

Background: AMPK is a heterotrimeric enzyme targeted for drug discovery (activation) in diabetes.
Results: Diversity of AMPK was studied by chemical capture and proteomic LC-MS.
Conclusion: AMPK of human and rat livers differs with respect to the β chain. Certain direct activators only activate AMPK containing the β1 form.
Significance: Interspecies divergence could compromise the translatability of preclinical data.
Keywords: AMP-activated Kinase (AMPK), Chemical Biology, Chemical Modification, Drug Discovery, Mass Spectrometry (MS)
Abstract
AMP-activated protein kinase (AMPK) is a heterotrimeric enzyme that senses and governs changes in the cellular energy balance represented by concentrations of AMP, ADP, and ATP. Each of its three chains (α, β, and γ) exists as either two or three subtypes, theoretically allowing up to 12 different forms of the complete enzyme. Tissue specificity in the distribution of AMPK subtypes is believed to underpin a range of biological functions for AMPK, a central regulator of metabolic function and response. It is of particular interest for drug discovery purposes to compare AMPK isoforms that are most prevalent in human liver and muscle with isoforms present in key preclinical species. To complement immunocapture/immunodetection methods, which for AMPK are challenged by sequence similarities and difficulties of obtaining accurate relative quantitation, AMPK was captured from lysates of a range of cells and tissues using the ActivX ATP probe. This chemical probe covalently attaches desthiobiotin to one or more conserved lysyl residues in the ATP-binding sites of protein kinases, including AMPK, while also labeling a wide range of ATP-utilizing proteins. Affinity-based recovery of labeled proteins followed by gel-based fractionation of the captured sample was followed by proteomic characterization of AMPK polypeptides. In agreement with transcript-based analysis and previous indications from immunodetection, the results indicated that the predominant AMPK heterotrimer in human liver is α1β2γ1 but that dog and rat livers mainly contain the α1β1γ1 and α2β1γ1 forms, respectively. Differences were not detected between the AMPK profiles of normal and diabetic human liver tissues.
Introduction
AMP-activated protein kinase (AMPK)3 gauges and governs available intracellular energy, reading its level by adjusting to changes in the respective concentrations of ATP, ADP, and AMP (1). Although the millimolar range intracellular concentrations of ATP exceed those of ADP and AMP by multiple orders of magnitude, differential affinities for the three nucleotides allow AMPK to monitor shifts in their relative abundance (2, 3). ADP and AMP both stimulate the enzyme to favor its active state, leading it to phosphorylate a panoply of protein substrates that include both enzymes and transcription factors (4). The net effect is to promote the activity of catabolic ATP-synthesizing pathways while down-modulating anabolic ATP-consuming pathways and facilitating the uptake of glucose into cells. As these activities can lead to the reduction of blood glucose levels in hyperglycemic individuals (5–8), direct pharmacological activation of AMPK has become a mechanism of interest for potentially treating type II diabetes and metabolic syndrome (8–15).
Mammalian AMPK is a heterotrimeric complex composed of a catalytic α-subunit and regulatory β- and γ-subunits (see Fig. 1) (16). Each subunit exists as either two (α1 and α2; β1 and β2) or three (γ1, γ2, and γ3) isoforms encoded by distinct genes, giving rise to a total of 12 possible AMPK α/β/γ-heterotrimers. Variable splicing of these gene products may add to the diversity of AMPK species in mammalian cells. The extent of sequence identity between subunit isoforms is 75% for the α-subunit and 71% for the β-subunit, respectively, whereas the three γ isoforms exhibit a wider range of mass and sequence.
FIGURE 1.
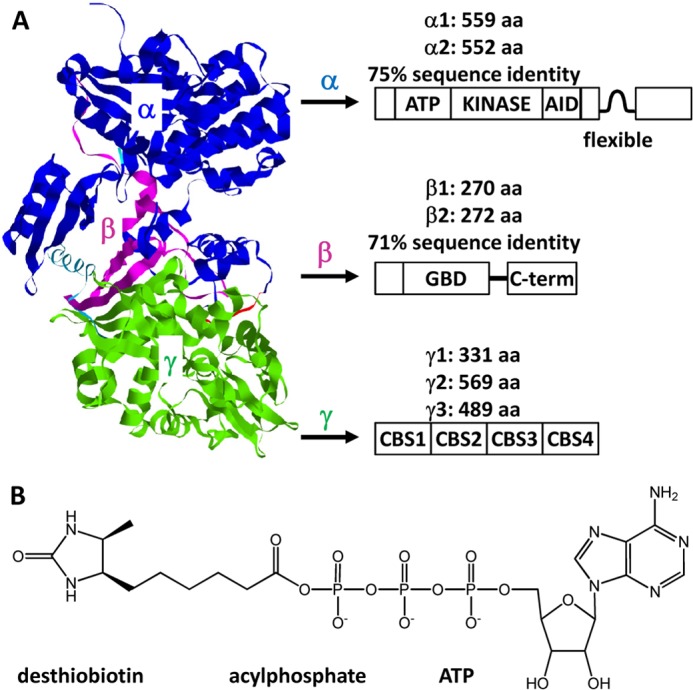
A, primary basis for diversity of the AMPK heterotrimer. The ribbon diagram of a mammalian AMPK structure is from Protein Data Bank code 2Y94 (16). Sequence information refers to canonical UniProtKB entries for human AMPK polypeptides. aa, amino acids; AID, autoinhibitory domain; GBD, glycogen-binding domain; CBS, cystathionine β-synthase sequence. B, structure of the ActivX ATP probe designed to couple desthiobiotin to conserved Lys in the ATP-binding loop of a protein kinase.
The catalytic α-subunit contains a classical serine/threonine protein kinase domain located near the N terminus with a conserved ATP-binding loop. Phosphorylation of Thr-172 in this loop by upstream kinases is essential for activation (11, 14). The C-terminal part of the β-subunit acts as a scaffold for its interaction with the α- and γ-subunits. In addition, the β-subunit contains a glycogen-binding domain. The main feature of the γ-subunit is four tandem repeats of the cystathionine β-synthase sequence that form two Bateman domains for selective binding to AMP, ADP, or ATP.
Different types of cells and tissues express distinct combinations of the AMPK subunit isoforms (12, 17–21). In an aspect relevant to drug discovery, there have also been indications of interspecies differences with respect to the tissue specificity of AMPK subtypes. Stephenne et al. (12) recently noted that human and rodent hepatocytes differ in their expression of AMPK subunit isoforms. They found that α1β2γ1 was the major isoform in human hepatocytes but that liver tissues of both rat and mouse predominantly expressed α2β1γ1. In developing tissue-specific therapeutic interventions and selecting preclinical species, understanding the relative abundance of AMPK isoforms in different tissues and species is likely to strengthen correlations of these isoform distributions with disease pathology.
