

Background: The mechanisms for association of altered proteostasis and inflammation are not known.
Results: The deficiency of either autophagy or p62 led to inflammasome hyperactivation.
Conclusion: Accumulation of misfolded proteins caused inflammasome activation by inducing generation of nonmitochondrial ROS and lysosomal damage.
Significance: Our results suggest that altered proteostasis results in inflammasome activation and thus provide mechanisms for the association of altered proteostasis with inflammatory disorders.
Keywords: Autophagy, Inflammation, Lysosomes, Macrophages, Protein Misfolding, Inflammasome, Proteostasis
Abstract
The association between altered proteostasis and inflammatory disorders has been increasingly recognized, but the underlying mechanisms are not well understood. In this study, we show that deficiency of either autophagy or sequestosome 1 (p62 or SQSTM) led to inflammasome hyperactivation in response to LPS and ATP in primary macrophages and in mice in vivo. Importantly, induction of protein misfolding by puromycin, thapsigargin, or geldanamycin resulted in inflammasome activation that was more pronounced in autophagy- or p62-deficient macrophages. Accumulation of misfolded proteins caused inflammasome activation by inducing generation of nonmitochondrial reactive oxygen species and lysosomal damage, leading to release of cathepsin B. Our results suggest that altered proteostasis results in inflammasome activation and thus provide mechanisms for the association of altered proteostasis with inflammatory disorders.
Introduction
The inflammatory response must be actively controlled to prevent “bystander” damage to tissues. Yet the mechanisms controlling excessive inflammatory responses are poorly understood (1). Acute inflammation resulting in septic shock, acute lung injury, and multiorgan dysfunction is a common cause of death in critically ill patients (2). Furthermore, chronic inflammation is a major underlying pathology in many systemic disorders, including lung diseases, metabolic disorders, and cancer (1). Macrophages are critical for pathogen recognition, initiation, and amplification of the inflammatory cascade mainly through regulation of key inflammatory mediators, such as TNF-α and IL-1β (3). The strong pro-inflammatory activity of IL-1β, its processing, and secretion are tightly regulated by the inflammasome (4).
The inflammasomes are molecular platforms activated upon cellular infection or stress, which trigger the activation of caspase 1 that carries out processing of pro-IL-1β and pro-IL-18 (4). Dysregulated inflammasome activation is associated with certain human heritable and acquired inflammatory diseases (4). Recent studies have identified multiple pathways for inflammasome activation (5). However, physiological mechanisms for suppression of inflammasome activity remain to be elucidated. There is emerging evidence that autophagy deficiency is associated with hyperactivation of the inflammasome (6–8). However, the mechanisms by which autophagy regulates inflammasome activity are not fully understood.
Protein biogenesis is an error-prone process, and newly synthesized proteins are constantly subjected to misfolding. Cells have developed a network of pathways aimed at maintaining protein homeostasis (proteostasis) (9). These include protein refolding systems, degradation pathways, and sequestration strategies. Because of the critical role of autophagy in maintaining proteostasis, it is conceivable that altered proteostasis in autophagy-deficient cells leads to hyperactivation of the inflammasome. In this study, we show that altered proteostasis triggers NALP3 inflammasome activation that is dependent on generation of ROS2 and cathepsin B release. In addition, we show that deficiency of either autophagy or p62 enhances inflammasome activation in response to protein misfolding-inducing agents.
EXPERIMENTAL PROCEDURES
Mice and Cell Culture
Atg7f/f-LysMCre+ and p62−/− mice were previously described (10). Mice were housed in a specific pathogen-free animal facility. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee. Bone marrow cells were cultured in DMEM containing 10% fetal bovine serum and supplemented with mouse macrophage colony-stimulating factor (M-CSF) or 10% conditioned media from L929 cell culture for 7 days to differentiate into macrophages. Primary human macrophages were differentiated from peripheral blood monocytes isolated from human blood via a double density technique using Ficoll Isopaque and 46% iso-osmotic Percoll centrifugation and were cultured with human M-CSF for 7 days.
Antibodies
The following antibodies were used: anti-caspase 1 p10 (Santa Cruz Biotechnology, sc-514); anti-caspase 1 p20 (Adipogen, AG-20B-0042-C100); anti-cathepsin B (R&D Systems, AF9650); anti-IL-1β (R&D Systems, MAB4011); anti-Atg7 (Rockland Immunochemicals, 600-401-487); anti-β-actin (Ambion, AM43020); anti-p62 (American Research Products, 03-GP62-C-1), and anti-Beclin1 (Novus, NB500-249). LC3 antibody was previously described (10).
