

Background: NF-κB activation and GRK5 up-regulation have been shown to be associated with heart disease.
Results: GRK5 induces NF-κB signaling pathway both in cardiomyocytes and in mouse hearts.
Conclusion: GRK5 triggers the binding of NF-κB (p50/p65) to DNA in the nucleus and promotes gene transcription including hypertrophic gene.
Significance: GRK5 up-regulation may lead to a significant increase in expression and activation of NF-κB seen in heart disease.
Keywords: G Protein-coupled Receptors (GPCR), Gene Expression, Gene Regulation, NF-Kappa B (NF-KB), Transcription
Abstract
G protein-coupled receptor kinase 5 GRK5 plays a key role in regulating cardiac signaling and its expression is increased in heart failure. GRK5 activity in the nucleus of myocytes has been shown to be detrimental in the setting of pressure-overload hypertrophy. The ubiquitous nuclear transcription factor κB (NF-κB) is involved in the regulation of numerous genes in various tissues, and activation of NF-κB has been shown to be associated with heart disease. Herein, we investigated whether GRK5 can specifically regulate the NF-κB signaling pathway in myocytes. We found that overexpression of GRK5 increased the levels of NF-κB -p50 and p65 in vitro and in vivo, whereas loss of GRK5 resulted in lower cardiac NF-κB levels. Furthermore, increased GRK5 expression induced the phosphorylation status of p65, increased the activity of a NF-κB reporter, and increased NF-κB DNA binding activity in cultured neonatal rat ventricular myocytes. Importantly, siRNA against GRK5 presented with the opposite results in neonatal rat ventricular myocytes as p65 and p50 were decreased, and there was a loss of NF-κB DNA binding activity. The influence of GRK5 on NF-κB expression and activity was dependent on its nuclear localization as overexpression of a mutant GRK5 that cannot enter the nucleus was devoid of NF-κB activation or DNA binding. Our study demonstrates that a novel pathological consequence of GRK5 up-regulation in the injured and failing heart is the induction of NF-κB expression and activity.
Introduction
G protein-coupled receptors (GPCRs),3 such as β-adrenergic receptors, are critical regulators of the function of the cardiovascular system. GPCRs undergo nodal regulation following agonist activation triggered by phosphorylation via a family of kinases known as GPCR kinases (GRKs) (1, 2). GRK phosphorylation of activated receptors triggers a process of desensitization that involves the loss of G protein signal through binding of β-arrestins to the phosphorylated receptor (1). Seven GRKs have been identified, and they all have distinct tissue distribution, subcellular localization, and undergo specific regulatory actions (2, 3). In the heart, GRK2 and GRK5 are the most abundant, and importantly, both have been shown to be up-regulated in failing human myocardium (1, 4). Animal models have led to the discovery that when GRK2 and GRK5 are elevated in the heart, they can drive pathological signaling and heart failure (HF) (5–8).
GRK5 is unique compared with GRK2 as it has a functional nuclear localization signal (NLS) (9), and indeed, it has been found in the nucleus of myocytes (6, 8, 10, 11). In fact, nuclear localization has been shown to be the determinant of cardiomyopathy after pressure overload where it has non-GPCR activity as a class II histone deacetylase kinase driving cardiac hypertrophic gene transcription (8, 12). Consistent with this, mice devoid of GRK5 in their hearts have an attenuated response to ventricular pressure overload, and HF is prevented in these mice with demonstrable lower histone deacetylase 5 phosphorylation (12).
Previously, we have explored potential mechanisms for GRK5 up-regulation in the heart and found that GRK5 gene expression and protein levels can be induced by NF-κB activity (13). This is a potentially interesting interaction because previous studies have shown that GRK5 can regulate NF-κB signaling at the protein level (14–18). In this study, we have uncovered both in vitro in myocytes and in vivo in the mouse heart, that GRK5 acting in the nucleus can drive expression and activity of NF-κB molecules. Thus, in addition to facilitating hypertrophic signaling through myocyte enhancer factor-2 (8), GRK5 can activate NF-κB, which has also been implicated in cardiac hypertrophic gene transcription (19, 20). Therefore, both systems (GRK5 and NF-κB) appear to co-regulate each other, linking these two potential pathological mediators that suggest novel means of therapeutic intervention against maladaptive cardiac hypertrophy and HF.
EXPERIMENTAL PROCEDURES
Maintenance of Primary Culture, Immunoblot Analysis, and Total Cellular Lysate Preparation
Ventricular cardiomyocytes from 1- to 2-day-old rat neonatal hearts (NRVMs) were prepared as we have published recently (2). Myocytes were cultured in Ham's F-10 supplemented with penicillin/streptomycin (100 units/ml) and 5% FBS at 37 °C in 5% CO2-humidified atmosphere for 2–3 days. NRVMs were cultured for 24 h in complete medium consisted of 85% Ham's F-10, 10% heat-inactivated horse serum, 5% FBS at 37 °C, in the presence of 5% CO2 and 95% ambient air followed by maintaining in 5% FBS containing medium for another 24 h. After 2 days, cells were infected or transfected and maintained in serum free medium for 48 h. In some experiments, NRVMs were infected with appropriate adenoviruses or siRNA. The cells were scraped from the dishes using ice-cold phosphate-buffered saline containing protease inhibitor mixture (1 tablet/10 ml) (Roche Applied Science) and lysed using lysis buffer. Protein concentrations were determined by a Pierce BCA protein assay kit. Proteins (15 μg) were separated on NuPAGE Novex 4–20% bis-tris gels (Invitrogen) and transferred to nitrocellulose membranes. The membranes were then analyzed by immunoblotting using specific antibodies to GRK5, p65, phospho-p65 (p-p65), p50, and GAPDH. Antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Cell Signaling Technology, Inc.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts (NE) were prepared from NRVMs as described previously (13). Protein concentrations were determined by a Pierce BCA protein assay kit. Double-stranded NF-κB consensus oligonucleotides (sense, 5′-GAG CGT GGG GAT CCC GGG AGT C-3′ and antisense, 5′-GAC TCC CGG GAT CCC CAC GCT C-3′) were used in EMSA. The oligonucleotides were end labeled using IRdye 700 and used as probes (Integrated DNA Technologies, Inc.). NE (7 μg) were incubated with IRdye-labeled NF-κB oligonucleotide in dark for 30 min at room temperature in reaction buffer (20 mm HEPES, pH 7.6, 75 mm KCl, 0.2 mm EDTA, 20% glycerol) and 1 μg of poly(dI-dC)-poly(dI-dC) (Li-Cor) as nonspecific competitor. For antibody-mediated super shift assay, 1 μg of p50 or p65 was incubated in reaction mixture for 30 min. followed by incubation with IRdye-labeled probe for another 30 min. Protein-DNA complexes were separated from free probe on 4% non-denaturing polyacrylamide gel. The image was visualized and scanned by the Odyssey infrared imaging system.