Activity-based chemical proteomics is a powerful tool to identify new drug targets and define the specificity of drug action in complex biological matrices (22–26). Under this approach, a chemical probe is designed to react with enzymes of a specific class irrespective of their substrate specificities. The same probe bears a fluorescent reporter group or affinity handle (often biotin) to facilitate the detection and/or capture of labeled proteins. Here we have used an activity-based probe to characterize the tissue- and species-specific expression of AMPK subunit isoforms. The approach integrates an activity-based desthiobiotin-ATP probe (ActivX ATP probe) for kinase labeling and affinity enrichment and subsequent mass spectrometric identification and relative quantification of subunit isoforms (Fig. 1). The probe consists of a modified biotin attached to the nucleotide by a labile acylphosphate bond (27, 28). It covalently modifies the conserved Lys residue(s) in the ATP-binding loop of protein kinases, also transferring the biotin-like moiety to other ATPases such as chaperones and the many metabolic kinases. We applied this method to studying AMPK isoform distribution in cell lines, hepatocytes, skeletal muscles, and heart tissues of different species. The expression of AMPK isoforms assessed by the chemical proteomics was compared with the relative mRNA levels of the corresponding isoforms measured by microarray.
EXPERIMENTAL PROCEDURES
Materials
Thermo Scientific Pierce Kinase Enrichment kits with the ActivX ATP probe were purchased from Thermo Fisher Scientific (Waltham, MA). The kits contained all materials to carry out kinase labeling and affinity pulldown. HEK293 and HepG2 cells were cultured in house. Recombinant human AMPK α2β2γ3 isoform was expressed as described (29). Tissues used in the study appear in Table 1. Primary rat hepatocytes were freshly isolated from male Wistar Han rats via a two-step, in situ liver perfusion method as described (30). Digestion and perfusion buffers were obtained in a Hepatocyte Isolation kit (Worthington, catalog number LK002060). Cryopreserved human and rat primary hepatocytes were purchased from Xenotech (Lenexa, KS), Zenbio (Research Triangle Park, NC), and BD Biosciences. Frozen human and rat skeletal muscles and hearts were from Biochain Institute (Hayward, CA). NuPAGE Novex Bis-Tris minigels were from Invitrogen. Sequence-grade modified trypsin was purchased from Promega (Madison, WI). Reprosil-Pur Basic C18 material (3 μm, 120 Å) was from Dr. Maisch GmbH (Ammerbuch-Entringen, Germany). All other chemicals were obtained from Sigma-Aldrich.
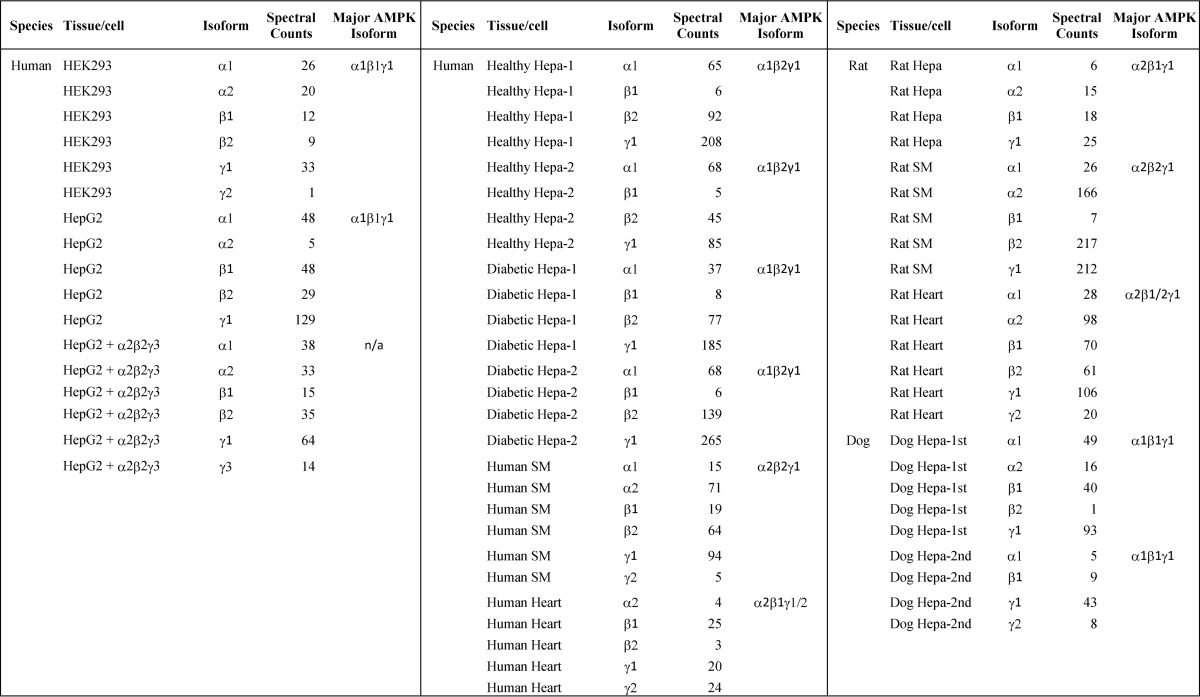
TABLE 1.
AMPK subunit isoforms identified in different species and tissues/cells
Spectral counts were used to measure relative levels of isoform expression. SM, skeletal muscle; Hepa, hepatocyte.
Gene Expression Analysis
Basal mRNA expression data for AMPK subunits in liver tissues across different in vivo species were generated by using corresponding Affymetrix GeneChip arrays (Human Genome U133 Plus 2.0, Rat Genome 230 2.0, and Canine Genome 2.0). mRNA from normal untreated tissue samples was subjected to microarray analysis. Data were normalized using the Affymetrix MAS5 method (31).