RNA-mediated Interference
Raw 264.7 cells were transduced with nontargeting control shRNA lentiviral transduction particles (SHC002V; Sigma) and Atg7 (SHCLNV, NM_019584; Sigma) or Beclin1 (SHCLNV, NM_028835; Sigma) mission shRNA lentiviral transduction particles, followed by selection using media containing puromycin (2 μg/ml). Primary BMDM were transfected with siRNA using Lipofectamine 2000 (Invitrogen) and siRNAs smart pools against nontargeting (control) and mouse NALP3 (Dharmacon; M-053455-01-0005).
Flow Cytometry
To detect total or mitochondrial ROS, cells were incubated at 37 °C with CM-H2DCFDA (10 μm; Invitrogen) for 30 min or MitoSOX (5 μm; Invitrogen) for 15 min in PBS, respectively, washed three times with PBS, and analyzed by flow cytometry. To measure mitochondrial membrane potential, cells were incubated with MitoTracker Deep Red (200 nm; Invitrogen) in culture media for 30 min at 37 °C, washed twice with PBS, and analyzed by flow cytometry. For evaluation of lysosomal damage, cells were incubated with DQ-BSA (10 μg/ml; Invitrogen) for 30 or 60 min at 37 °C or incubated with Lysotracker Red (75 nm; Invitrogen) for 2 h at 37 °C, washed three times with PBS, and analyzed by flow cytometry. All flow cytometry experiments were performed using LSRFortessaTM Cell Analyzer (BD Biosciences).
Immunofluorescence
Cells were plated on type I collagen-coated glass coverslips in a 6-well plate. After treatment, cells were washed twice with PBS, fixed with 4% paraformaldehyde overnight at 4 °C, permeabilized with 0.2% Triton X-100, blocked with 10% normal goat serum, incubated first with Lamp1 antibody (Pharmingen, 553792) and then with goat anti-rat IgG conjugated to Alexa Fluor 488, and mounted using the Prolong Gold Antifade reagent with DAPI. Images were acquired using a Zeiss Axiovert 200 M microscope.
In Vivo Sepsis Model
Atg7f/f and Atg7Δ mice (male, 8–10 weeks old) were injected intraperitoneally with PBS or 10 mg/kg of LPS from Escherichia coli serotype (0111:B4; Sigma). Two hours following injection, blood was collected from the inferior vena cava. p62+/+ and p62−/− mice (male, 8–10 weeks old) were injected intraperitoneally with 1 mg/kg of LPS, followed 2 h later by intraperitoneal injection of 100 μl of PBS or ATP (100 mm), and blood was collected from inferior vena cava 15 min after the second injection (n = 3–6 mice/group).
Statistical Analysis
Statistical significance was evaluated by Student's t test for two groups. All data are representative of at least three independent experiments.
RESULTS
Deficiency of Autophagy Leads to Inflammasome Hyperactivation in Macrophages
Recently, it has been reported that deficiency in autophagy proteins is linked to inflammasome activation (6, 7, 11). Several mechanisms have been proposed, but the underlying direct mechanisms are not well understood. We investigated the mechanisms of how deficiency of autophagy regulates inflammasome activation. Inflammasome activation was evaluated by measuring IL-1β production in culture media and by immunodetecting cleaved caspase 1 in cell lysates. In macrophage cell line Raw 264.7, LPS treatment was sufficient to induce robust inflammasome activation that could be inhibited by specific caspase 1 inhibitor Z-YVAD. In these cells, knockdown of autophagy-related gene 7 (Atg7) or Beclin1 or inhibition of autophagy with 3-methyladenine enhanced inflammasome activation in response to LPS (Fig. 1, A–C). In contrast, production of IL-6 or TNF-α was not affected by Beclin1 knockdown (data not shown). We extended our observations to primary macrophages and generated myeloid-specific Atg7-deficient mice (10). We crossed mice bearing Atg7flox/flox alleles with LysM-Cre transgenic mice, which express Cre recombinase in a myeloid specific manner, including macrophages and neutrophils. We then compared Atg7flox/flox LysM-Cre-positive mice, henceforth referred to as Atg7Δ, to Atg7flox/flox littermates, i.e. Atg7f/f. To induce inflammasome activation, macrophages were treated with LPS or primed with LPS and then stimulated with ATP, a potent activator of NALP3 inflammasome (12). We observed stronger caspase 1 activation and more IL-1β production in BMDM of Atg7Δ mice than in Atg7f/f (Fig. 1D). Importantly, deficiency of autophagy did not affect pro-IL-1β expression in BMDM or in Raw 264.7 cells (data not shown), supporting the conclusion that increased IL-1β production is not simply caused by transcriptional increase of pro-IL-1β but rather by increased inflammasome activity. Finally, Atg7Δ mice had increased serum levels of IL-1β after intraperitoneal injection of LPS (Fig. 1E). These results are consistent with recent reports that autophagy deficiency can induce hyperactivation of inflammasome in vitro and in vivo (6, 13).
FIGURE 1.

