

Background: B. subtilis GS catalyzes the production of glutamine, a key metabolite in nitrogen assimilation.
Results: B. subtilis GS forms a dodecamer that undergoes large intersubunit conformational changes during catalysis.
Conclusion: B. subtilis GS structures reveal a heretofore unseen active site restructuring that is linked to a novel feedback regulatory mechanism.
Significance: The GS specific regulatory/catalytic mechanism may be used to target Gram-positive pathogens.
Keywords: Cooperativity, Enzyme Catalysis, Enzyme Inactivation, Enzyme Mechanisms, Enzyme Structure, Glutamine
Abstract
Glutamine synthetase (GS), which catalyzes the production of glutamine, plays essential roles in nitrogen metabolism. There are two main bacterial GS isoenzymes, GSI-α and GSI-β. GSI-α enzymes, which have not been structurally characterized, are uniquely feedback-inhibited by Gln. To gain insight into GSI-α function, we performed biochemical and cellular studies and obtained structures for all GSI-α catalytic and regulatory states. GSI-α forms a massive 600-kDa dodecameric machine. Unlike other characterized GS, the Bacillus subtilis enzyme undergoes dramatic intersubunit conformational alterations during formation of the transition state. Remarkably, these changes are required for active site construction. Feedback inhibition arises from a hydrogen bond network between Gln, the catalytic glutamate, and the GSI-α-specific residue, Arg62, from an adjacent subunit. Notably, Arg62 must be ejected for proper active site reorganization. Consistent with these findings, an R62A mutation abrogates Gln feedback inhibition but does not affect catalysis. Thus, these data reveal a heretofore unseen restructuring of an enzyme active site that is coupled with an isoenzyme-specific regulatory mechanism. This GSI-α-specific regulatory network could be exploited for inhibitor design against Gram-positive pathogens.
Introduction
Glutamine is a key metabolite in nitrogen assimilation because it is not only incorporated into proteins but also serves as the direct nitrogen donor for the synthesis of nearly 25% of all nitrogen-containing compounds (1). Glutamine is synthesized from ammonium, ATP, and glutamate by the enzyme glutamine synthetase (GS).2 Underscoring its essential role in nitrogen metabolism and assimilation, GS is found in all organisms and is tightly regulated (2–3). GS enzymes are all large homo-oligomeric machines, in which each active site is formed between subunit interfaces (3–13). There are three different classes of GS enzymes, categorized as GSI, GSII, and GSIII (14). GSI enzymes are specific to prokaryotes and archaea and have thus far all been shown to be dodecamers (3, 7–14). GSII enzymes are decamers consisting of two stacked pentamer rings and are found in eukaryotes and soil-dwelling bacteria (4, 5). GSIII proteins are harbored in certain anaerobic bacteria and cyanobacteria and, like GSI enzymes, are dodecamers but are formed from hexamer-hexamer interactions different from those that form GSI oligomers (6).
Despite their distinct oligomeric arrangements, the active site and residues involved in catalysis are conserved in GS proteins, suggesting similar overall mechanisms. This enzymatic reaction can be divided into two steps, which are facilitated by either Mg2+ or Mn2+ (3, 15). In the first step, the ϵ-oxygen of the substrate glutamate attacks the γ-phosphorus of ATP to form a γ-glutamyl phosphate intermediate (16–21). In the second step, ammonia, formed by the deprotonation of an ammonium ion, attacks the reaction intermediate to produce glutamine (Fig. 1A). Because of their central role in metabolism, GSs have emerged as important chemotherapeutic targets. For example, the herbicide glufosinate-ammonium, which inhibits plant GS, has recently become commercially available, and research is under way for the development of compounds that selectively target the Mycobacterium tuberculosis GS (22). The design of pathogen-specific drugs would be greatly facilitated by structural and biochemical studies revealing any unique catalytic or regulatory mechanisms utilized by these enzymes. However, our understanding of bacterial GS isoenzyme structure and function has been limited.
FIGURE 1.

Structure and enzymatic mechanism of B. subtilis GSI-α. A, top, a scheme depicting the GS reaction mechanism. Bottom left, secondary structural elements of the GSI-α subunit. For reference, the Glu flap is colored blue, and the Tyr loop is colored magenta. Ribbon diagrams figures were made using PyMOL (47). Bottom right, the GS active site is illustrated as a bifunnel with entrance and exit portals. The glutamate and ATP substrates enter and bind at opposite ends of the bifunnel, and the respective products leave via the same portals. B, a hexamer of the dodecameric B. subtilis GS viewed down the molecular 6-fold axis. C, the GS dodecamer rotated by 90º relative to Fig. 1B. In this figure, the “bottom” hexamer is shown as a surface to highlight the interhexamer contacting α14-α15 helices. D, close-up view of an active site created by neighboring subunits. Key active site loops that line the active site are labeled and colored differently. A bound glutamate is shown in CPK. E, comparison of GS dodecamers of the B. subtilis GSI-α and S. typhimurium GSI-β proteins. Shown in different colors (B. subtilis in blue and S. typhimurium in red) are the active site Tyr179 loops that line the dodecameric pore (labeled). F, superimposition of a B. subtilis subunit (blue) onto a S. typhimurium subunit (red), highlighting their strong structural correspondence except for the Tyr179 loop. The location of the pore is labeled in B and E for reference.
GSI is the major bacterial enzyme, and phylogenetic analysis reveals two GSI subdivisions, GSI-α and GSI-β (14). There are no GSI-α structures thus far described. GSI-β structures are available for the Escherichia coli, M. tuberculosis, and Salmonella typhimurium proteins, and the current understanding of GS catalysis is largely based on early studies of structures obtained from soaking various substrates/products into GSI-β crystals (3, 7–13). These studies suggested that relatively small structural alterations in active site loops are involved in catalysis (3, 10). GSI-α enzymes are found in low G + C Gram-positive bacteria and some thermophilic bacteria. The best studied GSI-α is that from the model Gram-positive bacterium Bacillus subtilis (23–32). Interestingly, despite the fact that GSI-α and GSI-β share 35–41% sequence identity, their mechanisms of regulation are distinct. GSI-β enzymes are regulated by adenylylation of an active site tyrosine, whereas GSI-α activity is subject to feedback inhibition by the product, Gln, and to a lesser extent, AMP (23, 32–36).
Notably, the GS·Gln feedback-inhibited form of B. subtilis GSI-α performs an unexpected role in controlling the DNA-binding activity of two global regulatory factors, GlnR and TnrA (29–31). GlnR and TnrA have similar N-terminal DNA binding domains with putative Mer-like motifs; however, they have distinct C-terminal domains, which are targeted by GS·Gln. GS·Gln forms a complex with TnrA that prevents it from binding DNA, hence shutting off transcription of genes encoding nitrogen catabolic and scavenging enzymes (29). By contrast, GS·Gln acts as a chaperone to stabilize GlnR·DNA complexes, which allows it to repress expression of genes such as glnA (encoding GS) (31). Thus, B. subtilis GS serves as an enzyme, a chaperone, and a DNA binding coeffector. To gain insight into the functions of this unique, multitasking GS protein, we obtained structures of all GS catalytic and regulatory states and performed biochemical and in vivo studies.