Lysate Preparation
Tissue lysate preparation, protein labeling, and affinity capture were carried out according to the manufacturer's manual (Thermo Kinase Enrichment kits) with minor revisions. Briefly, frozen tissues (∼250 mg) were cut into small pieces and homogenized in 2.0 ml of ice-cold cell lysis buffer (25 mm sodium phosphate, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 5% glycerol, 1 mm EDTA, protease and phosphatase inhibitors). The homogenate was cooled on ice for 5 min and then sonicated for 30 s. The primary hepatocyte, HepG2, and HEK293 cell pellets (107 each) were resuspended in 2 ml of the cell lysis buffer and subsequently homogenized for 2 min using a tip sonicator. The tissue and cell lysates were centrifuged at 14,000 × g for 10 min at 4 °C. The supernatant was collected and exchanged to the kinase reaction buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 0.1% Triton X-100) using a Zeba spin desalting column following the manufacturer's protocol. To the column eluates placed on ice were added protease/phosphatase inhibitors and 1 m MgCl2 (final concentration of 20 mm).
Chemical Labeling and Affinity Enrichment
The samples were treated with freshly prepared desthiobiotin-ATP reagent (20 μm) for 10 min at room temperature. After excess reagent was removed using the Zeba desalting column, biotinylated proteins were captured by incubation with 150 μl of the 50% streptavidin-agarose resin for 1 h in a rotator. The beads were washed with 4 × 0.4 ml of the lysis buffer followed by a final wash with 0.4 ml of PBS buffer. The bound proteins were eluted by boiling in 2 × 100 μl of NuPAGE loading buffer for 5 min.
SDS-PAGE Separation and In-gel Digestion
To reduce disulfide bonds, the samples were incubated with 5 mm dithiothreitol (DTT) for 1 h at 60 °C. The resulting –SH groups were subsequently alkylated with 15 mm iodoacetamide for 30 min at room temperature in the dark. Half of the aliquot was concentrated in a SpeedVac and loaded to a 4–12% NuPAGE Novex Bis-Tris precast minigel. After Coomassie Blue staining, the gel was subsequently cut into 10 slices and washed with 10 mm ammonium bicarbonate in 50% acetonitrile (ACN) and 100% ACN sequentially. The gel slices were incubated with sequence-grade modified trypsin (1 μg/slice; Promega) in 25 mm ammonium bicarbonate at 37 °C overnight. The ∼60-μl digestion was quenched by adding 40 μl of 4% formic acid in ACN. The peptides were concentrated in a SpeedVac and subjected to LC-MS/MS analysis as described below.
LC-MS/MS Analysis and Peptide Identification
An aliquot (7 μl) of each sample was loaded onto a PicoFrit column (New Objective, Woburn, MA) packed with reversed-phase Reprosil-Pur Basic C18 particles (75 μm × 10 cm) and coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher, Waltham, MA). Peptides were separated at a flow rate of 0.25 μl/min using a 120-min linear gradient ranging from 8 to 35% B (mobile phase A, 0.1% formic acid, 2% ACN; mobile phase B, 98% ACN, 0.1% formic acid). Electrospray voltage was 2.3 kV. The instrumental method consisted of a full MS scan (scan range, 300–1650 m/z with 60,000 full-width half-maximum resolution at m/z 400; target value, 1 × 106) followed by data-dependent collision-induced dissociation scans of the 20 most intense precursor ions. Peptide precursor ions were selected with an isolation window of 2.0 Da. Target ion quantities for MS and MS2 were 1 × 106 and 5 × 104, respectively. Dynamic exclusion was implemented with a repeat count of 1 and exclusion duration of 60 s. The mass spectra were searched against the human, dog, or rat International Protein Index database using Proteome Discoverer 1.3 (Thermo Fisher) with the Mascot search engine (32) with a false discovery rate of 0.1%. The mass accuracy was set to 10 ppm for precursor and 0.5 Da for fragment ions. The search parameters took into account two missed cleavages for trypsin, static modification of S-carboxamidomethylation at Cys (+57.0215 Da), and differential modifications of oxidation on Met (+15.9949 Da) and desthiobiotinylation on Lys (+196.1212 Da).
RESULTS
We sought to develop a universal approach to capture the respective populations of AMPK α-, β-, and γ-subunits in several tissues of multiple species. The strategy integrated chemical labeling of kinases, subsequent affinity enrichment, and the use of mass spectrometry-based proteomics for polypeptide identification and relative quantification (Fig. 2). An essential aspect was to lyse tissues and cells under conditions that retained the quaternary structure of AMPK heterotrimers so that covalent delivery of biotin (actually desthiobiotin) to the α (kinase) subunit of AMPK resulted in biotinylated heterotrimers that could be affinity-captured intact. After the capture step, AMPK (one of the many proteins labeled by this probe) was disrupted, and subunits were identified as independent polypeptides. This allowed inferences about heterotrimer composition when one isoform of a subunit predominated but left uncertainty when the populations were mixed.
FIGURE 2.
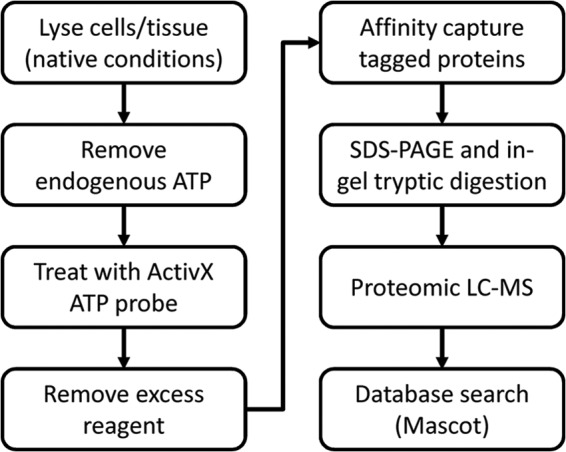
Flowchart of covalent labeling and affinity capture of kinases using the desthiobiotin-ATP probe followed by identification and relative quantitation of AMPK isoforms in cells or tissues.
As noted, the ActivX ATP probe modifies a broad population of ATP-binding proteins (kinases, transporter proteins, chaperones, etc.). Lysates were allowed to react with desthiobiotin-ATP reagent, excess reagent was removed using a desalting column, and the biotin-tagged proteins were captured using streptavidin beads. Captured proteins were then further fractionated by SDS-PAGE after which the gel lane was sectioned and proteins in each section were digested with trypsin. The resulting peptides were identified by nano-LC and mass spectrometry.
To examine its effectiveness, we first applied the protocol to HEK293 cells. This cell line contains multiple AMPK isoforms (33, 34). Fig. 3 shows SDS-PAGE fractionation of desthiobiotin-tagged proteins captured by the streptavidin beads. Overall, more than 600 proteins were identified with ≥2 unique peptides from 10 gel slices (data not shown). Consistent with earlier reports (35), most of the top hits were ATP-binding proteins such as heat shock proteins, kinases, and nucleotide-binding proteins, confirming selectivity of the probe. Proteins that naturally contain biotin were also identified.