RNA Isolation and Reverse Transcriptase Real Time (q)PCR
NRVMs were cultured for 24 h in complete medium then changed to 5% FBS containing medium for another 24 h. Cells in 5% medium were then maintained in serum-free medium in the presence of either appropriate adenoviruses or siRNA for 48 h. Total RNA was isolated from these cells and from heart tissues obtained from mouse models by the one-step method described previously (21) (TRIzol; Invitrogen). RNA was treated with DNase to remove any contaminating DNA, and 500 ng were reversed transcribed using I-script cDNA synthesis kit from Bio-Rad. Validated primer sets directed against GRK5 (forward, 5′-CAA GGA GCT GAA TGT GTT CGG AC-3′ and reverse, 5′-GCT GCT TCC AGT GGA GTT TGA AT-3′) and p65 (forward, 5′-CAA GTG CCT TAA TAG CAG GGC AAA-3′ and reverse, 5′-AGA GCT AGA AAG AGC AAG AGT CCA AT-3′) along with the constitutively expressed 18S rRNA (forward, 5′-ACC GCA GCT AGG AAT AAT GGA-3′ and reverse, 5′-GCC TCA GTT CCG AAA ACC A-3′) were used for qPCR amplification. The Bio-Rad detection system (MyIQ) was employed using the DNA binding dye SYBR green for the quantitative detection of PCR products. The cycle threshold was set at a level where the exponential increase in PCR amplification was approximately parallel between all samples. Data were normalized by analyzing the ratio of the target cDNA concentrations to that of 18S rRNA. We have compared expression of 18S rRNA to that of GAPDH and have found both to be unaffected by adGRK5 or GRK5 siRNA treatment.
Transfection of Myocytes with GRK5 siRNA
NRVMs were cultured for 24 h in complete medium then changed to 5% FBS containing medium for another 24 h. Cells in 5% medium were then maintained in serum free medium in the presence or absence of GRK5 siRNA (30 nm) for 72 h using HiPerFect as transfecting reagent from Qiagen. Both RNA and total lysates were prepared from these cells. Lysates were used to determine proteins levels by immunoblot. cDNAs obtained from RNA were analyzed by real time quantitative PCR using the primers for GRK5, p65, p50, and 18s. Rat GRK5 siRNA was obtained from Dharmacon, Inc.
Infection of NRVMs with Recombinant Adenoviruses Expressing GFP (AdGFP) and GRK5 (AdGRK5)
NRVMs were cultured for 24 h in complete medium and then changed to 5% FBS containing medium for another 24 h. Cells in 5% medium were then maintained in serum-free medium and were infected with recombinant adenoviruses. Briefly, the cells were incubated for overnight with recombinant adenoviruses expressing GRK5 (AdGRK5) (multiplicity of infection of 200) or with GFP (AdGFP) (multiplicity of infection of 200) or with GRK5 missing nuclear localization sequence (AdGRK5-NLS). The next day, media were removed and changed to serum-free media and incubated for another 24 h. Cells were harvested and lysed followed by immunoblot analysis, EMSA, and RT-qRT-PCR.
Luciferase Activity
NRVMs were cultured for 24 h in complete medium then changed to 5% FBS containing medium for another 24 h. Cells in 5% medium were then maintained in serum-free medium in the presence of recombinant adenoviruses expressing NF-κB·luciferase reporter (NF-κB·Luc) or GFP (AdGFP) or GRK5 (AdGRK5) or GRK5-NLS (AdGRK5-NLS) or in some combination for 24 h. The next day, media were removed and changed to serum-free media and incubated for another 24 h. Cells were harvested 48 h after infection in passive lysis buffer (Promega). The samples were prepared according to the manufacturer's instructions (Promega) and measured by using a Tecan plate reader.
Transverse Aortic Constriction (TAC)
TAC was performed essentially as described previously (22). Briefly, mice were sedated in an isoflurane sedation box (induction, 3%; maintenance, 1.5%) and anesthetized to a surgical plane with an intraperitoneal dose of ketamine (50 mg/kg) and xylazine (2.5 mg/kg). Anesthetized mice were intubated, and a midline cervical incision was made to expose the trachea and carotid arteries. A blunt 20-gauge needle was inserted into the trachea and connected to a volume-cycled rodent ventilator on supplemental oxygen at a rate of 1 liter/min and respiratory rate of 140 bpm/min. Aortic constriction was performed by tying a 7–0 nylon suture ligature against a 27-gauge needle, which was promptly removed to yield a constriction of 0.4 mm in diameter. Pressure gradients were determined by in vivo hemodynamics, and mice with gradients >30 mmHg were used.