EXPERIMENTAL PROCEDURES
Protein Purification, Crystallization, and Structure Determination
An artificial gene (codon optimized for E. coli expression) encoding the B. subtilis GS was obtained from Genscript Corp. (Piscataway, NJ) and subcloned into pET15b for protein expression. Gel filtration of GS species indicated a dodecameric oligomer. The His tag was removed for all structural and biochemical studies. Crystals were grown via hanging drop vapor diffusion at room temperature. B. subtilis apo-GS crystals were obtained by mixing the protein (40 mg/ml) at a 1:1 ratio with 40% 4-methyl-2,4-pentanediol and 200 mm MgSO4 and inverting the drop over the reservoir solution. These crystals contain a dodecamer in the crystallographic asymmetric unit (ASU). To produce GS·glutamate·AMPPCP crystals, glutamate and AMPPCP were added to GS (at 40 mg/ml) to final concentrations of 5 mm, and the solution was mixed 1:1 with a reservoir of 15% PEG 8000, 0.1 m Hepes, pH 7.5, and 10 mm MgCl2. These crystals contain a hexamer in the ASU, and the dodecamer is generated via symmetry.
GS·Met-Sox-P·ADP was produced by mixing GS with 5 mm MgCl2, 5 mm ATP, and 5 mm l-methionine-S-sulfoximine (Met-Sox) for 1 h. This solution was then combined 1:1 with a crystallization reagent of 10% PEG 4000, 0.1 m Hepes, pH 7.5. These crystals contain a dodecamer in the ASU. Two crystal forms of GS·Gln were obtained. Form 1 was grown by mixing the GS·glutamine complex 1:1 with 40% 4-methyl-2,4-pentanediol and 100 mm MgCl2. Form 2 was obtained using 10% PEG 8000, 0.1 m Tris, pH 8.0, 5 mm MgCl2 as a crystallization solution. Form 1 contains a dodecamer in the ASU, whereas form 2 contains a hexamer (with the dodecamer generated by crystal symmetry). All x-ray intensity data were collected at 100 K at the Advanced Light Source beamline 8.3.1. The space group and cell parameters of each crystal are shown in Tables 1 and 2. Phases were determined first for the apo-GS form by molecular replacement, using the M. tuberculosis GS dodecamer as the search model. Several nonconserved loops were removed from the search model before molecular replacement. Refinement was carried out using the Crystallography and NMR Software package (CNS) and PHENIX (37, 38). The models were all constructed with the modeling program O (39).
TABLE 1.
Selected crystallographic data for GS structures
| Structure | Apo-GS | GS·glutamate·AMPPCP | GS·transition state |
|---|---|---|---|
| Resolution (Å) | 120.2–3.10 | 120.4–2.87 | 117.1–2.58 |
| Space group | P1 | C2221 | P1 |
| Cell constants (Å) | a = 110.0 | a = 138.9 | a = 110.0 |
| b = 138.4 | b = 240.9 | b = 141.6 | |
| c = 138.7 | c = 207.6 | c = 142.1 | |
| Cell constants (degrees) | α = 119.8 | α = 90.0 | α = 60.3 |
| β = 90.2 | β = 90.0 | β = 67.4 | |
| γ = 93.9 | γ = 90.0 | γ = 76.2 | |
| Overall Rsym (%)a | 11.5 (43.6)b | 5.0 (30.9) | 5.76 (20.1) |
| Overall I/σ(I) | 12.1 (1.8) | 13.8 (2.5) | 12.2 (3.4) |
| No. of total reflections | 186475 | 239473 | 364805 |
| % Complete | 97.8 (98.2) | 92.1 (92.0) | 90.0 (90.4) |
| Refinement statistics | |||
| Rwork/Rfree (%)c | 21.7/26.7 | 19.5/23.6 | 20.3/23.2 |
| r.m.s. deviation | |||
| Bond angles (degrees) | 1.30 | 1.33 | 1.12 |
| Bond lengths (Å) | 0.008 | 0.010 | 0.022 |
a Rsym = ΣΣ|Ihkl − Ihkl(j)|/ΣIhkl, where Ihkl(j) is observed intensity, and Ihkl is the final average value of intensity.
b Values in parentheses are for the highest resolution shell.
c Rwork = Σ‖Fo| − |Fc‖/Σ|Fo| and Rfree = Σ‖Fo| − |Fc‖/Σ|Fo|, where all reflections belong to a test set of 5% randomly selected data.
TABLE 2.
Selected crystallographic data for GS-Gln structures
| Form 1 (P1) | Form 2 (C2) | |
|---|---|---|
| Structural data | ||
| Resolution (Å) | 119.0–2.95 | 61.9–2.90 |
| Space group | P1 | C2 |
| Cell constants (Å) | a = 112.0 | a = 209.0 |
| b = 136.5 | b = 138.9 | |
| c = 137.7 | c = 144.7 | |
| Cell constants (degrees) | α = 119.8 | α = 90.0 |
| β = 90.2 | β = 125.7 | |
| γ = 93.4 | γ = 90.0 | |
| Overall Rsym (%)a | 10.3 (37.3)b | 8.0 (37.7) |
| Overall I/σ (I) | 12.2 (2.0) | 13.3 (2.1) |
| No. of total reflections | 274,737 | 175,345 |
| Percentage complete | 96.1 (96.0) | 98.8 (91.6) |
| Refinement statistics | ||
| Rwork/Rfree (%)c | 20.0/25.8 | 19.6/25.6 |
| r.m.s. deviation | ||
| Bond angles (degrees) | 1.12 | 1.20 |
| Bond lengths (Å) | 0.022 | 0.009 |
a Rsym = ΣΣ|Ihkl − Ihkl(j)|/ΣIhkl, where Ihkl(j) is observed intensity and Ihkl is the final average value of intensity.
b Values in parentheses are for the highest resolution shell.
c Rwork = Σ‖Fo| − |Fc‖/Σ|Fo| and Rfree = Σ‖Fo| − Fc‖/Σ|Fo|, where all reflections belong to a test set of 5% randomly selected data.
Enzyme, Kinetic, and in Vivo Assays
The glnA(R62A) mutant was constructed, and overexpression and purification of the mutant R62A GS were performed as described previously (40). Methods for the cultivation of bacteria in the minimal medium of Neidhardt et al. (41) have been described previously (42). All cultures contained 0.5% glucose, whereas the final concentration of all nitrogen sources was 0.2%. β-Galactosidase was assayed in crude extracts prepared from cells grown to mid-log growth phase (42). The reported β-galactosidase levels were corrected for the endogenous activity present in cells containing the promoterless lacZ fusion vectors integrated into the amyE gene. One unit of β-galactosidase activity produced 1 nmol of o-nitrophenol min−1. The kinetic constants for glutamate, ATP, and hydroxylamine were measured by assaying the production of γ-glutamylhydroxamate (32). The ammonium Km values were determined by measuring the production of inorganic phosphate that resulted from the hydrolysis of ATP as described (43). The inhibition of the Mg2+-dependent biosynthetic reaction by glutamine, AMP, and Met-Sox were determined with glutamate and ATP concentrations of 150 and 18 mm, respectively.
