376775a0 [DOI] [PubMed] [Google Scholar]
- 10. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261 (5123): 921–923. [DOI] [PubMed] [Google Scholar]
- 11. Casserly I, Topol E (2004) Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet 363 (9415): 1139–1146 10.1016/S0140-6736(04)15900-X [DOI] [PubMed] [Google Scholar]
- 12. Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, et al. (2007) Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 298 (11): 1300–1311 10.1001/jama.298.11.1300 [DOI] [PubMed] [Google Scholar]
- 13. Song Y, Stampfer MJ, Liu S (2004) Meta-analysis: apolipoprotein E genotypes and risk for coronary heart disease. Ann Intern Med 141 (2): 137–147. [DOI] [PubMed] [Google Scholar]
- 14. Mahley RW, Weisgraber KH, Huang Y (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A 103 (15): 5644–5651 10.1073/pnas.0600549103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kehoe P, Wavrant-De Vrieze F, Crook R, Wu WS, Holmans P, et al. (1999) A full genome scan for late onset Alzheimer’s disease. Hum Mol Genet 8 (2): 237–245. [DOI] [PubMed] [Google Scholar]
- 16. Bertram L, Blacker D, Mullin K, Keeney D, Jones J, et al. (2000) Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science 290 (5500): 2302–2303 10.1126/science.290.5500.2302 [DOI] [PubMed] [Google Scholar]
- 17. Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, et al. (2000) Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 290 (5500): 2303–2304 10.1126/science.290.5500.2303 [DOI] [PubMed] [Google Scholar]
- 18. Myers A, Holmans P, Marshall H, Kwon J, Meyer D, R et al (2000) Susceptibility locus for Alzheimer’s disease on chromosome 10. Science 290 (5500): 2304–2305 10.1126/science.290.5500.2304 [DOI] [PubMed] [Google Scholar]
- 19. Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, et al. (2003) Results of a high-resolution genome screen of 437 Alzheimer’s disease families. Hum Mol Genet 12 (1): 23–32. [DOI] [PubMed] [Google Scholar]
- 20. Farrer LA, Bowirrat A, Friedland RP, Waraska K, Korczyn AD, Baldwin CT (2003) Identification of multiple loci for Alzheimer disease in a consanguineous Israeli-Arab community. Hum Mol Genet 12 (4): 415–422. [DOI] [PubMed] [Google Scholar]
- 21. Liang X, Schnetz-Boutaud N, Bartlett J, Allen MJ, Gwirtsman H, et al. (2008) No association between SNP rs498055 on chromosome 10 and late-onset Alzheimer disease in multiple datasets. Ann Hum Genet 72 (Pt 1): 141–144 10.1111/j.1469-1809.2007.00394.x [DOI] [PubMed] [Google Scholar]
- 22. Liang X, Slifer M, Martin ER, Schnetz-Boutaud N, et al. (2009) Genomic convergence to identify candidate genes for Alzheimer disease on chromosome 10. Hum Mutat 30 (3): 463–471 10.1002/humu.20953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKhann G, Drachman D, Folstein M, Katzman R, Price D, et al. (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34 (7): 939–944. [DOI] [PubMed] [Google Scholar]
- 24. Vina J, Lloret A (2010) Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. J Alzheimers Dis 20 Suppl 2S527–533 10.3233/JAD-2010-100501 [DOI] [PubMed] [Google Scholar]
- 25. Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial analysis of gene expression. Science 270 (5235): 484–487. [DOI] [PubMed] [Google Scholar]
- 26. Li YJ, Xu P, Qin X, Schmechel DE, Hulette CM, et al. (2006) A comparative analysis of the information content in long and short SAGE libraries. BMC Bioinformatics 7: 504 10.1186/1471-2105-7-504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin ER, Bass MP, Hauser ER, Kaplan NL (2003) Accounting for linkage in family-based tests of association with missing parental genotypes. Am J Hum Genet 73 (5): 1016–1026 10.1086/378779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin ER, Monks SA, Warren LL, Kaplan NL (2000) A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am J Hum Genet 67 (1): 146–154 10.1086/302957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin ER, Bass MP, Gilbert JR, Pericak-Vance MA, Hauser ER (2003) Genotype-based association test for general pedigrees: the genotype-PDT. Genet Epidemiol 25 (3): 203–213 10.1002/gepi.10258 [DOI] [PubMed] [Google Scholar]