Deficiency of autophagy leads to inflammasome hyperactivation in macrophages. A and B, Raw 264.7 cells were transduced with lentiviruses containing nontargeting (Control), Atg7, or Beclin1 shRNA. Cells were incubated with LPS (100 ng/ml) for 16 h in the presence or absence of the caspase 1 inhibitor Z-YVAD (20 μm). C, Raw 264.7 cells were incubated with LPS (100 ng/ml) for 16 h in the presence or absence of 3-methyladenine (3-MA) (5 mm) or DMSO. D, BMDM from Atg7f/f or Atg7Δ mice were left untreated, primed with LPS, or primed with LPS and then treated with ATP (5 mm) for 30 min (left panel) or 0–60 min (right panel). E, Atg7f/f or Atg7Δ mice were injected intraperitoneally with PBS or LPS (10 mg/kg), and blood was collected from the inferior vena cava 2 h after injection, n = 3–6 mice/group. IL-1β was measured in culture media or serum using ELISA, and cell lysates were analyzed by immunoblotting. Data are mean ± S.D., n = 3; *, p < 0.05; **, p < 0.001.
Altered Proteostasis Triggers NALP3 Inflammasome Activation in Macrophages
Autophagy is a major pathway for degradation of intracellular contents such as damaged organelles, protein aggregates, and misfolded proteins (14, 15). Thus, we hypothesized that altered proteostasis results in inflammasome activation that will be exaggerated in autophagy-deficient cells because of reduced capacity to handle misfolded proteins. To test this hypothesis, we investigated if induction of misfolded proteins in wild type cells could enhance inflammasome activation. We treated LPS-primed BMDM with various stimuli of protein misfolding-inducing agents, including puromycin, thapsigargin, or geldanamycin. Macrophages were primed with LPS to induce expression of pro-IL-1β. Cells were then incubated for 4 h with reagents that induce ER stress and/or misfolded proteins. Puromycin prematurely stops protein translation and thus leads to generation of truncated peptides (16). ER stress was induced by treatment with thapsigargin, which inhibits ER calcium pump (17). Geldanamycin is an inhibitor of the chaperone HSP90 and thus reduces protein folding (18). Induction of altered proteostasis, by all of the above agents, caused a significant increase in aggregation of ubiquitinated proteins (Fig. 2A), which is an early event in the cytosol in response to altered proteostasis (10), and resulted in inflammasome activation in mouse BMDM (Fig. 2A) and in primary monocyte-derived human macrophages (Fig. 2B). In support of inflammasome-specific effect, production of IL-6 was not induced in response to protein misfolding-inducing agents (data not shown). siRNA knockdown of NALP3 markedly attenuated caspase 1 activation and IL-1β production in response to puromycin or the classic NALP3 activator ATP in LPS-primed BMDM (Fig. 2C). NALP3 inflammasome activation is dependent on K+ efflux (19, 20). High concentrations of K+ significantly abrogated puromycin- or ATP-induced IL-1β production (Fig. 2D). These results suggested that NALP3 has a critical role in inflammasome activation by altered proteostasis.
FIGURE 2.

Induction of protein misfolding triggers NALP3 inflammasome activation in macrophages. A and B, LPS-primed BMDM (A) or LPS-primed monocyte-derived human macrophages (B) were treated for 4 h with puromycin (15 μg/ml), thapsigargin (5 μm), or geldanamycin (10 μm) or for 30 min with ATP (5 mm). BMDM were also analyzed by immunofluorescence microscopy using ubiquitin antibody (left panel in A). C, LPS-primed BMDM transfected with nontargeting (Control) or NALP3 siRNA were treated with puromycin (15 μg/ml) or ATP (5 mm). D, LPS-primed BMDM were treated with puromycin or ATP in the presence or absence of 130 mm KCl. IL-1β was measured in culture media using ELISA, and cell lysates were analyzed by immunoblotting. Data are mean ± S.D., n = 3; *, p < 0.05; **, p < 0.001. Scale bar, 10 μm.