Statistics
All values in the text and figures are presented as mean ± S.E. from at least three independent experiments from given n sizes. Statistical significance of multiple treatments was determined by one-way analysis of variance followed by the Bonferroni post hoc test when appropriate. Statisical significance between two groups was determined using the two-tailed Student's t test. p values of <0.05 were considered significant.
RESULTS
Overexpression of GRK5 Increases Both mRNA and Protein Levels of p50 and p65 in NRVMs
To determine the role of GRK5 in NF-κB signaling in terms of expression and activity in cultured myocytes, NRVMs were infected with either AdGFP or AdGRK5, and cells lysates were obtained for immunoblotting or RNA isolated for qRT-PCR. Overexpression of GRK5 induced the up-regulations of NF-κB subunits p50 and p65 (Fig. 1, A–C). Furthermore, mRNA levels of p50 and p65 were increased (Fig. 1, D and E). Overexpression of GRK5 was also confirmed after AdGRK5 infection of myocytes by measuring both proteins (Fig. 1A) and mRNA (data not shown). Importantly, phosphorylation of p65 (p-p65) was induced in cultured myocytes infected with AdGRK5 (Fig. 1A, middle panel). It has been reported that phosphorylation of p65 increases the transcriptional activity of NF-κB by strengthening the interaction between p65 and the transcriptional coactivator CBP/p300 in the nucleus (23).
FIGURE 1.
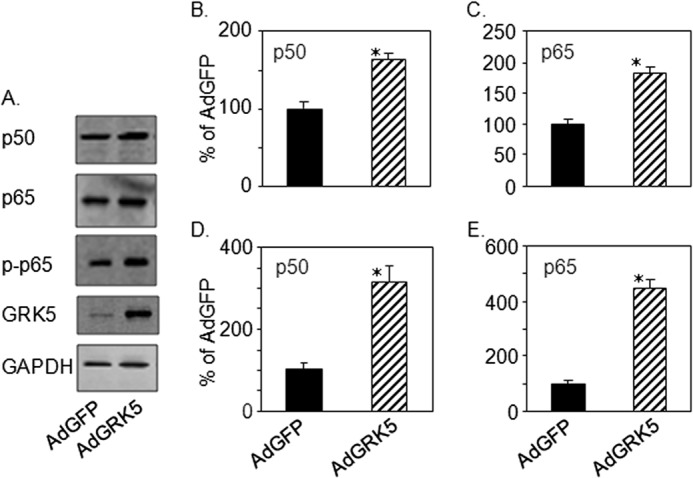
GRK5 induces NF-κB expression in cultured myocytes. NRVMs were infected with AdGFP or AdGRK5 (see “Experimental Procedures”). A, total lysates (15 μg) were analyzed by immunoblot for GRK5, p65, phosho-p65 (p-p65), p50, and GAPDH. B and C, quantitation of immunoblots in A. D and E, cDNAs obtained from myocyte RNA were analyzed by real time qPCR (see “Experimental Procedures”). Expression of mRNA is shown in % of AdGFP for p50 (D) and p65 (E). The data presented above are the means ± S.E.; *, p < 0.05 versus AdGFP (t test) (n = 3 separate experiments).
Reduction in the levels of p50 and p65 after treatment of myocytes with GRK5 siRNA
The data presented above suggest that GRK5 acts to mediate induction of NF-κB expression in myocytes. To directly demonstrate the mechanistic action of GRK5 on NF-κB signaling, we treated NRVMs with siRNA against rat GRK5. Cells treated with GRK5 siRNA showed significant reduction of p50 and p65 protein levels compared with levels found in cells treated with control siRNA (Fig. 2, A, C, and D). Interestingly, reduction in the level phosphorylated p65 (p-p65) was also observed in cultured myocytes after GRK5 knockdown (Fig. 2A). Our data indicate that GRK5 is required for the phosphorylation of p65 (Figs. 1A and 2A). Loss of GRK5 expression (Fig. 2, A and B) also led to a reduction of p50 and p65 mRNA in NRVMs (data not shown) demonstrates that the mechanistic action of GRK5 occurs at the level of transcription.
FIGURE 2.
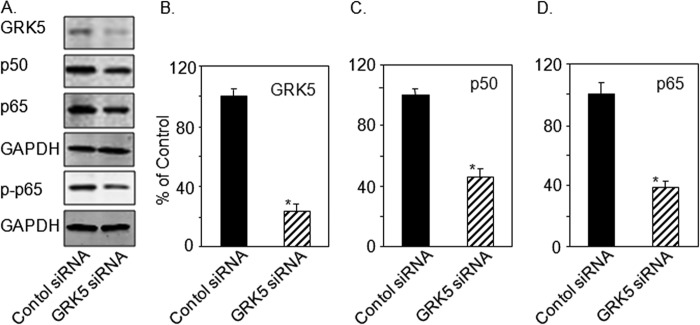
Lowering of NF-κB levels by GRK5 silencing in cultured NRVMs. NRVMs were transfected with GRK5 siRNA (30 nm) or control siRNA (see “Experimental Procedures”). A, total lysates were prepared and analyzed by immunoblot for GRK5, p65, p-p65, p50, and GAPDH. B–D are quantitations of immunoblots in A. The data presented above are the means ± S.E., *, p < 0.005 versus control (t test) (n = 3 separate experiments).