FIGURE 3.
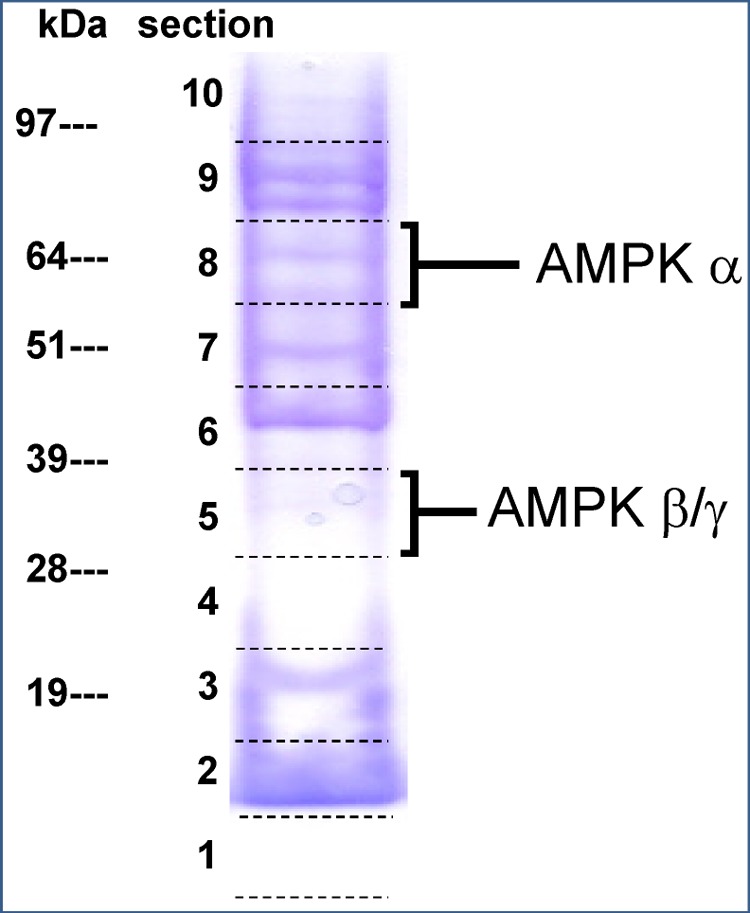
SDS-PAGE fractionation of biotin-tagged proteins captured from HEK293 cells. AMPK α1 and α2 isoforms were identified in slice 8, and isoforms of β- and γ-subunits were identified in slice 5.
The α1 and α2 isoforms were identified in gel slice 8 (Fig. 3), whereas β1, β2, γ1, and γ2 were all identified in slice 5. All these polypeptides migrated to positions consistent with their respective masses. Mapping identified peptide segments onto subunit isoform sequences (Fig. 4) indicated that both isoforms of the α- and β-subunits were present with similar abundances and that the predominant γ isoform was γ1 (Table 1). The fact that multiple isoforms were identified supported the recently published view that the ActivX ATP probe pulls down protein kinases without pronounced bias between different kinases or between their active and inactive states (36). In addition, mass spectrometry identified the desthiobiotin-tagged peptide VAVKILNR. Its MS/MS spectrum unambiguously placed the modification on the lysyl residue, the assignment being justified by the characteristic mass shift of +196.1 Da for y5, y6, and y7 ions (Fig. 5). Sequence alignment confirmed that this is a conserved peptide located in the kinase ATP-binding loop of both forms of the α-subunit in all mammalian species. This result confirmed expectations that the chemical probe covalently and specifically reacted at the ATP-binding site of the α-subunit.
FIGURE 4.

AMPK isoforms identified in HEK293 cells. The peptides highlighted in green or purple are isoform-unique. The sequences marked in yellow are common to subunit isoforms. The sequence printed in red contains the site of covalent modification by the ActivX ATP probe.
FIGURE 5.

MS/MS spectrum of the biotinylated peptide VAVK*ILNR (asterisk denotes modification) located in the ATP-binding pocket of the human and rat α-subunits. The conserved lysyl residue is covalently labeled with the desthiobiotin moiety.
After further testing with HepG2 cells, the method was applied without further modification to a number of tissues from human, dog, and rat. Fig. 6 shows heat maps demonstrating peptide coverage of different AMPK isoforms in hepatocytes of healthy individuals and diabetic patients.
FIGURE 6.
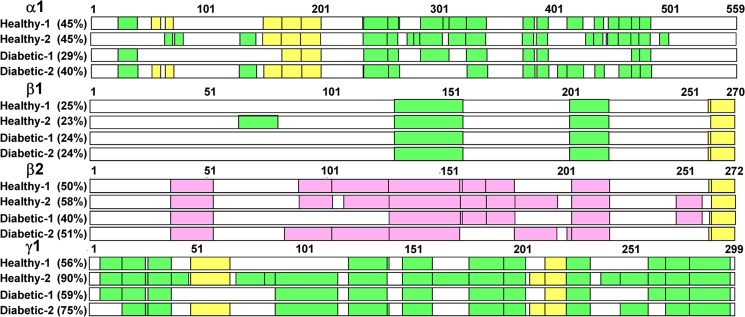
Proteomic peptide coverage of different AMPK isoforms in hepatocytes of healthy individuals and diabetic patients. Peptide sequences identical in subunit isoforms are marked in yellow. Peptide sequences unique to a single isoform are marked in green or purple.
Relative abundance of the subunit isoforms was assessed by comparing the “spectral counts” of the isoform-unique peptides (Table 1) (37–46). For instance, in the sample designated healthy hepatocyte-1, β1 and β2 isoforms were both identified with spectral counts of 6 and 92 (1:15), respectively. Therefore, β2 was the predominant β-subunit in human liver by a very large factor. Likewise, β2 was the major isoform in human skeletal muscle but a minor isoform in human heart.
Spectral counting has widely been utilized for the relative quantification of proteins in multiple biological samples (37–46), but its application to the relative quantification of isoforms remains to be validated. In theory, the respective peak intensities of highly homologous peptides should be an ideal measure of the relative abundance of subunit isoforms provided that these peptides are co-eluted and identified in the same gel slice. In light of this, we calculated the peak intensity ratio of APPILPPHLLQVILNK (a β1 peptide) and the co-eluting SPPILPPHLLQVILNK (a β2 peptide) and plotted it against the spectral count ratio for β1 and β2 isoforms. As shown in Fig. 7, spectral counts and peak intensity were well correlated for most of the samples examined.