Cooperativity Assays
To assay for GS cooperativity, mixed GS oligomers were produced. The two proteins that were used to produce the mixed dodecamer were the WT, active protein and an inactive GS(E304A/A305G) mutant. The mutant also contained an N-terminal 35 residue extension, which included a His tag, followed by a tobacco etch virus cleavage site and a hydrophilic region. This extension permitted the detection of mixed dodecamer populations by SDS-PAGE. The GS(E304A/A305G) protein was expressed as for WT and purified via Ni-NTA chromatography. The N-terminal His tag of WT GS was cleaved via thrombin, and the cleaved protein was isolated as the flow-through of a Ni-NTA column. This untagged WT protein was then mixed with the His-tagged GS(E304A/A305G) protein. The mixed sample was dialyzed in buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, and 2 m urea for 12 h. This urea treatment resulted in dodecamer dissolution and monomer formation. The sample was then dialyzed into a buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, and 5 mm MgCl2 for 16 h with buffer changes at 4 and 8 h to allow dodecamer formation. The sample was subjected to Ni-NTA purification; the imidazole-eluted fractions were isolated and treated with tobacco etch virus protease to remove the His tag from the GS(E304A-A305G) protein. The digested sample was dialyzed into buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, and 5 mm MgCl2 overnight and applied to a Ni-NTA column to remove any His tag peptide and uncleaved protein. The samples were run on SDS-PAGE to ensure that both proteins were present. The enzymatic activities of the mixed dodecamer were measured by following the production of γ-glutamylhydroxamate (19). The Mg2+-dependent forward reaction was assayed in a final volume of 0.5 ml containing 0.05 m imidazole, pH 7.0, 100 mm sodium glutamate, 40 mm hydroxylamine, 100 mm MgCl2, and 18 mm ATP. The Mn2+-dependent forward reaction was measured in a final volume of 0.5 ml containing 0.05 m imidazole, pH 7.0, 100 mm sodium glutamate, 40 mm hydroxylamine, 7.5 mm MnCl2, and 7.5 mm ATP. The Mn2+-dependent reverse (transferase) reaction was assayed in 0.5 ml containing 20 mm sodium arsenate, 50 mm hydroxylamine, 40 mm glutamine, 2 mm ADP, 25 mm Tris, pH 7.5, 0.33 mm MnCl2, and 0.4 m K2SO4. The purified enzymes were pretreated with 0.4 m K2SO4 before addition to the transferase reaction buffer (19). For the biosynthetic reactions, 1 unit of GS activity corresponds to the ATP-dependent formation of 1 nmol of γ-glutamylhydroxamate min−1. In the transferase reaction, 1 unit of GS activity is the ADP-dependent formation of 1 nmol of γ-glutamylhydroxamate min−1.
Circular Cichroism (CD) and ITC Studies
CD measurements in the far UV regions (190–260 nm) were performed with pre- and post-urea-treated WT GS, GS(E304A/A305G) and the mixed complex at 25 °C with a 1-mm path length. Each sample solution contained 300 mm NaF and 10 mm Tris, pH 7.5. The temperature of the sample was controlled with a PTC-348 WI Peltier block. ITC experiments were performed using a VP-ITC system with GS loaded in the sample cell and either AMP or AMPPCP loaded in the syringe. The nucleotides were titrated into the sample cell with GS at 25 °C, and the resulting isotherms were fitted with the Origin software.
RESULTS AND DISCUSSION
Structure Determinations of B. subtilis GS and Its Complexes
To deduce the structural mechanism of GSI-α catalysis and its regulation, de novo crystals were obtained of the apo-, substrate-bound, glutamine-bound, and transition state complex (Tables 1 and 2). The transition state structure was obtained by reacting GS with ATP and Met-Sox (see “Experimental Procedures”). GS phosphorylates Met-Sox in the presence of ATP to form the transition state analog, Met-Sox-P, which causes irreversible inhibition of the enzyme (44). The B. subtilis apo-GS structure was solved first by molecular replacement using the M. tuberculosis GSI-β structure, which is 41% identical to B. subtilis GS (see “Experimental Procedures”). The structure, refined to Rwork/Rfree values of 21.7%/26.7% 3.1 Å (Table 1), contains 444 residues/subunit and is composed of 19 β-strands and 15 α-helices. The subunit can be divided by helix α3 into a larger C-terminal domain and a smaller, N-terminal region (Fig. 1A). Two hexameric rings are formed from six GS subunits and stack to form a dodecamer (Fig. 1, B and C). The hexameric rings are held together primarily by interactions between C-terminal helices α14 and α15 and α14′ and α15′ (where a prime indicates the neighboring subunit).
Each subunit has the classical GS bifunnel structure, whereby key residues that mediate catalysis are located on loops that encapsulate the active site (3, 45, 46) (Fig. 1A). The active site regions are mainly located in the C-terminal domain, with the N-terminal domain of the adjacent subunit contributing one loop. Previous studies on GSI-β enzymes led to the designation of key active site loops (8). Based on homology with GSI-β proteins, the corresponding GSI-α active site loops within the C-domain are the Glu flap (B. subtilis residues 301–306), the Tyr loop (residues 365–373), the Asn loop (residues 231–242), and the Tyr179 loop (residues 148–158). The N-terminal domain provides the Asp50′ loop (also called the “latch”), which contributes a key catalytic aspartic acid. In B. subtilis, this loop consists of residues 52–66, and the catalytic aspartic acid corresponds to Asp53′ (Fig. 1D).
Analysis of the Apo-GS Structure
The Glu flap and latch are arguably the most critical GS active site loops because they contribute residues that are central for catalysis. The aspartic acid from the Asp50′ loop abstracts a proton from ammonium to create ammonia, and the Glu flap harbors the catalytic base (Glu304 in B. subtilis GS), which abstracts a proton from the transition state to form glutamine and ADP. In the apo-GSI-α structure, the Glu flap is almost entirely disordered; only residues 301 and 306 are visible, and they display B-factors (average) > 100 Å2 (Table 3). Glu flap flexibility is important because it permits facile substrate entry and product release. The Tyr loop, which contains the tyrosine that is adenylylated in GSI-β enzymes, is also disordered. A corresponding tyrosine (Tyr373) is found in the B. subtilis GSI-α; however, studies have shown that it is not adenylylated, consistent with a different GSI-α regulatory mechanism (23). Aside from the Glu flap and Tyr loop, clear density is observed for the other active site regions in the GSI-α apo-structure, and with the exception of the Tyr179 loop, the catalytic regions of the GSI-α enzyme are similar in length to GSI-β proteins. In GSI-β enzymes, the Tyr179 loop contains 36 residues and extends from the active site into the central dodecamer channel, whereas in the GSI-α structure, this region consists of a short, 11-residue loop (Fig. 1, E and F) (3). When the Tyr179 region is excluded, the Cα atoms of the B. subtilis GS subunit can be superimposed onto those of S. typhimurium (Protein Data Bank entry 1f52) and M. tuberculosis (Protein Data Bank entry 2bvc) with root mean square (r.m.s.) deviations of 1.3 and 1.5 Å, respectively. The altered structures of the Tyr179 loop in GSI-α compared with GSI-β lead to different pore structures for these enzymes. The function(s), if any, of the pores, however, is not known (Fig. 1E).
TABLE 3.
B-Factor (average) for GS structure Glu flap residues and substrates
| Structure | Glu flap B(av) | Substrate B(av) |
|---|---|---|
| Å2 | Å2 | |
| Apo | >100 | NAa |
| Glutamate-bound | 85 | 86.7 |
| Transition state | 24.0 | 20.1 |
| Glutamine-bound | 35.9 | 33.0 |
a Not applicable.
GSI-α-Substrate and Transition State Structures Reveal That Large Conformational Changes Are Required for Catalysis
To gain insight into the enzymatic mechanism employed by GSI-α enzymes, we determined the structure of GSI-α bound to the substrates glutamate and AMPPCP (which is a non-hydrolyzable ATP analog) to 2.87 Å (Table 1). The substrate glutamates and AMPPCP molecules bind at each end of the bifunnel. The glutamate substrates are located under the Glu flap. Although better ordered than the apo-structure the Glu flap residues in this structure, as well as the glutamate substrates, display high B-factors (B-factors (average) = 86.7 Å2 for the glutamate substrates and 85.0 Å2 for Glu flap residues) (Table 3). This suggests that the active site may become protected by Glu flap closure only during catalysis. The glutamate substrate peptide carboxyl and amide groups interact with Arg298 and His245, whereas its side chain carboxyl interacts with a Mg2+ ion. The adenine moiety of the AMPPCP is sandwiched between the side chains of Arg331 and Tyr201, whereas hydrogen bonds to the adenine N6, N1, and N7 atoms are provided by the carbonyl oxygen of Ile328 and the side chains of Ser249 and Ser329. Interestingly, the phosphate groups of AMPPCP point away from the active site and are only contacted weakly by Lys44. In fact, density is only observed for the α- and β-phosphates of the AMPPCP molecules. Hence, AMPPCP does not appear to bind GS productively, which is consonant with ITC data showing that GS binds AMPPCP with 100-fold lower affinity than AMP (Fig. 2). Comparisons of the substrate-bound and apo-GSI-α structures revealed that not only are the subunits and their corresponding active sites essentially unchanged upon substrate binding, but the dodecameric arrangement remains unaffected as well (superimposition of Cα atoms = 0.50–0.60 Å/subunit and 0.60 Å for all Cα atoms of the dodecamers) (Fig. 3, A–D).