Expression p50 and p65 in GRK5 Transgenic (Tg GRK5) and GRK5 Knock-out (KO) Mice Basally and with the Aortic Constriction
To correlate the above findings in cultured myocytes to the heart in vivo, we used our available GRK5 KO mice and cardiac-specific GRK5 overexpressing transgenic mice (Tg GRK5). Interestingly, Tg GRK5 mice had increased levels of p50 and p65 mRNA in myocardium compared with non-transgenic littermate control (NLC) mice (Fig. 3, A and B). Interestingly, when pressure overload hypertrophic stress was applied to NLC and Tg GRK5 mice via TAC, p50 and p65 mRNA were significantly increased, which is consistent with results in NRVMs (Fig. 1). Interestingly, TAC induced p50 and p65 even more robustly in Tg GRK5 hearts (Fig. 3, A and B). Overexpression of GRK5 was also confirmed in the heart tissues of Tg GRK5 mice as well as in mice after TAC (Fig. 3C). Importantly, increased levels of p-p65 were observed in the heart tissues of Tg GRK5 mice and more robustly increased in mice after TAC (Fig. 3D). Similarly, the levels of mRNA expression of p50 and p65 were analyzed in heart tissues obtained from littermate WT control and GRK5 KO mice under sham conditions or 2 weeks of after TAC. Data in Fig. 3, E and F, show that mRNA expression of both p50 and p65 were significantly decreased in cardiac specific GRK5 KO mice as compared with WT. Moreover, TAC induction in GRK5 KO mice was not able to increase expression of p50 and p65 as compared with WT mice (Fig. 3, E and F). Similarly, p-p65 levels were markedly decreased in GRK5 KO mice compared with WT mice (Fig. 3G). There was also a reduction of p-p65 in TAC induced GRK5 KO as compared with WT mice after TAC (Fig. 3G). Thus, it appears that levels of GRK5 directly determine NF-κB levels including p-p65 in myocytes.
FIGURE 3.
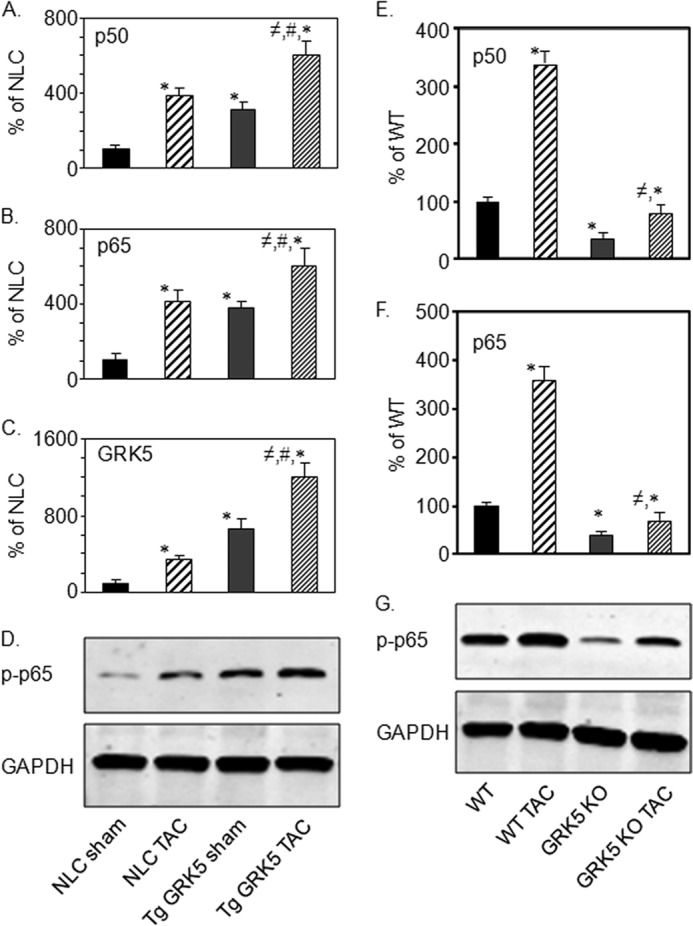
In vivo effects of ventricular pressure overload and GRK5 on cardiac p50 and p65 expression. Heart tissue from NLC mice and transgenic mice with cardiac-specific overexpression of GRK5 (Tg GRK5) under sham conditions or 2 weeks after TAC were collected, and RNA was prepared. cDNAs obtained from RNA were analyzed by real time qPCR, and expression of mRNA is shown in % of NLC for p50 (A); p65 (B); and GRK5 (C). The data presented are the mean ± S.E., *, p < 0.05 versus NLC sham; ≠, p < 0.05 versus NLC TAC; #, p < 0.05 versus Tg GRK5 sham (one-way analysis of variance, Bonferroni's multiple comparison test) (n = 4). D, total lysates were prepared from heart tissues of these mice and were analyzed by immunoblot for p-p65 and GAPDH (n = 4). E and F, heart tissue from WT mice and GRK5 KO mice under sham conditions or 2 weeks of after TAC were collected, and RNA was prepared. cDNAs obtained from RNA were analyzed by real time quantitative PCR and expression of mRNA is shown in arbitrary units for p50 (E) and p65 (F). Data presented above are means ± S.E. *, p < 0.05 versus WT; ≠, p < 0.05 versus WT TAC (one-way analysis of variance, Bonferroni's multiple comparison test) (n = 4). G, total lysates were prepared from the heart tissues of these mice and were analyzed by immunoblot for p-p65 and GAPDH (n = 4).