Deficiency of Autophagy or p62, in Macrophages, Enhances Inflammasome Activation in Response to Altered Proteostasis
We then investigated if inflammasome activation induced by altered proteostasis was exaggerated in autophagy-deficient cells because of their reduced capacity to handle misfolded proteins. Compared with wild type, LPS-primed BMDM from Atg7Δ mice exhibited increased IL-1β production in a dose-dependent response to puromycin (Fig. 3A). Consistent with inflammasome activation, puromycin-induced IL-1β production was significantly reduced by treatment of Z-YVAD, a caspase 1 inhibitor, in both Atg7f/f and Atg7Δ macrophages (Fig. 3B). Enhanced inflammasome activation in Atg7Δ macrophages was also observed when misfolded proteins were generated by geldanamycin treatment (Fig. 3C). Because these data suggested that altered proteostasis enhanced inflammasome activation in autophagy-deficient cells, we reasoned that the deficiency of another important proteostatic mechanism might also lead to inflammasome activation. p62 is an important adaptor protein in cellular proteostasis (21). We found that, in p62−/− mice, BMDM had exaggerated response to inflammasome activators, including ATP and valinomycin (Fig. 4, A and B). We noticed a slight but significant increase in pro-IL-1β expression in the p62−/− BMDM in response to LPS (Fig. 4A, right panel). This increase, however, could not account for the marked increase of IL-1β noted in p62−/− BMDM, which is more likely due to increased inflammasome activity. Importantly, caspase 1 inhibitor Z-YVAD prevented IL-1β production caused by p62 deficiency, confirming that the increase in IL-1β in p62−/− BMDM was primarily caused by increased caspase 1 activation. Furthermore, following in vivo injection of LPS and ATP, p62−/− mice had higher levels of serum IL-1β compared with wild type mice (Fig. 4C). These results suggested that p62 deficiency, similar to autophagy deficiency, led to exaggerated inflammasome activation. We then tested the response of p62−/− macrophages to agents that induced misfolded proteins. Induction of inflammasome activation with puromycin was significantly increased in a dose-dependent manner, as evidenced by increased IL-1β production and caspase 1-cleaved product (Fig. 4, D and E). Similar results were obtained using thapsigargin or geldanamycin (Fig. 4, F and G). To confirm the specificity of inflammasome activation by these agents, IL-1β was significantly reduced by treatment of Z-YVAD, a caspase 1 inhibitor (Fig. 4, E–G). These results suggested that inflammasome was hyperactivated in p62-deficient macrophages in vitro and in vivo.
FIGURE 3.
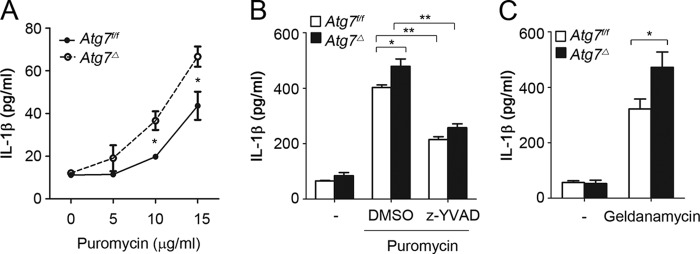
Deficiency of autophagy enhances inflammasome activation in response to protein misfolding-inducing agents. LPS-primed BMDM from Atg7f/f or Atg7Δ mice were treated with increasing concentrations of puromycin for 4 h (A) or incubated for 4 h in the presence or absence of puromycin (15 μg/ml) and caspase 1 inhibitor Z-YVAD (20 μm) (B), or geldanamycin (10 μm) (C). IL-1β was measured in culture media using ELISA. Data are mean ± S.D., n = 3; *, p < 0.05; **, p < 0.001.
FIGURE 4.

Inflammasome hyperactivation in p62-deficient macrophages. LPS-primed BMDM from p62+/+ or p62−/− mice were incubated with ATP (5 mm) for 30 min in the presence or absence of caspase 1 inhibitor Z-YVAD (20 μm) (A) or incubated for 2 h with valinomycin (10 μm) (B). p62+/+ or p62−/− mice were injected intraperitoneally with LPS (1 mg/kg) followed 2 h later by intraperitoneal injection of PBS or ATP (100 mm), and blood was collected after 15 min from the inferior vena cava. n = 3–6 mice/group (C). LPS-primed BMDM from p62+/+ or p62−/− mice were treated for 4 h with increasing concentrations of puromycin (D) or incubated for 4 h in the presence or absence of puromycin (15 μg/ml) and Z-YVAD (E), thapsigargin (5 μm) (F), or geldanamycin (10 μm) (G). IL-1β was measured in culture media or serum using ELISA, and cell lysates were analyzed by immunoblotting. Data are mean ± S.D., n = 3; *, p < 0.05; **, p < 0.001.
Inflammasome Activation by Altered Proteostasis Is Dependent on Nonmitochondrial ROS
We investigated the role of ROS in inflammasome activation by altered proteostasis. In LPS-primed BMDM, levels of ROS were increased after treatment with puromycin, thapsigargin, or geldanamycin, similar to that observed with ATP (Fig. 5A). Importantly, ROS levels were higher in LPS-primed p62−/− BMDM than in p62+/+ BMDM, following their incubation with puromycin or thapsigargin, but not with ATP (Fig. 5B). ROS scavenger N-acetylcysteine (NAC) inhibited inflammasome activation by ATP, puromycin, or thapsigargin (Fig. 5, C and D). Similar results were obtained using other ROS scavengers, including glutathione monoethyl ester (Fig. 5C). These results suggested that increased ROS by protein misfolding-inducing agents contribute to inflammasome activation. Recent studies suggest a role for mitochondrial ROS in inflammasome activation in autophagy-deficient cells (6, 13). Therefore, we investigated a potential role for mitochondrial ROS in induced inflammasome activation by midfolded proteins. These studies were done in LPS-primed BMDM from p62−/− or Atg7Δ mice and their wild type controls in response to ATP or puromycin. Surprisingly, treatment of LPS-primed BMDM with ATP but not with puromycin increased the percentages with loss of mitochondrial membrane potential, detected with MitoTracker Red staining (Fig. 6, A and B). In addition, treatment of LPS-primed BMDM with ATP, but not with puromycin or thapsigargin, resulted in an increase in mitochondrial ROS (Fig. 6C). Furthermore, mitochondrion-specific ROS scavenger mTempo suppressed ATP- but not puromycin-induced inflammasome activation (Fig. 6, D and E). These results suggested that inflammasome activation by altered proteostasis was not caused by mitochondrial damage; it was triggered by nonmitochondrial ROS.