FIGURE 2.

Isothermal titration calorimetry thermograms of AMP and AMPPCP binding to GS. A, AMP binding to GS. Kd = 7.5 ± 0.3 μm. B, AMPPCP binding to GS. Kd = 667 ± 21 μm.
FIGURE 3.

B. subtilis GS active site architecture, substrate binding, and transition state conformational changes. A, close-up of key active site loop regions in the apo-GS (cyan) and glutamate-bound GS structures (yellow). The apo-Glu flap is mostly disordered, whereas in the glutamate-bound structure, the Glu flap backbone atoms are resolved. No large structural changes are noted between key loops. B, initial Fo − Fc map of the GS·transition state complex contoured at 3.5 σ calculated before the addition of Met-Sox-P, ADP, and metal ions (to 2.58 Å). C, close-up of the active site regions around key loops (as in A) after superimposition of the apo-state (cyan) and transition state (magenta) complexes. Large structural changes in the loops occur upon transition state formation, especially in the Asp50′ loop. D, superimposition of the apo-GS dodecamer (cyan) onto the glutamate·AMPPCP-bound (yellow) and Met-Sox-P·ADP-bound (magenta) forms. E, stereo view showing superimposition of the substrate-bound form of GS (yellow) and the transition state complex (magenta), highlighting critical active site loop rearrangements that provide proper active site architecture for catalysis during formation of the transition state. Most notable are the complete ejection of Arg62′ and Glu65′, which permit adjustment of Asp53′ and Arg316 into the pocket for catalysis. The extent of the structural changes is indicated by arrows and distances in Å.
Key active site residues are conserved in all classes of GS, suggesting a similar overall catalytic mechanism. According to this reaction scheme, ATP and glutamate first bind in the active site at each end of the bifunnel. The bound ATP γ-phosphate is polarized by the metal ions and an arginine residue (corresponding to B. subtilis GSI-α Arg316), which facilitates nucleophilic attack by the bound glutamate substrate to yield γ-glutamyl phosphate (Fig. 3A). The next step in the reaction involves proton abstraction of ammonium by the Asp50′ aspartic acid to create the ammonia. The ammonia then reacts with the γ-glutamyl phosphate to form the tetrahedral transition state. The final step is dissociation of the phosphate from the intermediate and concomitant abstraction of the proton from the γ-amino group of the intermediate by the Glu flap glutamate (B. subtilis Glu304).
GSI-β structures have defined the residues and specific active site architecture optimal for catalysis (3). At first glance, the minimal shifts in the active site regions of the apo- and substrate-bound B. subtilis GS structures appear to suggest that GSI-α catalysis may also involve only moderate active site loop movements. However, close examination of the apo- and substrate-bound GSI-α active sites reveal that they lack an optimal organization of catalytic residues. This is largely due to the deep insertion of the latch, in particular residues Arg62′ and Glu65′, into the neighboring active site of each subunit. Arg62′ blocks and prevents the correct positioning of the catalytic Asp53′ as well as Ser56′, which stabilizes the Glu flap. Moreover, the side chain of Glu65′ overlaps the location of the key catalytic arginine, Arg316, which assists in phosphoryl transfer. In fact, in the apo- and substrate-bound GSI-α structures, the side chain of Arg316 is located entirely outside the active site.
The non-optimal active site organization in the apo- and substrate-bound GSI-α structures suggests that either a different catalytic mechanism may be at play in GSI-α and GSI-β proteins or that significant structural changes must occur during catalysis. To address this question, we generated a GSI-α·transition state complex by reacting the protein with Met-Sox, ATP, and Mg2+ and crystallized and solved the structure to Rwork/Rfree values of 20.3%/23.2% to 2.58 Å (see “Experimental Procedures”) (Table 1). Met-Sox-P mimics the GS transition state as its methyl group occupies the ammonium substrate binding site (8). Thus, the Met-Sox-P adduct forms a stable complex with the enzyme that precludes the last step, which is release of the phosphate from the intermediate and concomitant abstraction of the proton from the intermediate γ-amino group by Glu flap residue Glu304. The structure revealed that the expected catalysis had occurred, leading to Met-Sox-P and ADP creation (Fig. 3B).
GS enzymes all utilize two Mg2+ ions to aid substrate binding, transfer of the phosphate group, and structural stability. In the GSI-α·transition state structure, a third metal ion is found, which probably functions in neutralizing the negative charge arising from the intermediate. A similarly located metal ion was identified in the M. tuberculosis GSI-β·Met-Sox-P and maize GSII·phosphinothricin structures (5, 8). Contacts to the adenine and ribose moieties of ADP in the transition state complex are essentially identical to those made to AMPPCP. In the transition state complex, however, the phosphate groups are rotated into the active site near the Met-Sox-P and are contacted by Arg321 and Arg331 as well as a Mg2+ ion. The β-phosphate lies close to the phosphate transferred to Met-Sox to form Met-Sox-P. There are numerous contacts from GS to the Met-Sox-P. Arg298 interacts with the carboxyl moiety, and the Met-Sox-P amide nitrogen interacts with the Glu134 side chain as well as the carbonyl of Gly241. The Met-Sox-P phosphate moiety is surrounded by multiple basic residues, including Arg316, Arg321, Arg335, and His245 (Figs. 3,C–E, and 4). Glu flap residue Glu304 contacts Asp53′ and is within close distance to the Met-Sox-P methyl group. The Glu304-Asp53′ interaction probably helps to stabilize and shield the Met-Sox-P from attack by bulk solvent as well as facilitating proton abstraction of the ammonium to form ammonia. The resultant ammonia is then in an ideal position to attack the γ-glutamyl phosphate intermediate. Hence, the GSI-α·Met-Sox-P·ADP complex mimics the tetrahedral adduct before the generation of glutamine and inorganic phosphate products.
FIGURE 4.
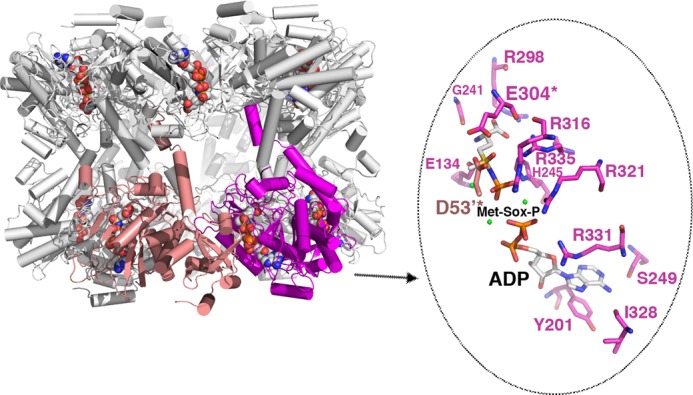
Structure of the GSI-α Met-Sox-P·ADP complex and close-up of contacts to the transition state. The structure of the dodecamer is displayed as ribbons with one subunit colored magenta and the neighboring subunit colored salmon. The Met-Sox-P and ADP molecules in each active site are shown as CPK. Note that there is a dodecamer in the crystallographic asymmetric unit, and each active site (formed between subunits) has generated the identical transition state with the same contacts. One active site (between the magenta and salmon subunits) is highlighted, and key residues that contact Met-Sox-P and ADP are shown as sticks (enlarged view to the right). The key catalytic residues Asp53 and Glu304 are labeled and highlighted with asterisks. Magnesium ions are colored green.