Induction of NF-κB Activities by GRK5 in Cultured Myocytes
To analyze the effect of GRK5 on NF-κB DNA binding activity, NRVMs were infected with either AdGRK5 or AdGFP and NE were prepared from cultured NRVMs after infection. NE from the infected myocytes was analyzed by EMSA using consensus NF-κB site as an IR-dye labeled probe. The gel was scanned using an Odyssey infrared imaging system. As can be seen in Fig. 4A, overexpression of GRK5 increased the NF-κB DNA binding activity as compared with AdGFP-infected cells. The data indicate that the induction of NF-κB proteins levels by GRK5 also increased its activity in heart cells. To determine the specificity of NF-κB binding activity nonspecific oligonucleotide and nonspecific antibody were added to the reaction mixture containing AdGRK5-infected NE (Fig. 4B). As can be seen, NE from AdGRK5 infected NRVMs bound to the NF-κB oligonucleotide (Fig. 4B, lane 2) and non-labeled (200-fold excess) NF-κB consensus oligonucleotide competed with IR-dye labeled NF-κB oligonucleotides for binding to nuclear proteins (Fig. 4B, lane 3) compared with non-labeled nonspecific oligonucleotide (Fig. 4B, lane 4). Antibodies to NF-κB proteins p50 and p65 added alone reduced the intensity of the NF-κB-nuclear protein complex as compared nonspecific antibody (Fig. 4B, lanes 5–7). This suggests the presence of p50 and p65 in the complex of nuclear proteins from AdGRK5-infected myocytes. Interestingly, NF-κB DNA binding activity was also decreased by the treatment of myocytes with GRK5 siRNA (Fig. 4C). To determine an additional role of nuclear GRK5 in the regulation of NF-κB signaling, the effect of GRK5-NLS (a mutant form of GRK5 that lacks the nuclear localization sequence) was tested for NF-κB DNA binding activity the requirement of GRK. We observed previously that GRK5 is required for the activation of myocyte enhancer factor-2 after hypertrophic stress (8). Similarly, in the present study, we found that nuclear localization sequence of GRK5 is required to activate NF-κB in cultured NRVMs (Fig. 4D).
FIGURE 4.
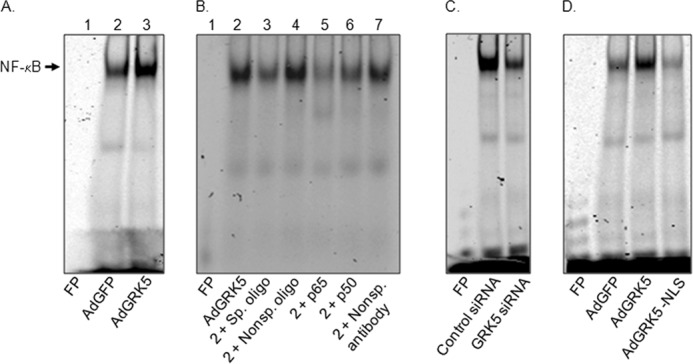
Effects of GRK5 on NF-κB DNA binding activity in cultured myocytes. A, EMSA was carried out for 7.5 μg of NE from cultured NRVMs infected with AdGFP or AdGRK5 to analyze DNA binding activity of NF-κB using IR dye-labeled oligonucleotide for consensus NF-κB sites (see “Experimental Procedures”). Lane 1, free probe (FP; no NE); lane 2, AdGFP; and lane 3, AdGRK5. B, competitive and antibody-mediated super-shift EMSA using NE from AdGRK5-infected (NRVMs) and labeled oligonucleotide for consensus NF-κB sites. For EMSA super-shift and competition assays, samples were incubated with specific antibodies to p50 or p65 and with 200-fold excess non-labeled consensus or non-labeled nonspecific oligonucletides, respectively, for 30 min prior to the addition of labeled nucleotides (see “Experimental Procedures”). Lane 1, FP (no NE); lane 2, NE alone; lane 3, NE + 200× specific oligonucleotide (non-labeled); lane 4, NE + 200X nonspecific oligonucleotide (non-labeled); lane 5, NE + p65; lane 6, NE + p50; and lane 7, NE + nonspecific (nonsp.) antibody. C, EMSA for NF-κB DNA binding activity in NE of myocytes treated with GRK5 siRNA or control siRNA. D, EMSA for NF-κB DNA binding activity in NE of myocytes infected with AdGFP, AdGRK5, or adenovirus expressing GRK5-NLS (AdGRK5-NLS). An arrow indicates specific NF-κB binding activity. The experiment shown here is a representative of three independent experiments that yielded comparable results.
Induction of NF-κB Luciferase (NF-κB·Luc) activity by GRK5
In addition to NF-κB DNA binding activity, the role of GRK5 in NF-κB using a reporter construct was assessed. NRVMs were infected with an adenovirus expressing notification of an NF-κB luciferase reporter (NF-κB·Luc) followed by incubation with either AdGRK5 or AdGRK5-NLS. As can be seen in Fig. 5, GRK5 positively induced NF-κB luciferase activity. Importantly, when GFP- and GRK5-expressing myocytes were treated with SC 514, a selective inhibitor of IκB kinase 2 that can inhibit NF-κB gene expression, we found significant reduction of GRK5-induced NF-κB activity. Interestingly, SC 514 is able to inhibit GRK5 expression in cultured myocytes (13). The effect of GRK5-NLS was analyzed for NF-κB·luc activity, and as shown in Fig. 5B, overexpression of a GRK5 devoid of nuclear sequence had no effect on NF-κB luciferase activity. These data suggest that NLS of GRK5 is important for the activation of NF-κB signaling in cultured myocytes.
FIGURE 5.