- 30. Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 (2): 263–265 10.1093/bioinformatics/bth457 [DOI] [PubMed] [Google Scholar]
- 31. Bhat A, Heath SC, Ott J (1999) Heterogeneity for multiple disease loci in linkage analysis. Hum Hered 49 (4): 229–231. [DOI] [PubMed] [Google Scholar]
- 32. Hodge SE (1994) What association analysis can and cannot tell us about the genetics of complex disease. Am J Med Genet 54 (4): 318–323 10.1002/ajmg.1320540408 [DOI] [PubMed] [Google Scholar]
- 33. Xu PT, Li YJ, Qin XJ, Kroner C, Green-Odlum A, et al. (2007) A SAGE study of apolipoprotein E3/3, E3/4 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Mol Cell Neurosci 36 (3): 313–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pericak-Vance MA, Grubber J, Bailey LR, Hedges D, West S, et al. (2000) Identification of novel genes in late-onset Alzheimer’s disease. Exp Gerontol 35 (9–10): 1343–1352. [DOI] [PubMed] [Google Scholar]
- 35. Zubenko GS, Hughes HB, Stiffler JS, Hurtt MR, Kaplan BB (1998) A genome survey for novel Alzheimer disease risk loci: results at 10-cM resolution. Genomics 50 (2): 121–128 10.1006/geno.1998.5306 [DOI] [PubMed] [Google Scholar]
- 36. Liang X, Martin ER, Schnetz-Boutaud N, Bartlett J, Anderson B, et al. (2007) Effect of heterogeneity on the chromosome 10 risk in late-onset Alzheimer disease. Hum Mutat 28 (11): 1065–1073 10.1002/humu.20567 [DOI] [PubMed] [Google Scholar]
- 37. Coronary Artery Disease (C4D) Genetics Consortium (2011) A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 43 (4): 339–344 10.1038/ng.782 [DOI] [PubMed] [Google Scholar]
- 38. Erdmann J, Willenborg C, Nahrstaedt J, Preuss M, Konig IR, et al. (2011) Genome-wide association study identifies a new locus for coronary artery disease on chromosome 10p11.23. Eur Heart J 32 (2): 158–168 10.1093/eurheartj/ehq405 [DOI] [PubMed] [Google Scholar]
- 39. Eriksson UK, Bennet AM, Gatz M, Dickman PW, Pedersen NL (2010) Nonstroke cardiovascular disease and risk of Alzheimer disease and dementia. Alzheimer Dis Assoc Disord 24 (3): 213–219 10.1097/WAD.0b013e3181d1b99b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43 (5): 436–441 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, et al. (2009) Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer’s disease. Mol Psychiatry 14 (11): 1004–1016 10.1038/mp.2009.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Akashi M, Higashi T, Masuda S, Komori T, Furuse M (2011) A coronary artery disease-associated gene product, JCAD/KIAA1462, is a novel component of endothelial cell-cell junctions. Biochem Biophys Res Commun 413 (2): 224–229 10.1016/j.bbrc.2011.08.073 [DOI] [PubMed] [Google Scholar]
- 43. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, et al. (2002) The human genome browser at UCSC. Genome Res 12 (6): 996–1006 10.1101/gr.229102.ArticlepublishedonlinebeforeprintinMay2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, et al. (2008) High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4 (10): e1000214 10.1371/journal.pgen.1000214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chowdhury R, Bois PR, Feingold E, Sherman SL, Cheung VG (2009) Genetic analysis of variation in human meiotic recombination. PLoS Genet 5 (9): e1000648 10.1371/journal.pgen.1000648 [DOI] [PMC free article] [PubMed] [Google Scholar]