Comparison of the transition state structure with the apo- and substrate-bound forms revealed dramatic conformational changes, consonant with the finding that the residues in the transition state structure are optimally positioned for catalysis. The largest structural changes are located on the active site loop regions (r.m.s. deviations of apo-/substrate-bound versus Met-Sox-bound subunits = 1.8–2.0 Å, whereas the r.m.s. deviations are 0.8 Å when the active site loops are not included). Moreover, superimposition of the transition state dodecamer onto the apo- or substrate-bound dodecamers results in r.m.s. deviations of 2.5–3.0 Å, indicating that the catalytically induced structural changes are transmitted between subunits, leading to significant alterations in the overall dodecamer structure (Fig. 3D). Although all of the GSI-α active site loops are altered upon transition state formation, residues in the Asp50′ loop undergo the most dramatic structural rearrangements (r.m.s. deviations = 3.7–4.0 Å for superimposition of the Asp50′ loops of the transition state and glutamate-bound structures). As noted, the Asp50′ loop is wedged into the active site in apo- and substrate-bound structures, preventing an optimal configuration of catalytic residues. The restructuring of the active site that takes place during catalysis results in a functional arrangement of catalytic residues (Fig. 3, C and E). These extensive structural changes include the unfolding of one turn of the helix from residue 53 to 59. The structural changes in the Asp50′ loop lead to the expulsion of Glu65′ and Arg62′ more than 10 Å from the active site to the solvent-exposed surface of the dodecamer (Fig. 3E). Concomitantly, the Arg316 loop moves by ∼10 Å, resulting in relocation of the Arg316 side chain into the position vacated by Glu65′. These alterations create an active site organization that allows GSI-α to employ the same general catalytic mechanism used by all characterized GS proteins (3).
Demonstration that B. subtilis GS Is Cooperative Using Mixed Oligomers
The structural changes that take place during GSI-α catalysis are found in all the active sites and result in an altered dodecamer structure, suggesting that the enzyme may be cooperative, whereby binding and catalysis at one active site is affected by adjacent subunits. Such cooperativity has been indicated by previous studies revealing active site coupling with E. coli GS (45). However, to directly assess GSI-α cooperativity, we developed a mixed dodecamer assay. Previous studies showed that single E304A and A305G mutations abrogated GS activity (40). Thus, in this assay, WT GS subunits were mixed with inactive, double GS(E304A/A305G) mutant subunits in the presence of 2 m urea, allowing subunit exchange. The mixture was then dialyzed into urea free buffer resulting in the formation of mixed oligomers. 2 m urea treatment did not cause denaturation as assessed by circular dichroism but broke up subunit-subunit interactions (see “Experimental Procedures”) (Fig. 5). To assay possible effects of urea treatment on GS function, we first tested the activity of WT GS and WT GS that had undergone the identical urea treatment as the mixed oligomer. As shown in Table 4, this procedure had no effect on GS enzyme activity. If GSI-α is not cooperative, a mixed WT GS·GS(E304A/A305G)-containing dodecamer should display either WT or only slightly reduced enzyme activity. By contrast, if the dodecamer is cooperative, then incorporation of even a few GS(E304A/A305G) subunits into the dodecamer should impair or abrogate activity for a cooperative enzyme. The mixture used in the assay depicted in Fig. 5 contained between a 3- and 4-fold excess of the WT protein (lower molecular weight band) compared with the mutant. Thus, this experiment provided a strong test case of cooperativity. Even under these conditions, where there is far less mutant than WT, the activity was abrogated, supporting the hypothesis that the dodecamer is cooperative (Table 4).
FIGURE 5.
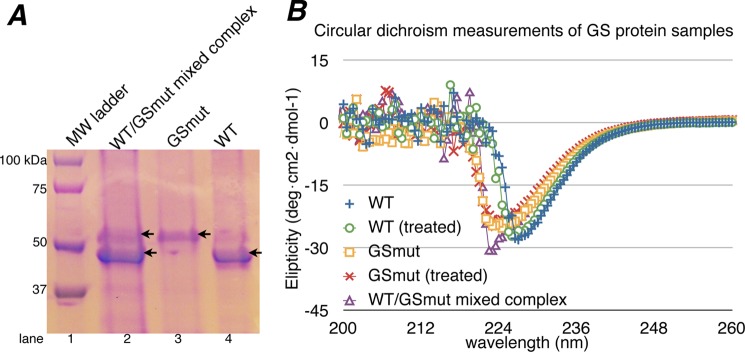
A, SDS-PAGE of GS proteins used for mixed dodecamer/cooperativity assays. Lanes 3 and 4, the E304A/A305G and WT protein samples, respectively. Lane 2, WT GS/GS(E304A-A305G) mixed complex after urea treatment, urea removal, and purification. B, circular dichroism measurements of GS proteins before and after urea treatment. Overall, the spectrum of treated and untreated WT GS, GS(E304A/A305G), and the mixed complex are indicative of well folded α-β proteins (note the peaks in the 220–230 nm range). This demonstrates that urea treatment did not affect the secondary structures or overall folds of the GS proteins.
TABLE 4.
Glutamine synthetase cooperativity assay
| Enzyme | Glutamine synthetase-specific activity |
||
|---|---|---|---|
| Forward reaction (biosynthetic) |
Reverse reaction (transferase)a | ||
| Mg2+-dependenta | Mn2+-dependenta | ||
| WT | 0.406 ± 0.035 | 0.187 ± 0.009 | 9.555 ± 0.490 |
| WT (treated)b | 0.413 ± 0.019 | 0.193 ± 0.012 | 9.643 ± 0.422 |
| GS(E304A/A305G) | 0.003 ± 0.001 | 0.005 ± 0.004 | 0.029 ± 0.007 |
| GS(E304A/A305G) (treated)b | 0.003 ± 0.001 | 0.005 ± 0.002 | 0.038 ± 0.014 |
| WT-E304A/A305G mixed complex | 0.005 ± 0.001 | 0.006 ± 0.004 | 0.037 ± 0.012 |
a 1 unit of GS activity corresponds to the ATP/ADP-dependent formation of 1 nmol of γ-glutamylhydroxamate min−1.
b Samples were treated with 2 m urea then dialyzed to remove urea (see “Experimental Procedures”).