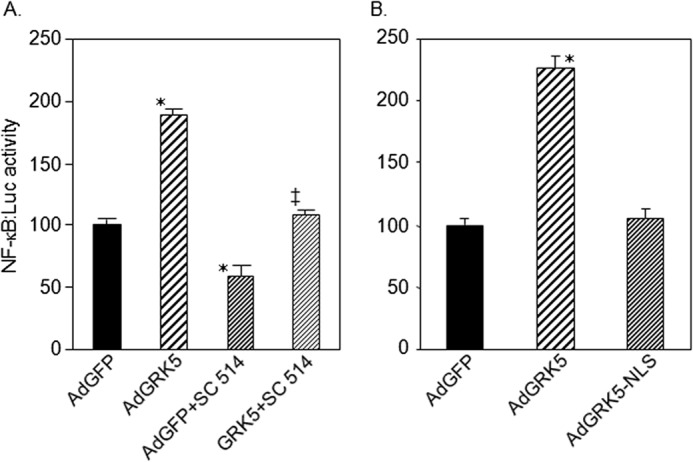
Induction of NF-κB luciferase (NF-κB·Luc) reporter activity by GRK5 in NRVMs. A, an adenovirus containing NF-κB-luciferase reporter (AdNF-κB·Luc) was used to infect NRVMs along with AdGFP or AdGRK5 and treated without or with the NF-κB inhibitor SC 514 (100 μm). NF-κB·Luc activity was measured by using Tecan plate reader. B, similar experiment but AdGRK5-NLS (virus lacks NLS (nuclear localization sequence)) used to show that NF-κB·Luc activity induced by GRK5 in cultured NRVMs requires nuclear localization. The data presented above represent mean ± S.E. of n = 3 independent experiments done in duplicate. *, p < 0.05 versus AdGFP; ‡, p < 0.05 versus AdGRK5 (one-way analysis of variance, Bonferroni's multiple comparison test).
DISCUSSION
Based on the data presented above, we describe a critical role for GRK5 in the regulation of cardiac NF-κB signaling pathway that occurs at the gene transcriptional level where the p50 and p65 subunits can directly bind to the DNA in the nucleus and promote various gene transcription events, including hypertrophic genes. GRKs are well recognized as critical regulators of GPCR signaling based on their roles on phosphorylating and desensitizing these receptors as demonstrated by numerous studies (24–26). As the most widely expressed and best characterized member of the GRK4 subfamily, GRK5 plays important roles in GPCR-mediated physiological processes, especially in the central nervous and cardiovascular systems (3). The data presented above uncovers a novel mechanism involved in the regulation of NF-κB signaling in cardiomyocytes. Because cardiac NF-κB is activated following myocardial stress and has been shown to be increased in HF, the role GRK5 plays in the NF-κB signaling pathway is significant. It is well known that GRK5 can be activated by hypertrophic stimuli as well as reactive oxygen species (13), and therefore, various forms of cardiac stress may presumably lead to GRK5-mediated NF-κB activation. Increased GRK5 appears to be a key pathological component of the post-stressed heart as it can localize to the nucleus where it can act as a class II histone deacetylase kinase promoting maladaptive hypertrophy and HF (8) through activation of fetal gene expression, including NF-κB. Activation of this transcription factor in turn up-regulates fetal gene expression, including GRK5 (13), which is up-regulated during hypertrophy/HF.
In other studies, interactions between GRK5 and NF-κB signaling components (albeit at the protein level) have been demonstrated (15–18), and we recently looked for the potential of this system that can be activated by PKC to regulate GRK5 expression in myocytes (13). NF-κB activation occurs through IκB degradation and subsequent nuclear translocation of the p65 subunit to regulate gene transcription (19, 20, 27). NF-κB plays important roles in cardiac hypertrophy and remodeling. In vitro studies have shown that NF-κB is required for hypertrophic growth of cardiomyocytes in response to GPCR agonists such as phenylephrine, endothelin-1, and angiotensin II (20, 27, 28). In vivo studies using different animals models of NF-κB inactivation by IκBα mutant overexpression, p65 silencing, or p50 deletion also showed reduced cardiac hypertrophy in response to aortic banding (29), chronic infusion of GPCR agonists (30, 31), and transgenic overexpression of myotrophin (32, 33). NF-κB has been implicated in cardiac hypertrophy, and the best evidence is from a transgenic mouse model with cardiac-specific expression of a mutant IκBα that acts as a super-repressor of NF-κB (34, 35). The expression of this mutant attenuated hypertrophic phenotypes induced by angiotensin II or isoproterenol infusion as well as aortic banding (30, 36). The fetal gene program was also abrogated in this model. Therefore, irrespective of whether these two molecules can interact at the protein level in the cytoplasm (15–18), GRK5 and NF-κB activity in the heart may lead to interrelated or parallel hypertrophic gene transcriptional paths.
Importantly, our previous study showed a putative nuclear link between those two molecules and pathways as we identified a putative NF-κB binding sequence within the 5′-flanking region of the GRK5 gene and both p50 and p65 NF-κB appear to interact with this region of the promoter in the nucleus of cardiomyocytes (13). It was then posited that this interaction of GRK5 could activate NF-κB signaling pathway. Herein, we found that overexpression of GRK5 increased p50 and p65 expression in vivo and in vitro. To strengthen this result, direct activation of NF-κB was induced by overexpression of GRK5 and was inhibited by lowering the levels of GRK5 in cultured myocytes through gene knockdown strategy. NF-κB is considered to be a redox-sensitive transcription factor, and reactive oxygen species is a potent stimulator of NF-κB; however, the precise steps sensitive to oxidative stress that may activate GRK5 up-regulation has not been determined clearly. In a previous study, it was shown that reactive oxygen species induces GRK5 levels in cultured myocytes (13). The potential role of endogenous GRK5 in the regulation of NF-κB signaling was supported by the finding that transfection with GRK5 siRNA reduced p50 and p65 expression as well as decreased NF-κB DNA binding activity in cultured myocytes. In addition, our in vivo study showed that NF-κB expression was induced in Tg GRK5 mice, and it was reduced in cardiac specific TgGRK5 KO mice. For further mechanistic study for the involvement of GRK5 in regulation of NF-κB signaling, we tested the effects of GRK5-NLS in cultured myocytes. Interestingly we found that GRK5-NLS had no effect on activation of NF-κB, which indicates that only nuclear localized GRK5 regulates NF-κB expression and activity.