FIGURE 7.
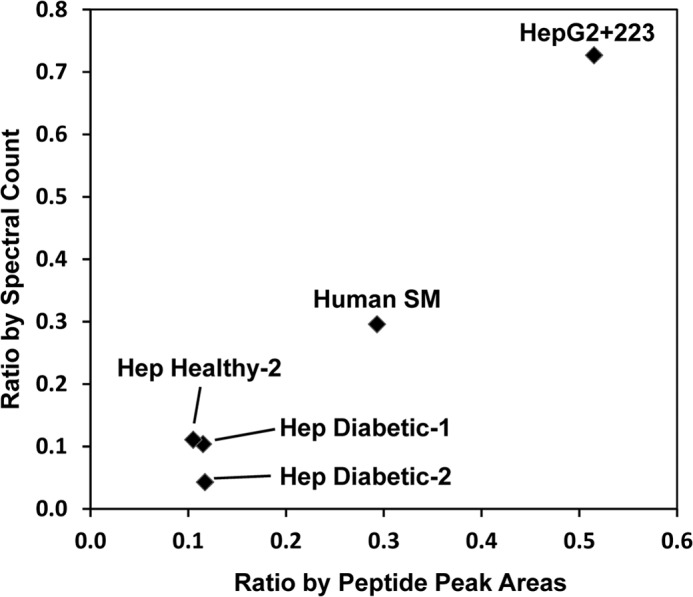
Correlation between relative spectral counts of the β1 and β2 isoforms and the relative peak intensities of peptides APPILPPHLLQVILNK (β1) and SPPILPPHLLQVILNK (β2). SM, skeletal muscle; Hep, hepatocyte.
The overall reproducibility of the proposed approach and the reliability of the analytical results were examined by processing four different human liver hepatocyte preparations at different dates, two from healthy individuals and two from diabetic patients. As shown in Table 1, although the individual peptides identified in each sample could differ, α1β2γ1 was identified as the predominant isoform in all human hepatocyte samples analyzed.
With the same approach, we analyzed AMPK isoforms in rat and dog tissues. The predominant AMPK isoforms from these studies are summarized in Table 1.
It is noteworthy that γ1 was the predominant isoform of its type in all tissues analyzed except in human heart where the γ2 isoform appeared to be equally abundant. The γ3 isoform was never detected in our proteomics experiments, although it was found in human skeletal muscles by both immunoblotting and mRNA analysis (18, 21, 47). To examine whether the failure to detect the γ3 isoform proteomically was attributable to its low abundance or was caused by dissociation of the isoform from the heterotrimeric complex, we spiked in 0.1 μg of the recombinant α2β2γ3 trimer to the HepG2 cell lysate and followed the entire protocol to enrich the AMPK complex. In addition to the γ1 isoform originally present in HepG2 cells, four peptides unique to the γ3 isoform were identified.
To compare transcriptomic with proteomic estimates of the expression of AMPK isoforms, mRNA microarray expression data from human, rat, and dog liver tissues were obtained from a Pfizer gene expression database. Fig. 8 shows the correlation between mRNA (relative expression) and proteomics data (spectral counting) across human (A), rat (B), and dog (C). Clearly, α1β2γ1, α2β1γ1, and α1β1γ1 mRNAs were the major isoforms expressed in human, rat, and dog liver tissues, respectively, and these results were generally consistent with hepatocyte proteomics data. There was good correlation between mRNA and protein expression for α- and β-subunit isoforms. However, both γ2 and γ3 mRNAs were detected in human and rat liver tissues by microarray analysis, but neither were detected at the protein level.
FIGURE 8.
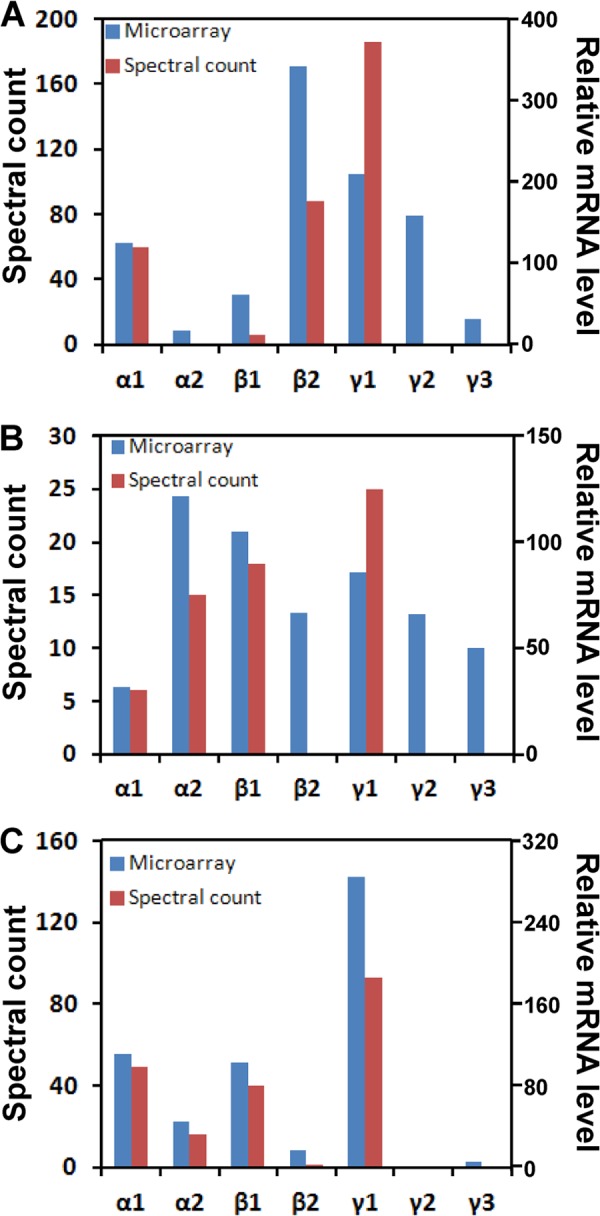
Relative expression levels of AMPK subunit isoform protein and mRNA in human (A), rat (B), and dog (C) liver tissues. The protein and mRNA expressions were assessed by number of spectra counts and by microarray analysis, respectively.
DISCUSSION
The central significance of AMPK as a metabolic regulator makes it an attractive potential target for drug action in diseases of positive malnutrition (5, 14, 48). Efforts are in hand to evolve prototypical activators into safe and effective drugs (49–51). Their presumptive action would be to divert cellular activity away from energy-storing pathways such as fatty acid synthesis in the direction of energy-refining and ATP-producing alternatives. Preclinical reports indicate the potential success of such agents (49).