Molecular Basis for GSI-α-specific Gln Feedback Inhibition
GSI-α enzymes are distinct from their GSI-β counterparts in that they are feedback-inhibited by the reaction product, Gln. Previous GSI-β-Gln structures showed no interactions between Gln and side chains of the Glu flap, and the bound glutamine and Glu flap were largely disordered in the structure (3). Indeed, this Glu flap flexibility was suggested as being important in facile product release following catalysis. The finding that GSI-α enzymes are feedback-inhibited by Gln suggests that differences must exist in GSI-Gln isoenzyme structures. It also indicates that important distinctions must be present between GSI-α-Glu and GS-α-Gln structures. Understanding the structural basis for GSI-α-Gln feedback inhibition is crucial, because this form of the enzyme serves as a key nexus in the Bacilli nitrogen regulation pathway (29–31). Thus, we next determined structures of B. subtilis GS·Gln-Mg2+ complexes. Two structures were obtained using different crystallization conditions (see “Experimental Procedures”) (Table 2). The two structures are identical and reveal that Gln binds within the Glu flap pocket in a similar location as the substrate glutamate (Fig. 6A). However, there are striking differences between the Gln-bound and Glu-bound states. Unlike the Glu-bound state, in the Gln-bound structure, both the side chain and backbone atoms of Glu flap residues are well ordered; the B-factors (average) for Glu flap residues in the Gln-bound structure are 35.9 Å2 compared with 85.0 Å2 for the glutamate-bound form, and the B-factors (average) for the glutamines are 33.0 Å2 (Table 3). In fact, although not directly comparable due to their differing resolutions, the B-factors of the Glu flap residues in the Gln-bound structures are on par with those in the transition state structure (which have B-factors (average) = 24.0 Å2 for Glu flap residues and 20.0 and 20.3 Å2 for ADP and Met-Sox-P, respectively) (Table 3).
FIGURE 6.
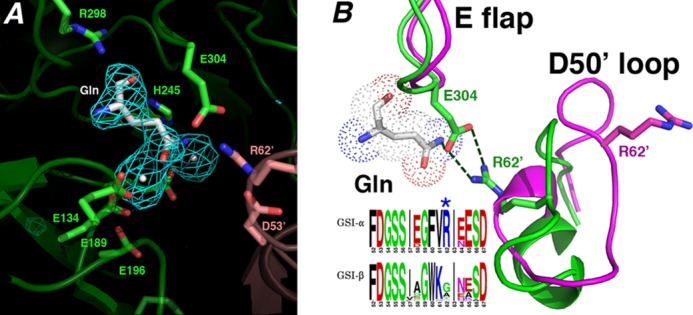
Structural basis for GSI-α-specific Gln feedback inhibition. A, initial Fo − Fc map calculated before the addition of glutamine and metal ions and contoured at 3.2 σ (to 2.95 Å). B, superimposition of active site loops of the glutamine-bound state (green) and the transition state complex (magenta). The Gln-bound active site loops adopt structures similar to apo- and substrate-bound except that the Glu flap (E flap) is well ordered, and the Glu flap residue Glu304 hydrogen-bonds to the Gln as well as the GSI-α-specific residue from the Asp50′ loop (D50′ loop), Arg62′. Notably, GSI-β enzymes are not feedback-inhibited by Gln. As shown by the logo, GSI-β enzymes all lack the key Arg62′ residue, explaining why they are not subject to Gln feedback inhibition (in the logo, residues are colored according to type, basic in blue, acidic in red, hydrophobic in black, and glycine/serine in green).
In the GSI-α-Gln structures, the Asp50′ loop adopts the same conformation as that observed in the apo- and substrate-bound GSI-α forms (Figs. 6B and 7). Remarkably, however, in the GS-Gln structure, Arg62 plays a central role in Gln binding and, hence, Gln feedback inhibition. Specifically, the Arg62′ side chain hydrogen-bonds with the key catalytic residue from the Glu flap, Glu304 (Fig. 6B). The stabilized Glu304 also makes a tight hydrogen bond to the glutamine Nϵ atom. This intimate hydrogen bond network ties the Glu flap closed, explaining its well ordered state in this structure (Fig. 6B). The vital role played by Arg62 in Gln feedback inhibition probably explains why, unlike GSI-β enzymes, the GSI-α Asp50′ loop adopts distinct structures in different states. Asp50′ loop insertion into the active site in GSI-α is critical for Gln feedback inhibition, but it can then be readily ejected in the absence of the Gln-specific hydrogen bond network to allow catalysis. In this way, the Asp50′ loop acts as a GSI-α-specific regulatory trigger.
FIGURE 7.
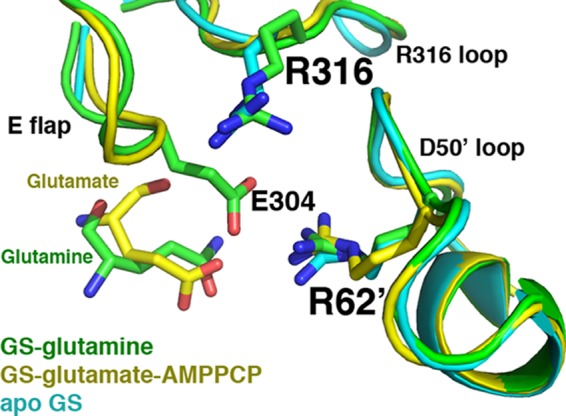
The GS·glutamine structure active site is similar to that of the apo- and substrate-bound forms. Superimposition of the apo-, glutamate-AMPPCP, and glutamine-bound GSI-α structures, focusing on key active site loops. The apo-structure is colored cyan, the glutamate-AMPPCP-bound structure is yellow, and the glutamine-bound structure is green. The active site loops adopt essentially the same conformation. The Glu flap in the apo-state is mostly disordered, whereas backbone atoms of the Glu flap in the substrate-bound state are visible (but not side chains), and in the glutamine-bound form, the Glu flap is as well ordered as in the transition state.
The finding that Glu304 directly interacts with Gln provides a molecular explanation for previous studies showing that E304D or E304A mutants were defective in Gln feedback inhibition (40). The intricate Arg62-Glu304-Gln hydrogen bond linkage also explains why GSI-α, but not GSI-β, enzymes are feedback-inhibited by Gln because sequence alignments of GS proteins show that Arg62 is completely conserved in GSI-α enzymes but not GSI-β enzymes, where this position is typically Gly or Ala (Fig. 6B). Thus, these structures demonstrate that B. subtilis GS uses a residue found only in GSI-α enzymes, Arg62, along with a conserved catalytic residue (Glu304) to mediate glutamine binding. The structures also reveal the basis for Gln feedback inhibition, because the Arg62′-Glu304-Gln hydrogen bond network locks the Glu flap in a closed state, preventing Gln release, substrate binding and catalysis.
The Role of Arg62 in GS Activity and Regulation; Kinetic, Enzymological, and in Vivo Studies
Our structures indicate that Arg62 plays a key role in GSI-α-specific Gln feedback inhibition. The structures also suggest that Arg62 is not involved in catalysis and, in fact, must be ejected from the active site during transition state formation. To test these structure-based hypotheses, an R62A mutant was constructed. The purified R62A enzyme had only small differences (less than 2-fold) in its steady-state kinetic constants compared with WT, indicating that it does not play a significant role in substrate binding or catalysis, consistent with our structural data (Table 5). Unlike the E304A substitution in GS, the R62A replacement did not increase the Km for ammonium (Table 5). This result is again consistent with the structural data and argues that Arg62 is not part of the ammonium substrate binding site. In contrast to E304A, which is highly resistant to Met-Sox inhibition, the R62A enzyme has a sensitivity to Met-Sox that is similar to that of WT GS (Table 6). These data, again, support the idea that Arg62 is not involved in the GS reaction mechanism. Strikingly, however, and consistent with our structural findings, the R62A mutant was highly resistant to Gln feedback inhibition (Table 6). The R62A substitution was also found to increase the resistance to AMP inhibition. We have argued previously that there is an interdependence between the glutamate (glutamine) and ATP (AMP) binding sites (40). The activities of the nitrogen transcription factors TnrA and GlnR are regulated by the feedback-inhibited form of GS (29–31). TnrA- and GlnR-regulated genes were shown previously to be expressed constitutively in mutants encoding feedback-resistant GS enzymes (29–31). The contribution of the GS Arg62 residue to the in vivo regulation of TnrA and GlnR was assessed by constructing a B. subtilis mutant containing an R62A substitution in the chromosomal GS gene (glnA) (see “Experimental Procedures”) (40). Gene regulation by TnrA and GlnR was abolished in cells encoding GS with an R62A mutation (Table 7). These results indicate that the glnA(R62A) mutant has the in vivo phenotypes expected for a strain encoding for feedback-resistant GS enzyme.