In conclusion, our findings describe a critical role of GRK5 in the regulation of cardiac NF-κB signaling pathway where GRK5 induces expression, phosphorylation, and DNA binding activity of NF-κB. This potentially strengthens the interaction of NF-κB -p65 with coactivator (e.g. CBP/p300) or other transcription factors resulting in transcriptional activation of NF-κB in the nucleus of the myocytes. We have demonstrated that both stimulatory and inhibitory effects of GRK5 on NF-κB signaling are associated with changes in the expression, interaction, phosphorylation, binding, and transcriptional activity of NF-κB. A recent report from our laboratory showed that decreasing myocyte GRK5, either completely as in the global GRK5 KO mice or in cardiac specific GRK5 KO resulted in decreased cardiac hypertrophy and prevented pathogenesis of HF (12). Our present study indicates that decreasing GRK5 levels could represent a novel approach to attenuate cardiac hypertrophy/HF through inactivation of NF-κB signaling pathway in the heart. As increased GRK5, acting as a Class II histone deacetylase kinase and activity of NF-κB is enhanced in cardiac hypertrophy, can promote maladaptive ventricular hypertrophy and HF, this is an especially significant finding. As such, the NF-κB signaling pathway may be mediated by GRK5, which may directly be involved in hypertrophy and heart disease through inducing hypertrophic gene expression. Thus, strategies to limit GRK5 expression after cardiac injury could limit maladaptive gene changes or activation of transcription factor such as NF-κB, including its target genes. Future studies can be designed to investigate the molecular mechanism for the regulatory roles of GRK5 in the NF-κB signaling pathway in vivo models of hypertrophy and HF.
Acknowledgments
We thank Jessica Ibetti for isolation of NRVMs, Kate Sydnes for technical assistance, and Dr. J. Kurt Chuprun for helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants PO1 HL 091799 (Project 1 to W. J. K. and Core B to E. G.) and PO1 HL 075443 (Project 2 to W. J. K.).
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- Tg
- transgenic
- GRK5-NLS
- GRK5 with missing nuclear localization sequence
- Tg GRK5
- GRK5 overexpressor mice
- NLC
- non-transgenic littermate control
- SC 514
- IκB kinase 2 inhibitor
- HF
- heart failure
- NE
- nuclear extract(s)
- Ad
- adenovirus
- NRVM
- neonatal rat ventricular myocyte
- qPCR
- quantitative PCR
- TAC
- transverse aortic constriction.
REFERENCES
- 1. Rockman H. A., Koch W. J., Lefkowitz R. J. (2002) Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212 [DOI] [PubMed] [Google Scholar]
- 2. Brinks H., Koch W. J. (2010) Targeting G protein-coupled receptor kinases (GRKs) in Heart Failure. Drug Discov. Today Dis. Mech. 7, e129-e134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Premont R. T., Gainetdinov R. R. (2007) Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69, 511–534 [DOI] [PubMed] [Google Scholar]
- 4. Huang Z. M., Gold J. I., Koch W. J. (2011) G protein-coupled receptor kinases in normal and failing myocardium. Front. Biosci. 16, 3047–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brinks H., Boucher M., Gao E., Chuprun J. K., Pesant S., Raake P. W., Huang Z. M., Wang X., Qiu G., Gumpert A., Harris D. M., Eckhart A. D., Most P., Koch W. J. (2010) Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ. Res. 107, 1140–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gold J. I., Martini J. S., Hullmann J., Gao E., Chuprun J. K., Lee L., Tilley D. G., Rabinowitz J. E., Bossuyt J., Bers D. M., Koch W. J. (2013) Nuclear translocation of cardiac g protein-coupled receptor kinase 5 downstream of select gq-activating hypertrophic ligands is a calmodulin-dependent process. PLoS One 8, e57324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koch W. J., Rockman H. A., Samama P., Hamilton R. A., Bond R. A., Milano C. A., Lefkowitz R. J. (1995) Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a β ARK inhibitor. Science 268, 1350–1353 [DOI] [PubMed] [Google Scholar]
- 8. Martini J. S., Raake P., Vinge L. E., DeGeorge B. R., Jr., DeGeorge B., Jr., Chuprun J. K., Harris D. M., Gao E., Eckhart A. D., Pitcher J. A., Koch W. J. (2008) Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 105, 12457–12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson L. R., Scott M. G., Pitcher J. A. (2004) G protein-coupled receptor kinase 5 contains a DNA-binding nuclear localization sequence. Mol. Cell Biol. 24, 10169–10179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yi X. P., Gerdes A. M., Li F. (2002) Myocyte redistribution of GRK2 and GRK5 in hypertensive, heart-failure-prone rats. Hypertension 39, 1058–1063 [DOI] [PubMed] [Google Scholar]