A central challenge in this project is the molecular complexity of AMPK, which potentially exists in up to 12 heterotrimeric forms. Intuition and objective data both suggest that AMPK subtypes are functionally distinct in terms of sensitivity to variations in the cellular energy status and other aspects related to control of their specific activities. This may include their susceptibility to phosphorylation by upstream kinases and dephosphorylation by protein phosphatases (52–55). Therefore, considerable evidence suggests that AMPK, nominally a single target, is in fact a family of related enzymes and that its members have distinct responsibilities in the control of cellular function. For the most part, however, current knowledge does not identify these specific roles.
The present chemoproteomic work adds a new line of investigation to efforts to study the tissue specificity of the expression of different AMPK subunits. It complements the main methods used previously: the analysis of mRNA and immunoprecipitation of AMPK.
Transcript-based methods are direct and fast but can only inspect expression at the message level of single subunits. They are also not certain to reflect the relative levels of abundance of polypeptides (56).
In contrast, immunocapture of AMPK can be designed as a multistage process that allows the identities of heterotrimers to be defined in full or in part (21). Challenges include the variability of antigen detection by immunoblotting methods and the need to distinguish between closely sequence-related variants of the AMPK subunits (12, 17, 18). Even so, antibody-based methods have provided the two most incisive demonstrations of distinct functions for specific AMPK subtypes.
In the first, Birk and Wojtaszewski (18) reported that human skeletal muscle possesses three AMPK complexes (α2β2γ1, α1β2γ1, and α2β2γ3) but that only α2β2γ3 is activated during high intensity exercise. More recently, Pinter et al. (57) reported specific localization of AMPK α2β2γ2 in the cytokinetic apparatus of human endothelial cells and showed that the relatively abundant α1 and γ1 polypeptides were concentrated in the cytoskeleton.
Diversity of this kind is probably crucial to achieving tissue-specific metabolic profiles and to the cell and tissue specificities of AMPK physiological functions as distinct AMPK isoforms may differ in their sensitivities to allosteric activation in response to various stimuli (52–55). The results support the intuition that different subtypes have specialized functions.
The present work explores the potential of capturing AMPK from complex mixtures and identifying its subunits by LC-MS-based proteomic analysis. Using the ActivX ATP probe (35), we captured AMPK (and many other ATP-binding proteins) from cell and tissue lysates, fractionated the resulting complex mixtures of polypeptides, digested the fractions, and obtained results representing the relative abundance of different AMPK component polypeptides. Like transcript analysis, this method does not address intact heterotrimers, but the principal composition of AMPK in cells or tissues in which a single alternative predominates can be inferred from the data. Comparisons of peak intensities from closely homologous peptides of subunit subtypes sharpen the estimates of relative abundance (Fig. 7).
Using a chemical probe to capture proteins that can exist in multiple forms (e.g. inactive and active forms of protein kinases) invites concerns about bias between the different forms of the target. A recent study of the ActivX ATP probe by Gygi and co-workers (36) found no evidence that either the active or inactive form of any of the kinases studied in cell lines related to breast cancer were preferentially recovered. By analogy with those results, the relative levels of different AMPK subunits derived from use of the ActivX ATP probe are likely to be valid. This is further supported by congruence in the present work between the chemoproteomics results and transcriptomics data (Fig. 8), a relationship that must not be taken for granted when data of only one of the two types are available (56).
An obvious question is whether AMPK labeling with the ActivX ATP probe can lead to recovery and characterization of intact heterotrimers. Possibly it can serve as a first enrichment step that could usefully be combined with the serial use of antibodies to different AMPK polypeptides as pioneered by other groups (18, 21).
The interspecies differences noted elsewhere (12) and in the present work between the AMPK subunit populations of human and rodent hepatocytes require interpretation. Final answers may not be at hand, but a major question can be framed and addressed.
For example, if different subtypes of AMPK (different AMPK heterotrimers) are presumed to contribute differently to the management of cellular functions, then such distinct roles could be realized not only by enzymological differences but also by differential subcellular localizations. Features of each kind are known. First, allosteric but non AMP-competitive activators that only stimulate β1-containing forms (34, 51) may emulate an unknown natural cellular component that restricts its effect to β1-AMPK-based signaling pathways. Second, AMPK interacts variably with cell membranes (through the α-N-myristoylated N terminus of the β-chain) (58) and glycogen (through the glycogen-binding domain also on the β-subunit) (59).
A provisional interpretation of the interspecies differences is that human and rat hepatocytes use similar signaling pathways but that each species physically implements these pathways at different levels according to its metabolic need. This proposal, the most conservative available, does not fit well with existing data. It predicts that human and rodent hepatocytes should respond to specific external stimuli or stresses through the same pathways even if the different component polypeptides of AMPK are represented in different ratios in the different species.
Available evidence favors the alternative, which is that the species have evolved AMPK-based signaling systems that are not directly orthologous. For example, liver-selective knock-out of the α2 polypeptide in mice left the animals without normal control of blood glucose levels (60), but it is unlikely that the minor (at most) complement of α2 in human hepatocytes (Fig. 8) carries the same function. A strong interspecies difference was also detected in studies of the effects of metformin in human, rat, and mouse hepatocytes (12).
Massive effort to date has begun to clarify the profound subtlety surrounding AMPK molecular diversity and specificity. All applicable biochemical tools will have to be deployed to continue to resolve the many unanswered questions. This work has shown the applicability of a chemical biology strategy to capturing and advancing the characterization of AMPK. When joined with additional strategies (immunocapture being the most obvious example), this method will continue to contribute to resolving the myriad functions and properties of AMPK.
Footnotes
- AMPK
- AMP-activated protein kinase
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ACN
- acetonitrile.