FIGURE 5.

Inflammasome activation by altered proteostasis is dependent on ROS. LPS-primed BMDM from p62+/+ or p62−/− mice were treated for 4 h with puromycin (15 μg/ml), thapsigargin (5 μm), or geldanamycin (10 μm) or for 15 min with ATP (5 mm), and ROS production was detected by flow cytometry using CM-H2DCFDA dye. Values are mean fluorescence intensity (MFI) ± S.D. A and B, in additional experiments, BMDM were incubated with puromycin, thapsigargin, or ATP in the presence or absence of ROS scavenger NAC (25 mm), GSH (10 mm), or APDC (50 μm) (C and D). IL-1β was measured in culture media using ELISA, and cell lysates were analyzed by immunoblotting. Data are mean ± S.D., n = 3; * p < 0.05; **, p < 0.001. The symbol ➢ indicates a nonspecific band.
FIGURE 6.

Inflammasome activation by altered proteostasis is dependent on nonmitochondrial ROS. A and B, LPS-primed wild type, p62−/−, or Atg7Δ BMDM were treated for 4 h with puromycin (15 μg/ml) or for 15 min with ATP (5 mm) and analyzed by flow cytometry using MitoTracker Deep Red. Numbers indicate percentages of cells with loss of mitochondrial membrane potential. C, unprimed or LPS-primed BMDM were treated for 4 h with puromycin (15 μg/ml), thapsigargin (5 μm), or for 15 min with ATP (5 mm). Mitochondrial ROS production was measured by flow cytometry using MitoSOX. Data are mean fluorescence intensity (MFI). D and E, parallel experiments to those of A and B were done in the presence or absence of mTempo (500 μm). IL-1β was measured in culture media using ELISA. Data are mean ± S.D., n = 3; *, p < 0.05; **, p < 0.001.
Accumulation of Misfolded Proteins Triggers Inflammasome Activation by Destabilizing Lysosomes
Recent studies suggest that NALP3 inflammasome is activated by lysosome rupture following phagocytosis of particles such as silica crystal and aluminum (22, 23). Inefficient clearance of these particles following phagocytosis leads to lysosomal damage and cathepsin B release, triggering inflammasome activation. Lysosomes play a key role in degradation of misfolded proteins (10). To investigate if accumulation of misfolded protein induced lysosomal damage, we evaluated lysosomal morphology in response to those agents using immunofluorescence with lysosome-associated membrane glycoprotein 1 (LAMP1) antibody. Chloroquine is a lysosomotropic agent that accumulates within lysosomes and inhibits lysosomal acidification (24). Therefore, we used chloroquine as a positive control for lysosomal damage. We observed that LAMP1-positive compartments became enlarged after stimulation with puromycin, thapsigargin, geldanamycin or chloroquine in LPS-primed BMDM (Fig. 7A). We quantified levels of lysosome by flow cytometry analysis using LysoTracker-Red. In LPS-primed BMDM, the fluorescence of LysoTracker Red was increased in response to puromycin or thapsigargin (data not shown). We measured the degradative activity of lysosomes by flow cytometry using DQ-BSA, a fluorogenic substrate for proteases. In LPS-primed BMDM, we observed a significant decrease in lysosomal activity following treatment with puromycin, thapsigargin, or chloroquine (Fig. 7B). These results suggested that protein misfolding-inducing agents activate the inflammasome by triggering lysosomal destabilization. Chloroquine is a strong inducer of lysosomal damage (25–27). Therefore, we speculated that chloroquine, similar to protein misfolding-inducing agents, may cause inflammasome activation. Indeed, we observed strong caspase 1 activation and IL-1β production in response to chloroquine in LPS-primed BMDM (Fig. 7C). Furthermore, puromycin- or thapsigargin-induced inflammasome activation was significantly enhanced by addition of chloroquine (Fig. 7D). Taken together, these results suggest that accumulation of misfolded proteins causes lysosomal destabilization and that impairment of lysosomal function contributes to inflammasome activation by altered proteostasis.