TABLE 5.
Enzymatic constants for wild-type and mutant GS proteins
The kinetic constants were determined for the Mg2+-dependent biosynthetic reaction. All assays were performed at least twice. The uncertainty is the S.E. from the nonlinear regression analysis of the data.
| Enzyme |
Km |
Vmax | |||
|---|---|---|---|---|---|
| Glutamate | ATP | Ammonium | Hydroxylamine | ||
| mm | μmol/min/mg | ||||
| Wild type | 27 ± 2 | 2.4 ± 0.1 | 0.18 ± 0.03 | 0.83 ± 0.07 | 3.7 ± 0.2 |
| R62A | 14 ± 2 | 2.3 ± 0.3 | 0.34 ± 0.05 | 1.4 ± 0.2 | 2.9 ± 0.1 |
| E304A | 3.4 ± 0.2 | 1.2 ± 0.1 | 32 ± 2 | 0.68 ± 0.09 | 1.3 ± 0.1 |
TABLE 6.
Sensitivities of wild-type and mutant GS proteins to inhibition
The IC50 is the inhibitor concentration that reduces enzymatic activity by 50%. The Mg2+-dependent biosynthetic reaction was used to measure the inhibition. All values are the averages of at least two determinations. The S.E. values were less than 10% for all values. The values for the wild-type and E304A enzymes are taken from Ref. 40.
| Enzyme | IC50 |
||
|---|---|---|---|
| Glutamine | AMP | Met-Sox | |
| mm | |||
| Wild type | 2.4 | 0.5 | 0.13 |
| R62A | >140 | 20 | 0.19 |
| E304A | >140 | >30 | 26 |
TABLE 7.
TnrA- and GlnR-dependent gene regulation
β-Galactosidase activity was determined in extracts of cells harvested during exponential growth in glucose minimal medium containing glutamate plus casamino acids or glutamate as the respective nitrogen sources for excess and limiting nitrogen conditions. Each value is the mean of at least two independent determinations and is reproducible to within 10% (S.E.) of the mean value.
| Relevant genotype | TnrA-dependent regulation; β-galactosidase activity from an amtB-lacZ fusion |
GlnR-dependent regulation; β-galactosidase activity from a glnRA-lacZ fusion |
||
|---|---|---|---|---|
| Excess nitrogen | Limited nitrogen | Excess nitrogen | Limited nitrogen | |
| nmol/min/mg | nmol/min/mg | |||
| Wild type | 14 | 140 | 17 | 30 |
| glnA(R62A) | 100 | 83 | 38 | 34 |
In conclusion, we have captured all key catalytic and regulatory states of the B. subtilis GSI-α enzyme. Surprisingly, these structures reveal heretofore unseen large structural changes during catalysis. GSI-α enzymes are also distinct among GSs in being feedback-inhibited by the product, Gln. Remarkably, our work demonstrates that the catalytic and regulatory features of GSI-α are linked. Specifically, Gln feedback inhibition arises from a hydrogen bond network between Gln, the active site Glu304, and a GSI-α-specific residue, Arg62′, from an adjacent subunit. Notably, Arg62′ is ejected from the active site as part of the GSI-α active site reorganization during catalysis. These key Arg62 roles were supported by biochemical and in vivo studies; mutation of Arg62 to alanine eliminates Gln feedback inhibition but does not affect catalysis. The extensive conformational changes that take place during catalysis also suggest a cooperative catalytic mechanism, which was supported by a mixed dodecamer enzyme assay. Due to its critical role in nitrogen metabolism, GS is an attractive and validated target for drug design against pathogenic bacteria. The isoenzyme-specific glutamine interaction network uncovered here presents a promising point of intervention in the development of highly specific inhibitors in the treatment of Gram-positive pathogenic bacteria.
Acknowledgments
We dedicate this work to our colleague and dear friend Dr. David Murray, who spearheaded the GS work as a graduate student and sadly passed away. We thank the Advanced Light Source (ALS) and their support staff. The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences, and Material Science Division of the United States Department of Energy at the Lawrence Berkeley National Laboratory.
This work was supported, in whole or in part, by National Institutes of Health Grants GM074815 (to M. A. S.) and GM051127 (to S. H. F.). This work was also supported by an M. D. Anderson Trust Fellowship and Burroughs Wellcome Career Development Award (to M. A. S.).
- GS
- glutamine synthetase
- ASU
- asymmetric unit
- AMPPCP
- adenosine 5′-(β,γ-methylene)triphosphate
- Ni-NTA
- nickel-nitrilotriacetic acid
- Met-Sox
- l-methionine-S-sulfoximine
- Met-Sox-P
- Met-Sox-phosphate
- ITC
- isothermal titration calorimetry
- r.m.s.
- root mean square.
REFERENCES
- 1. Magasanik B. (1993) The regulation of nitrogen utilization in enteric bacteria. J. Cell. Biochem. 51, 34–40 [DOI] [PubMed] [Google Scholar]
- 2. Pesole G., Bozzetti M. P., Lanave C., Preparata G., Saccone C. (1991) Glutamine synthetase gene evolution. A good molecular clock. Proc. Natl. Acad. Sci. U.S.A. 88, 522–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eisenberg D., Gill H. S., Pfluegl G. M., Rotstein S. (2000) Structure-function relationships of glutamine synthetases. Biochim. Biophys. Acta 1477, 122–145 [DOI] [PubMed] [Google Scholar]
- 4. Krajewski W. W., Collins R., Holmberg-Schiavone L., Jones T. A., Karlberg T., Mowbray S. L. (2008) Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 375, 217–228 [DOI] [PubMed] [Google Scholar]
- 5. Unno H., Uchida T., Sugawara H., Kurisu G., Sugiyama T., Yamaya T., Sakakibara H., Hase T., Kusunoki M. (2006) Atomic structure of plant glutamine synthetase. A key enzyme for plant productivity. J. Biol. Chem. 281, 29287–29296 [DOI] [PubMed] [Google Scholar]
- 6. van Rooyen J. M., Abratt V. R., Belrhali H., Sewell T. (2011) Crystal structure of the type III glutamine synthetase. Surprising reversal of the inter-ring interface. Structure 19, 471–483 [DOI] [PubMed] [Google Scholar]
- 7. Almassy R. J., Janson C. A., Hamlin R., Xuong N. H., Eisenberg D. (1986) Novel subunit-subunit interactions in the structure of glutamine synthetase. Nature 323, 304–309 [DOI] [PubMed] [Google Scholar]
- 8. Krajewski W. W., Jones T. A., Mowbray S. L. (2005) Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. U.S.A. 102, 10499–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nilsson M. T., Krajewski W. W., Yellagunda S., Prabhumurthy S., Chamarahally G. N., Siddamadappa C., Srinivasa B. R., Yahiaoui S., Larhed M., Karlén A., Jones T. A., Mowbray S. L. (2009) Structural basis for inhibition of Mycobacterium tuberculosis glutamine synthetase by novel ATP-competitive inhibitors. J. Mol. Biol. 393, 504–513 [DOI] [PubMed] [Google Scholar]
- 10. Liaw S. H., Eisenberg D. (1994) Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes. Biochemistry 33, 675–681 [DOI] [PubMed] [Google Scholar]
- 11. Liaw S. H., Jun G., Eisenberg D. (1994) Interactions of nucleotides with fully unadenylylated glutamine synthetase from Salmonella typhimurium. Biochemistry 33, 11184–11188 [DOI] [PubMed] [Google Scholar]
- 12. Liaw S. H., Kuo I., Eisenberg D. (1995) Discovery of the ammonium substrate site on glutamine synthetase, a third cation binding site. Protein Sci. 4, 2358–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gill H. S., Eisenberg D. (2001) The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry 40, 1903–1912 [DOI] [PubMed] [Google Scholar]