REFERENCES
- 1. Hardie D. G., Ross F. A., Hawley S. A. (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gowans G. J., Hawley S. A., Ross F. A., Hardie D. G. (2013) AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 18, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardie D. G., Carling D., Gamblin S. J. (2011) AMP-activated protein kinase: also regulated by ADP? Trends Biochem. Sci. 36, 470–477 [DOI] [PubMed] [Google Scholar]
- 4. Steinberg G. R., Kemp B. E. (2009) AMPK in health and disease. Physiol. Rev. 89, 1025–1078 [DOI] [PubMed] [Google Scholar]
- 5. Fogarty S., Hardie D. G. (2010) Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim. Biophys. Acta 1804, 581–591 [DOI] [PubMed] [Google Scholar]
- 6. Hardie D. G., Sakamoto K. (2006) AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology 21, 48–60 [DOI] [PubMed] [Google Scholar]
- 7. Viollet B., Guigas B., Leclerc J., Hébrard S., Lantier L., Mounier R., Andreelli F., Foretz M. (2009) AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol. 196, 81–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Viollet B., Mounier R., Leclerc J., Yazigi A., Foretz M., Andreelli F. (2007) Targeting AMP-activated protein kinase as a novel therapeutic approach for the treatment of metabolic disorders. Diabetes Metab. 33, 395–402 [DOI] [PubMed] [Google Scholar]
- 9. Boyle J. G., Salt I. P., McKay G. A. (2010) Metformin action on AMP-activated protein kinase: a translational research approach to understanding a potential new therapeutic target. Diabet. Med. 27, 1097–1106 [DOI] [PubMed] [Google Scholar]
- 10. Hardie D. G. (2007) AMP-activated protein kinase as a drug target. Annu. Rev. Pharmacol. Toxicol. 47, 185–210 [DOI] [PubMed] [Google Scholar]
- 11. Hardie D. G. (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stephenne X., Foretz M., Taleux N., van der Zon G. C., Sokal E., Hue L., Viollet B., Guigas B. (2011) Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 54, 3101–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Viollet B., Guigas B., Sanz Garcia N., Leclerc J., Foretz M., Andreelli F. (2012) Cellular and molecular mechanisms of metformin: an overview. Clin. Sci. 122, 253–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang B. B., Zhou G., Li C. (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 9, 407–416 [DOI] [PubMed] [Google Scholar]
- 15. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arad M., Seidman C. E., Seidman J. G. (2007) AMP-activated protein kinase in the heart: role during health and disease. Circ. Res. 100, 474–488 [DOI] [PubMed] [Google Scholar]
- 18. Birk J. B., Wojtaszewski J. F. (2006) Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. J. Physiol. 577, 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheung P. C., Salt I. P., Davies S. P., Hardie D. G., Carling D. (2000) Characterization of AMP-activated protein kinase γ-subunit isoforms and their role in AMP binding. Biochem. J. 346, 659–669 [PMC free article] [PubMed] [Google Scholar]
- 20. Mahlapuu M., Johansson C., Lindgren K., Hjälm G., Barnes B. R., Krook A., Zierath J. R., Andersson L., Marklund S. (2004) Expression profiling of the γ-subunit isoforms of AMP-activated protein kinase suggests a major role for γ3 in white skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 286, E194–E200 [DOI] [PubMed] [Google Scholar]
- 21. Wojtaszewski J. F., Birk J. B., Frøsig C., Holten M., Pilegaard H., Dela F. (2005) 5′AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. J. Physiol. 564, 563–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zanders E. D. (2012) Overview of chemical genomics and proteomics. Methods Mol. Biol. 800, 3–10 [DOI] [PubMed] [Google Scholar]
- 23. Wierzba K., Muroi M., Osada H. (2011) Proteomics accelerating the identification of the target molecule of bioactive small molecules. Curr. Opin. Chem. Biol. 15, 57–65 [DOI] [PubMed] [Google Scholar]
- 24. Van Summeren A., Renes J., van Delft J. H., Kleinjans J. C., Mariman E. C. (2012) Proteomics in the search for mechanisms and biomarkers of drug-induced hepatotoxicity. Toxicol. in Vitro 26, 373–385 [DOI] [PubMed] [Google Scholar]
- 25. Miao Q., Zhang C. C., Kast J. (2012) Chemical proteomics and its impact on the drug discovery process. Expert Rev. Proteomics 9, 281–291 [DOI] [PubMed] [Google Scholar]
- 26. Drewes G. (2012) Chemical proteomics in drug discovery. Methods Mol. Biol. 803, 15–21 [DOI] [PubMed] [Google Scholar]
- 27. Cravatt B. F., Wright A. T., Kozarich J. W. (2008) Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 77, 383–414 [DOI] [PubMed] [Google Scholar]
- 28. Patricelli M. P., Nomanbhoy T. K., Wu J., Brown H., Zhou D., Zhang J., Jagannathan S., Aban A., Okerberg E., Herring C., Nordin B., Weissig H., Yang Q., Lee J. D., Gray N. S., Kozarich J. W. (2011) In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 18, 699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rajamohan F., Harris M. S., Frisbie R. K., Hoth L. R., Geoghegan K. F., Valentine J. J., Reyes A. R., Landro J. A., Qiu X., Kurumbail R. G. (2010) Escherichia coli expression, purification and characterization of functional full-length recombinant α2β2γ3 heterotrimeric complex of human AMP-activated protein kinase. Protein Expr. Purif. 73, 189–197 [DOI] [PubMed] [Google Scholar]
- 30. Berry M. N., Friend D. S. (1969) High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J. Cell Biol. 43, 506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hubbell E., Liu W. M., Mei R. (2002) Robust estimators for expression analysis. Bioinformatics 18, 1585–1592 [DOI] [PubMed] [Google Scholar]
- 32. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 33. Hwang G. W., Tobita M., Takahashi T., Kuge S., Kita K., Naganuma A. (2010) siRNA-mediated AMPKα1 subunit gene PRKAA1 silencing enhances methylmercury toxicity in HEK293 cells. J. Toxicol. Sci. 35, 601–604 [DOI] [PubMed] [Google Scholar]
- 34. Hawley S. A., Fullerton M. D., Ross F. A., Schertzer J. D., Chevtzoff C., Walker K. J., Peggie M. W., Zibrova D., Green K. A., Mustard K. J., Kemp B. E., Sakamoto K., Steinberg G. R., Hardie D. G. (2012) The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336, 918–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patricelli M. P., Szardenings A. K., Liyanage M., Nomanbhoy T. K., Wu M., Weissig H., Aban A., Chun D., Tanner S., Kozarich J. W. (2007) Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–358 [DOI] [PubMed] [Google Scholar]