FIGURE 7.

Altered proteostasis activates inflammasome by destabilizing lysosomes. A and B, unprimed or LPS-primed BMDM were treated for 4 h with puromycin (Puro; 15 μg/ml), thapsigargin (Tg; 5 μm), geldanamycin (Gel; 10 μm), or chloroquine (CQ; 50 μm). Con, control. BMDM were then analyzed by immunofluorescence microscopy using LAMP-1 antibody (A) or incubated with DQ-BSA (10 μg/ml) and analyzed by flow cytometry (B). C and D, LPS-primed BMDM were treated for 4 h with chloroquine (50 μm) (C) or puromycin (15 μg/ml) or thapsigargin (5 μm) in the presence or absence of chloroquine (D). Sup, culture supernatant. Culture media were assessed for IL-1β production by ELISA and for cathepsin B and caspase 1 release by immunoblot analysis using cathepsin B and caspase 1 antibodies, and the cell lysates were analyzed by immunoblotting. Data are mean ± S.D., n = 3; *, p < 0.05; **, p < 0.001. Scale bar, 10 μm.
Accumulation of Misfolded Proteins Causes Lysosomal Cathepsin B Release
Enlarged and destabilized lysosomes are more prone to rupture and to leak hydrolytic enzymes (28). Release of lysosomal cysteine protease cathepsin B has been shown to trigger inflammasome activation (23). We found increased cathepsin B in culture supernatants of cells treated with chloroquine or protein misfolding-inducing agents (Figs. 7C and 8A). Consistent with altered proteostasis as the underlying mechanism, cathepsin B release by puromycin was significantly increased in Atg7Δ or p62−/− BMDM, compared with wild type (Fig. 8B). To confirm the role of cathepsin B in inflammasome activation by altered proteostasis, we used the cathepsin B inhibitor, CA-074-Me. Inflammasome activation in response to protein misfolding-inducing agents was attenuated by concomitant inhibition of cathepsin B in wild type, Atg7Δ, or p62−/− BMDM (Fig. 8C and data not shown). However, the treatment did not abolish the difference between the control and the knock out, suggesting that cathepsin B release might not be the only mechanism involved. ROS may lead to destabilization of the lysosomal membrane permeabilization, thereby causing the release of cathepsins and other hydrolases (29, 30). We reasoned that increased ROS due to altered proteostasis might lead to enhanced lysosomal cathepsin B release. In support of this notion, cathepsin B release, caused by protein misfolding-inducing agents, was attenuated by ROS scavengers NAC and GSH (Fig. 8, D and E, and data not shown). Taken together, these results suggest that inflammasome activation by altered proteostasis is mediated by an increase in nonmitochondrial ROS, leading to lysosomal damage and cathepsin B release.
FIGURE 8.

Altered proteostasis causes lysosomal cathepsin B release. A and B, LPS-primed wild type, Atg7Δ, or p62−/−; BMDM were treated for 4 h with puromycin (Puro; 15 μg/ml), thapsigargin (Tg; 5 μm), and geldanamycin (Gel; 10 μm). C, LPS-primed BMDM were treated with puromycin or thapsigargin in the presence or absence of Z-YVAD (20 μm) or cathepsin B inhibitor CA-074-Me (CA-074; 10 μm). D, LPS-primed BMDM were treated for 4 h with puromycin (15 μg/ml) in the presence or absence of NAC (25 mm) or APDC (50 μm). Lys, lysate; Sup, culture supernatant. E, LPS-primed BMDM were treated for 4 h with puromycin (15 μg/ml), thapsigargin (5 μm), or chloroquine (50 μm) in the presence or absence of Z-YVAD (20 μm) or NAC (25 mm). Culture media were assessed for IL-1β production by ELISA and for cathepsin B by immunoblot analysis using cathepsin B antibody, and the cell lysates were analyzed by immunoblotting. Data are mean ± S.D.; n = 3, *, p < 0.05; **, p < 0.001.
DISCUSSION
Autophagy is a housekeeping mechanism that controls the cellular homeostasis by clearing damaged organelles and preventing accumulation of misfolded or aggregated proteins (14). Altered proteostasis can occur due to the accumulation of misfolded proteins arising from disruption in the protein homeostatic machinery such as that of autophagy or ubiquitin-proteasome pathway. Polyubiquitin-binding protein p62 is involved in autophagy and the ubiquitin-proteasome pathway. Therefore, we reasoned that the p62-deficient model would be informative for studying the consequences of altered protein homeostatic machinery. In this study, we have shown that accumulation of misfolded proteins triggers inflammasome activation that is enhanced by deficiency of either autophagy or p62 (Fig. 9). Therefore, these findings link the roles of autophagy and p62 in proteostasis to inflammation and the innate immune response.
FIGURE 9.