- 14. Brown J. R., Masuchi Y., Robb F. T., Doolittle W. F. (1994) Evolutionary relationships of bacterial and archaeal glutamine synthetase genes. J. Mol. Evol. 38, 566–576 [DOI] [PubMed] [Google Scholar]
- 15. Deuel T. F., Prusiner S. (1974) Regulation of glutamine synthetase from Bacillus subtilis by divalent cations, feedback inhibitors, and l-glutamine. J. Biol. Chem. 249, 257–264 [PubMed] [Google Scholar]
- 16. Midelfort C. F., Rose I. A. (1976) A stereochemical method for detection of ATP terminal phosphate transfer in enzymatic reactions. Glutamine synthetase. J. Biol. Chem. 251, 5881–5887 [PubMed] [Google Scholar]
- 17. Wedler F. C., Boyer P. D. (1972) Substrate binding and reaction intermediates of glutamine synthetase (Escherichia coli W) as studied by isotope exchanges. J. Biol. Chem. 247, 984–992 [PubMed] [Google Scholar]
- 18. Wedler F. C., Horn B. R. (1976) Catalytic mechanisms of glutamine synthetase enzymes. Studies with analogs of possible intermediates and transition states. J. Biol. Chem. 251, 7530–7538 [PubMed] [Google Scholar]
- 19. Deuel T. F., Stadtman E. R. (1970) Some kinetic properties of Bacillus subtilis glutamine synthetase. J. Biol. Chem. 245, 5206–5213 [PubMed] [Google Scholar]
- 20. Purich D. L. (1998) Advances in the enzymology of glutamine synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 72, 9–42 [DOI] [PubMed] [Google Scholar]
- 21. Deuel T. F., Ginsburg A., Yeh J., Shelton E., Stadtman E. R. (1970) Bacillus subtilis glutamine synthetase. Purification and physical characterization. J. Biol. Chem. 245, 5195–5205 [PubMed] [Google Scholar]
- 22. Harth G., Horwitz M. A. (1999) An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes. Extracellular proteins as potential novel drug targets. J. Exp. Med. 189, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fisher S. H. (1999) Regulation of nitrogen metabolism in Bacillus subtilis. Vive la difference! Mol. Microbiol. 32, 223–232 [DOI] [PubMed] [Google Scholar]
- 24. Fisher S. H., Sonenshein A. L. (1984) Bacillus subtilis glutamine synthetase mutants pleiotropically altered in glucose catabolite repression. J. Bacteriol. 157, 612–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schreier H. J., Brown S. W., Hirschi K. D., Nomellini J. F., Sonenshein A. L. (1989) Regulation of Bacillus subtilis glutamine synthetase gene expression by the product of the glnR gene. J. Mol. Biol. 210, 51–63 [DOI] [PubMed] [Google Scholar]
- 26. Wray L. V., Jr., Ferson A. E., Rohrer K., Fisher S. H. (1996) TnrA, a transcription factor required for global nitrogen regulation in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 93, 8841–8845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferson A. E., Wray L. V., Jr., Fisher S. H. (1996) Expression of the Bacillus subtilis gabP gene is regulated independently in response to nitrogen and amino acid availability. Mol. Microbiol. 22, 693–701 [DOI] [PubMed] [Google Scholar]
- 28. Fisher S. H., Wray L. V., Jr. (2002) Bacillus subtilis 168 contains two differentially regulated genes encoding l-asparaginase. J. Bacteriol. 184, 2148–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wray L. V., Jr., Zalieckas J. M., Fisher S. H. (2001) Bacillus subtilis glutamine synthetase controls gene expression through a protein-protein interaction with transcription factor TnrA. Cell 107, 427–435 [DOI] [PubMed] [Google Scholar]
- 30. Wray L. V., Jr., Fisher S. H. (2008) Bacillus subtilis GlnR contains an autoinhibitory C-terminal domain required for the interaction with glutamine synthetase. Mol. Microbiol. 68, 277–285 [DOI] [PubMed] [Google Scholar]
- 31. Fisher S. H., Wray L. V., Jr., (2008) Bacillus subtilis glutamine synthetase regulates its own synthesis by acting as a chaperone to stabilize GlnR-DNA complexes. Proc. Natl. Acad. Sci. U.S.A. 105, 1014–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wray L. V., Jr., Fisher S. H. (2005) A feedback-resistant mutant of Bacillus subtilis glutamine synthetase with pleiotropic defects in nitrogen-regulated gene expression. J. Biol. Chem. 280, 33298–33304 [DOI] [PubMed] [Google Scholar]
- 33. Reitzer L. (2003) Nitrogen assimilation and global regulation in Escherichia coli. Annu. Rev. Microbiol. 57, 155–176 [DOI] [PubMed] [Google Scholar]
- 34. Shapiro B. M., Kingdon H. S., Stadtman E. R. (1967) Regulation of glutamine synthetase. VII. Adenylyl glutamine synthetase. A new form of the enzyme with altered regulatory and kinetic properties. Proc. Natl. Acad. Sci. U.S.A. 58, 642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shapiro B. M., Stadtman E. R. (1968) 5′-adenylyl-O-tyrosine. The novel phosphodiester residue of adenylylated glutamine synthetase from Escherichia coli. J. Biol. Chem. 243, 3769–3771 [PubMed] [Google Scholar]
- 36. Mehta R., Pearson J. T., Mahajan S., Nath A., Hickey M. J., Sherman D. R., Atkins W. M. (2004) Adenylylation and catalytic properties of Mycobacterium tuberculosis glutamine synthetase expressed in Escherichia coli versus mycobacteria. J. Biol. Chem. 279, 22477–22482 [DOI] [PubMed] [Google Scholar]
- 37. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography and NMR system. A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 38. Afonine P. V., Grosse-Kunsteve R. W., Echols N., Head J. J., Moriarty N. E., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 40. Wray L. V., Jr., Fisher S. H. (2010) Functional roles of the conserved Glu304 loop of Bacillus subtilis glutamine synthetase. J. Bacteriol. 192, 5018–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neidhardt F. C., Bloch P. L., Smith D. F. (1974) Culture medium for enterobacteria. J. Bacteriol. 119, 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Atkinson M. R., Wray L. V., Jr., Fisher S. H. (1990) Regulation of histidine and proline degradation enzymes by amino acid availability in Bacillus subtilis. J. Bacteriol. 172, 4758–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gawronski J. D., Benson D. R. (2004) Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal. Biochem. 327, 114–118 [DOI] [PubMed] [Google Scholar]
- 44. Weisbrod R. E., Meister A. (1973) Studies on glutamine synthetase from Escherichia coli. Formation of pyrrolidone carboxylate and inhibition by methionine sulfoximine. J. Biol. Chem. 248, 3997–4002 [PubMed] [Google Scholar]
- 45. Rhee S. G., Chock P. B., Wedler F. C., Sugiyama Y. (1981) Subunit interaction in unadenylated glutamine synthetase from Escherichia coli. J. Biol. Chem. 256, 644–648 [PubMed] [Google Scholar]
- 46. Fisher S. H., Wray L. V., Jr. (2009) Novel trans-acting Bacillus subtilis glnA mutations that deprepress glnRA expression. J. Bacteriol. 191, 2485–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]