- 36. McAllister F. E., Niepel M., Haas W., Huttlin E., Sorger P. K., Gygi S. P. (2013) Mass spectrometry based method to increase throughput for kinome analyses using ATP probes. Anal. Chem. 85, 4666–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 38. Balgley B. M., Wang W., Song T., Fang X., Yang L., Lee C. S. (2008) Evaluation of confidence and reproducibility in quantitative proteomics performed by a capillary isoelectric focusing-based proteomic platform coupled with a spectral counting approach. Electrophoresis 29, 3047–3054 [DOI] [PubMed] [Google Scholar]
- 39. Braisted J. C., Kuntumalla S., Vogel C., Marcotte E. M., Rodrigues A. R., Wang R., Huang S. T., Ferlanti E. S., Saeed A. I., Fleischmann R. D., Peterson S. N., Pieper R. (2008) The APEX quantitative proteomics tool: generating protein quantitation estimates from LC-MS/MS proteomics results. BMC Bioinformatics 9, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cooper B., Feng J., Garrett W. M. (2010) Relative, label-free protein quantitation: spectral counting error statistics from nine replicate MudPIT samples. J. Am. Soc. Mass Spectrom. 21, 1534–1546 [DOI] [PubMed] [Google Scholar]
- 41. Piersma S. R., Fiedler U., Span S., Lingnau A., Pham T. V., Hoffmann S., Kubbutat M. H., Jiménez C. R. (2010) Workflow comparison for label-free, quantitative secretome proteomics for cancer biomarker discovery: method evaluation, differential analysis, and verification in serum. J. Proteome Res. 9, 1913–1922 [DOI] [PubMed] [Google Scholar]
- 42. Borg J., Campos A., Diema C., Omeñaca N., de Oliveira E., Guinovart J., Vilaseca M. (2011) Spectral counting assessment of protein dynamic range in cerebrospinal fluid following depletion with plasma-designed immunoaffinity columns. Clin. Proteomics 8, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Houston N. L., Lee D. G., Stevenson S. E., Ladics G. S., Bannon G. A., McClain S., Privalle L., Stagg N., Herouet-Guicheney C., MacIntosh S. C., Thelen J. J. (2011) Quantitation of soybean allergens using tandem mass spectrometry. J. Proteome Res. 10, 763–773 [DOI] [PubMed] [Google Scholar]
- 44. Mosley A. L., Sardiu M. E., Pattenden S. G., Workman J. L., Florens L., Washburn M. P. (2011) Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes. Mol. Cell. Proteomics. 10, M110.000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neilson K. A., Ali N. A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., Lee A., van Sluyter S. C., Haynes P. A. (2011) Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 [DOI] [PubMed] [Google Scholar]
- 46. Vogel C., Marcotte E. M. (2012) Label-free protein quantitation using weighted spectral counting. Methods Mol. Biol. 893, 321–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frøsig C., Jørgensen S. B., Hardie D. G., Richter E. A., Wojtaszewski J. F. (2004) 5′-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 286, E411–E417 [DOI] [PubMed] [Google Scholar]
- 48. Gruzman A., Babai G., Sasson S. (2009) Adenosine monophosphate-activated protein kinase (AMPK) as a new target for antidiabetic drugs: a review on metabolic, pharmacological and chemical considerations. Rev. Diabet. Stud. 6, 13–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cool B., Zinker B., Chiou W., Kifle L., Cao N., Perham M., Dickinson R., Adler A., Gagne G., Iyengar R., Zhao G., Marsh K., Kym P., Jung P., Camp H. S., Frevert E. (2006) Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 3, 403–416 [DOI] [PubMed] [Google Scholar]
- 50. Pang T., Zhang Z. S., Gu M., Qiu B. Y., Yu L. F., Cao P. R., Shao W., Su M. B., Li J. Y., Nan F. J., Li J. (2008) Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J. Biol. Chem. 283, 16051–16060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scott J. W., van Denderen B. J., Jorgensen S. B., Honeyman J. E., Steinberg G. R., Oakhill J. S., Iseli T. J., Koay A., Gooley P. R., Stapleton D., Kemp B. E. (2008) Thienopyridone drugs are selective activators of AMP-activated protein kinase β1-containing complexes. Chem. Biol. 15, 1220–1230 [DOI] [PubMed] [Google Scholar]
- 52. McConell G. K., Lee-Young R. S., Chen Z. P., Stepto N. K., Huynh N. N., Stephens T. J., Canny B. J., Kemp B. E. (2005) Short-term exercise training in humans reduces AMPK signalling during prolonged exercise independent of muscle glycogen. J. Physiol. 568, 665–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nielsen J. N., Mustard K. J., Graham D. A., Yu H., MacDonald C. S., Pilegaard H., Goodyear L. J., Hardie D. G., Richter E. A., Wojtaszewski J. F. (2003) 5′-AMP-activated protein kinase activity and subunit expression in exercise-trained human skeletal muscle. J. Appl. Physiol. 94, 631–641 [DOI] [PubMed] [Google Scholar]
- 54. Wadley G. D., Lee-Young R. S., Canny B. J., Wasuntarawat C., Chen Z. P., Hargreaves M., Kemp B. E., McConell G. K. (2006) Effect of exercise intensity and hypoxia on skeletal muscle AMPK signaling and substrate metabolism in humans. Am. J. Physiol. Endocrinol. Metab. 290, E694–E702 [DOI] [PubMed] [Google Scholar]
- 55. Yu M., Stepto N. K., Chibalin A. V., Fryer L. G., Carling D., Krook A., Hawley J. A., Zierath J. R. (2003) Metabolic and mitogenic signal transduction in human skeletal muscle after intense cycling exercise. J. Physiol. 546, 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cooney J. M., Barnett M. P., Brewster D., Knoch B., McNabb W. C., Laing W. A., Roy N. C. (2012) Proteomic analysis of colon tissue from interleukin-10 gene-deficient mice fed polyunsaturated fatty acids with comparison to transcriptomic analysis. J. Proteome Res. 11, 1065–1077 [DOI] [PubMed] [Google Scholar]
- 57. Pinter K., Jefferson A., Czibik G., Watkins H., Redwood C. (2012) Subunit composition of AMPK trimers present in the cytokinetic apparatus: implications for drug target identification. Cell Cycle 11, 917–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oakhill J. S., Chen Z.-P., Scott J. W., Steel R., Castelli L. A., Ling N., Macaulay S. L., Kemp B. E. (2010) β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. U.S.A. 107, 19237–19241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Polekhina G., Gupta A., Michell B. J., van Denderen B., Murthy S., Feil S. C., Jennings I. G., Campbell D. J., Witters L. A., Parker M. W., Kemp B. E., Stapleton D. (2003) AMPK β subunit targets metabolic stress sensing to glycogen. Curr. Biol. 13, 867–871 [DOI] [PubMed] [Google Scholar]
- 60. Viollet B., Foretz M., Guigas B., Horman S., Dentin R., Bertrand L., Hue L., Andreelli F. (2006) Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J. Physiol. 574, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]