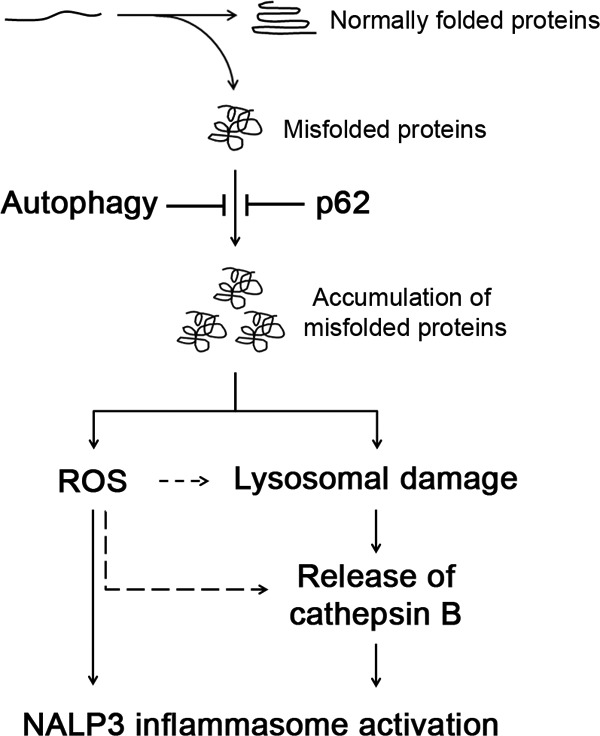
Autophagy and p62 protect against inflammasome activation by maintaining proteostasis. Deficiency of either autophagy or p62 results in failure of degradation of misfolded proteins. Accumulation of misfolded proteins triggers inflammasome activation by ROS generation and lysosomal damage, leading to release of the protease cathepsin B.
Several studies have shown that NALP3 inflammasome activation is triggered by increased generation of ROS and suppressed by ROS scavengers. However, the role of ROS in inflammasome activation in general and the source of ROS implicated in such a role are more controversial (31, 32). Altered proteostasis leads to increased ROS. This increase is because the unfolded protein response is associated with up-regulation of chaperones involved in disulfide bond formation in the ER (Ero1p and Erv2p). These enzymes use oxidation/reduction reactions, with molecular oxygen as the final electron recipient leading to increased ROS formation (17, 33). In this study, we showed that altered proteostasis enhanced the production of ROS in LPS-primed BMDM. Mitochondrial ROS were increased in response to ATP but not to altered proteostasis. In addition, inflammasome activation, by altered proteostasis, was suppressed by ROS scavengers but not by mTEMPO, a scavenger specific for mitochondrial ROS. These results suggest that activation of inflammasome by altered proteostasis is mediated by ROS, but in contrast to ATP activation, the source of ROS is not from mitochondria. Another cellular source of ROS is an NADPH-oxidative system. It has been reported that particles such as silica, asbestos, and monosodium orate activate the inflammasome activation by ROS, which are generated by NADPH oxidase during their phagocytosis (3). We found that APDC, an inhibitor of NADPH oxidase, had no effect on caspase 1 activation or cathepsin B release induced by altered proteostasis. Taken together, these findings suggest that inflammasome activation by altered proteostasis is triggered by nonmitochondrial ROS.
Lysosomes play an important role in protein homeostasis. Here, we have shown that altered proteostasis-induced inflammasome activation was significantly enhanced by inhibition of the lysosomal degradation pathway by chloroquine. Importantly, protein misfolding-inducing agents led to lysosomal destabilization and cathepsin B release. However, it is unclear whether or not accumulation of misfolded proteins induces the lysosomal destabilization directly or indirectly. One possible mechanism is that misfolded proteins, by overwhelming the capacity of the degradation within the lysosome, directly damage them. Another possibility is that increased ROS due to accumulation of misfolded proteins led to lysosomal damage. We found a strong correlation between caspase 1 activation and cathepsin B release in response to various stimuli, which induce altered proteostasis, including puromycin, thapsigargin, geldanamycin, or chloroquine. Importantly, inhibition of caspase 1 did not affect the cathepsin B release (Fig. 8E), suggesting that cathepsin B release is upstream event of caspase 1 activation. Furthermore, cathepsin B inhibition attenuated NALP3 inflammasome activation, suggesting a causal relationship.
In summary, we have shown that altered proteostasis induced NALP3 inflammasome activation through mechanisms involving ROS generation and lysosome destabilization. Altered proteostasis enhanced inflammasome activation in autophagy or p62-deficient cells. Our study provides an important mechanistic explanation for association between altered proteostasis and inflammation.
Acknowledgments
Atg7f/f and p62−/− mice were obtained from Drs. M. Komatsu and T. Ishii, respectively. We thank Joel M. Sederstrom for expert assistance.