- 11. Zhang Y., Matkovich S. J., Duan X., Gold J. I., Koch W. J., Dorn G. W., 2nd. (2011) Nuclear effects of G-protein receptor kinase 5 on histone deacetylase 5-regulated gene transcription in heart failure. Circ. Heart Fail 4, 659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gold J. I., Gao E., Shang X., Premont R. T., Koch W. J. (2012) Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ. Res. 111, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Islam K. N., Koch W. J. (2012) Involvement of nuclear factor κB (NF-κB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression. J. Biol. Chem. 287, 12771–12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parmar K. M., Larman H. B., Dai G., Zhang Y., Wang E. T., Moorthy S. N., Kratz J. R., Lin Z., Jain M. K., Gimbrone M. A., Jr., García-Cardeña G. (2006) Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Invest. 116, 49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patial S., Luo J., Porter K. J., Benovic J. L., Parameswaran N. (2010) G-protein-coupled-receptor kinases mediate TNFα-induced NFκB signalling via direct interaction with and phosphorylation of IκBα. Biochem. J. 425, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patial S., Shahi S., Saini Y., Lee T., Packiriswamy N., Appledorn D. M., Lapres J. J., Amalfitano A., Parameswaran N. (2011) G-protein coupled receptor kinase 5 mediates lipopolysaccharide-induced NFκB activation in primary macrophages and modulates inflammation in vivo in mice. J. Cell Physiol. 226, 1323–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sorriento D., Ciccarelli M., Santulli G., Campanile A., Altobelli G. G., Cimini V., Galasso G., Astone D., Piscione F., Pastore L., Trimarco B., Iaccarino G. (2008) The G-protein-coupled receptor kinase 5 inhibits NFκB transcriptional activity by inducing nuclear accumulation of IκBα. Proc. Natl. Acad. Sci. U.S.A. 105, 17818–17823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sorriento D., Santulli G., Fusco A., Anastasio A., Trimarco B., Iaccarino G. (2010) Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-κB-dependent hypertrophic gene expression. Hypertension 56, 696–704 [DOI] [PubMed] [Google Scholar]
- 19. Gordon J. W., Shaw J. A., Kirshenbaum L. A. (2011) Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ. Res. 108, 1122–1132 [DOI] [PubMed] [Google Scholar]
- 20. Purcell N. H., Tang G., Yu C., Mercurio F., DiDonato J. A., Lin A. (2001) Activation of NF-κ B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 98, 6668–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Islam K. N., Mendelson C. R. (2008) Glucocorticoid/glucocorticoid receptor inhibition of surfactant protein-A (SP-A) gene expression in lung type II cells is mediated by repressive changes in histone modification at the SP-A promoter. Mol. Endocrinol. 22, 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akhter S. A., Luttrell L. M., Rockman H. A., Iaccarino G., Lefkowitz R. J., Koch W. J. (1998) Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280, 574–577 [DOI] [PubMed] [Google Scholar]
- 23. Zhong H., Voll R. E., Ghosh S. (1998) Phosphorylation of NF-κ B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1, 661–671 [DOI] [PubMed] [Google Scholar]
- 24. Pitcher J. A., Freedman N. J., Lefkowitz R. J. (1998) G protein-coupled receptor kinases. Annu. Rev. Biochem. 67, 653–692 [DOI] [PubMed] [Google Scholar]
- 25. Moore C. A., Milano S. K., Benovic J. L. (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 69, 451–482 [DOI] [PubMed] [Google Scholar]
- 26. Gainetdinov R. R., Premont R. T., Bohn L. M., Lefkowitz R. J., Caron M. G. (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev Neurosci. 27, 107–144 [DOI] [PubMed] [Google Scholar]
- 27. Hirotani S., Otsu K., Nishida K., Higuchi Y., Morita T., Nakayama H., Yamaguchi O., Mano T., Matsumura Y., Ueno H., Tada M., Hori M. (2002) Involvement of nuclear factor-κB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation 105, 509–515 [DOI] [PubMed] [Google Scholar]
- 28. Cook S. A., Novikov M. S., Ahn Y., Matsui T., Rosenzweig A. (2003) A20 is dynamically regulated in the heart and inhibits the hypertrophic response. Circulation 108, 664–667 [DOI] [PubMed] [Google Scholar]
- 29. Li Y., Ha T., Gao X., Kelley J., Williams D. L., Browder I. W., Kao R. L., Li C. (2004) NF-κB activation is required for the development of cardiac hypertrophy in vivo. Am. J. Physiol. Heart Circ. Physiol. 287, H1712–1720 [DOI] [PubMed] [Google Scholar]
- 30. Freund C., Schmidt-Ullrich R., Baurand A., Dunger S., Schneider W., Loser P., El-Jamali A., Dietz R., Scheidereit C., Bergmann M. W. (2005) Requirement of nuclear factor-κB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 111, 2319–2325 [DOI] [PubMed] [Google Scholar]
- 31. Kawano S., Kubota T., Monden Y., Kawamura N., Tsutsui H., Takeshita A., Sunagawa K. (2005) Blockade of NF-κB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovascular research 67, 689–698 [DOI] [PubMed] [Google Scholar]
- 32. Gupta S., Young D., Maitra R. K., Gupta A., Popovic Z. B., Yong S. L., Mahajan A., Wang Q., Sen S. (2008) Prevention of cardiac hypertrophy and heart failure by silencing of NF-κB. J. Mol. Biol. 375, 637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Young D., Popovic Z. B., Jones W. K., Gupta S. (2008) Blockade of NF-κB using IκB α dominant-negative mice ameliorates cardiac hypertrophy in myotrophin-overexpressed transgenic mice. J. Mol. Biol. 381, 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dorn G. W., 2nd, Force T. (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest. 115, 527–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luedde M., Katus H. A., Frey N. (2006) Novel molecular targets in the treatment of cardiac hypertrophy. Recent Pat. Cardiovasc. Drug Discov. 1, 1–20 [DOI] [PubMed] [Google Scholar]
- 36. Zelarayan L., Renger A., Noack C., Zafiriou M. P., Gehrke C., van der Nagel R., Dietz R., de Windt L., Bergmann M. W. (2009) NF-κB activation is required for adaptive cardiac hypertrophy. Cardiovasc. Res. 84, 416–424 [DOI] [PubMed] [Google Scholar]