This work was supported, in whole or in part, by National Institutes of Health grants from NHLBI and NIAID and Grants AI036211, CA125123, and RR024574 (to the Cytometry and Cell Sorting Core at Baylor College of Medicine).
- ROS
- reactive oxygen species
- ER
- endoplasmic reticulum
- NAC
- N-acetylcysteine
- Z
- benzyloxycarbonyl
- BMDM
- bone marrow-derived macrophage
- APDC
- (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylic acid.
REFERENCES
- 1. Medzhitov R. (2008) Origin and physiological roles of inflammation. Nature 454, 428–435 [DOI] [PubMed] [Google Scholar]
- 2. Cohen J. (2002) The immunopathogenesis of sepsis. Nature 420, 885–891 [DOI] [PubMed] [Google Scholar]
- 3. Dostert C., Pétrilli V., Van Bruggen R., Steele C., Mossman B. T., Tschopp J. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schroder K., Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 5. Atianand M. K., Rathinam V. A., Fitzgerald K. A. (2013) SnapShot: Inflammasomes. Cell 153, 272–272.e1 [DOI] [PubMed] [Google Scholar]
- 6. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saitoh T., Fujita N., Jang M. H., Uematsu S., Yang B. G., Satoh T., Omori H., Noda T., Yamamoto N., Komatsu M., Tanaka K., Kawai T., Tsujimura T., Takeuchi O., Yoshimori T., Akira S. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 [DOI] [PubMed] [Google Scholar]
- 8. Sorbara M. T., Girardin S. E. (2011) Mitochondrial ROS fuel the inflammasome. Cell Res. 21, 558–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mu T. W., Ong D. S., Wang Y. J., Balch W. E., Yates J. R., 3rd, Segatori L., Kelly J. W. (2008) Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 134, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu X. D., Ko S., Xu Y., Fattah E. A., Xiang Q., Jagannath C., Ishii T., Komatsu M., Eissa N. T. (2012) Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J. Biol. Chem. 287, 19687–19698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Menu P., Mayor A., Zhou R., Tardivel A., Ichijo H., Mori K., Tschopp J. (2012) ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 3, e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mariathasan S., Weiss D. S., Newton K., McBride J., O'Rourke K., Roose-Girma M., Lee W. P., Weinrauch Y., Monack D. M., Dixit V. M. (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 [DOI] [PubMed] [Google Scholar]
- 13. Zhou R., Yazdi A. S., Menu P., Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 14. Levine B., Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kraft C., Peter M., Hofmann K. (2010) Selective autophagy: ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 12, 836–841 [DOI] [PubMed] [Google Scholar]
- 16. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsai B., Ye Y., Rapoport T. A. (2002) Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 3, 246–255 [DOI] [PubMed] [Google Scholar]
- 19. Kanneganti T. D., Lamkanfi M., Kim Y. G., Chen G., Park J. H., Franchi L., Vandenabeele P., Núñez G. (2007) Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26, 433–443 [DOI] [PubMed] [Google Scholar]
- 20. Pétrilli V., Papin S., Dostert C., Mayor A., Martinon F., Tschopp J. (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589 [DOI] [PubMed] [Google Scholar]
- 21. Seibenhener M. L., Babu J. R., Geetha T., Wong H. C., Krishna N. R., Wooten M. W. (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24, 8055–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., Wright S. D., Hornung V., Latz E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Steinman R. M., Mellman I. S., Muller W. A., Cohn Z. A. (1983) Endocytosis and the recycling of plasma membrane. J. Cell Biol. 96, 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shintani T., Klionsky D. J. (2004) Autophagy in health and disease: a double-edged sword. Science 306, 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoon Y. H., Cho K. S., Hwang J. J., Lee S. J., Choi J. A., Koh J. Y. (2010) Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 51, 6030–6037 [DOI] [PubMed] [Google Scholar]
- 27. Fan W., Tang Z., Chen D., Moughon D., Ding X., Chen S., Zhu M., Zhong Q. (2010) Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 6, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ono K., Kim S. O., Han J. (2003) Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor α-induced cell death. Mol. Cell. Biol. 23, 665–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boya P., Kroemer G. (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451 [DOI] [PubMed] [Google Scholar]
- 30. Johansson A. C., Appelqvist H., Nilsson C., Kågedal K., Roberg K., Ollinger K. (2010) Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 15, 527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bauernfeind F., Bartok E., Rieger A., Franchi L., Núñez G., Hornung V. (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van de Veerdonk F. L., Smeekens S. P., Joosten L. A., Kullberg B. J., Dinarello C. A., van der Meer J. W., Netea M. G. (2010) Reactive oxygen species-independent activation of the IL-1β inflammasome in cells from patients with chronic granulomatous disease. Proc. Natl. Acad. Sci. U.S.A. 107, 3030–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malhotra J. D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S. W., Kaufman R. J. (2008) Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U.S.A. 105, 18525–18530 [DOI] [PMC free article] [PubMed] [Google Scholar]