

Background: E. coli BirA is a both transcriptional repressor of the biotin operon and the biotin protein ligase.
Results: Structural and sequence integrity of the wing domain is important in both biotinylation and regulatory activities of BirA.
Conclusion: DNA-binding domain wing coordinates the two functions of BirA.
Significance: This is the first example of a wing domain that has a role in an enzymatic activity.
Keywords: AMP, Bacterial Genetics, Bacterial Metabolism, Enzyme Catalysis, Enzyme Mutation, Microbiology, Promoters, Transcription Factors, Transcription Regulation, Transcription Target Genes
Abstract
The BirA biotin protein ligase of Escherichia coli belongs to the winged helix-turn-helix (wHTH) family of transcriptional regulators. The N-terminal BirA domain is required for both transcriptional regulation of biotin synthesis and biotin protein ligase activity. We addressed the structural and functional role of the wing of the wHTH motif in both BirA functions. A panel of N-terminal deletion mutant proteins including a discrete deletion of the wing motif were unable to bind DNA. However, all the N-terminal deletion mutants weakly complemented growth of a ΔbirA strain at low biotin concentrations, indicating compromised ligase activity. A wing domain chimera was constructed by replacing the BirA wing with the nearly isosteric wing of the E. coli OmpR transcription factor. Although this chimera BirA was defective in operator binding, it was much more efficient in complementation of a ΔbirA strain than was the wing-less protein. The enzymatic activities of the wing deletion and chimera proteins in the in vitro synthesis of biotinoyl-5′-AMP differed greatly. The wing deletion BirA accumulated an off pathway compound, ADP, whereas the chimera protein did not. Finally, we report that a single residue alteration in the wing bypasses the deleterious effects caused by mutations in the biotin-binding loop of the ligase active site. We believe that the role of the wing in the BirA enzymatic reaction is to orient the active site and thereby protect biotinoyl-5′-AMP from attack by solvent. This is the first evidence that the wing domain of a wHTH protein can play an important role in enzymatic activity.
Introduction
The BirA biotin protein ligase of Escherichia coli is a transcriptional repressor that regulates the expression of the biotin operon (bioABFCD) in response to the availability of both biotin and the key metabolic protein that requires biotinylation for enzymatic function (1, 2) (Fig. 1, B and C). BirA is a unique transcriptional regulator in that it synthesizes its own regulatory ligand biotinoyl-5′-AMP (Bio-5′-AMP).2 Binding of Bio-5′-AMP by BirA results in dimerization of the protein and is required for bio operator binding (3). E. coli BirA is the best studied member of the biotin protein ligase (BPL) family, the members of which fall into two groups: group I ligases lack an N-terminal DNA-binding domain, whereas group II ligases (such as E. coli BirA) possess an N-terminal DNA-binding domain (4). The addition of the DNA-binding domain to the core catalytic region gives the Group II ligases their dual function as repressors and ligases. E. coli BirA is a 35.3-kDa protein that binds the 40-bp operator that controls both the bioA and bioBFCD promoters (5). The protein is composed of three distinct domains (6) (Fig. 1D): the N-terminal domain; the central catalytic core, which carries out the two-step ligation reaction; and the C-terminal domain. The N-terminal domain contains an N-terminal winged helix-turn-helix (wHTH) structure. The core domain, which has a structure and sequence that is well conserved throughout the BPL family, binds biotin and ATP to catalyze Bio-5′-AMP synthesis (Fig. 1A). BirA also catalyzes transfer of the adenylate biotin moiety to form an amide linkage with a specific lysine residue of a cognate acceptor protein. Finally, the C-terminal domain plays roles in dimerization, catalytic function, and DNA binding (7, 8). However, although BirA appears modular in the extant crystal structures, mutations in one domain of the protein often strongly influence functions residing in other domains.
FIGURE 1.

The biotin protein ligase reaction, the E. coli biotin operon regulation, and the three-dimensional structure of monomeric BirA protein. A shows the BPL activity of BirA. B and C show the general model of bio operon regulation. Green ovals denote BirA, tailed blue ovals are AccB, black dots represent biotin, and black dots with red pentagons denote biotinoyl-adenylate (Bio-5′-AMP). B shows the transcriptionally repressed state, whereas C shows the two modes of derepression of bio operon transcription engendered by either biotin limitation or excess unbiotinylated AccB acceptor protein. D shows the most recent BirA structure (Protein Data Bank 1HXD) in which the protein is complexed with biotinol-5′-AMP, an ester analog of Bio-5′-AMP (10). The N and C termini are denoted by N and C, respectively. Note that some loops were not visible in the crystals because of their high mobility and are shown as breaks in the chain. The figure was modified from Ref. 30.
The BirA wHTH structure (Fig. 1D) is well separated from the other domains of the protein in all of the extant crystal structures (6, 9, 10). Thus, it would seem straightforward to delete the E. coli BirA N-terminal domain and thereby convert the protein from a group II ligase to a group I ligase, a protein that would lack regulatory function but retain full ligase activity. However, the first BirA N-terminal deletion mutant was found to have compromised enzymatic activity as well as lack DNA binding ability (11). This deletion mutant protein catalyzed Bio-5′-AMP synthesis in vitro but bound both biotin and Bio-5′-AMP extremely poorly, although at saturating Bio-5′-AMP concentrations the second partial reaction, transfer of biotin to the cognate acceptor protein, proceeded normally.
In this paper, we address the function of the wing motif in DNA binding and ligase activity. Despite many attempts by us and other workers, no crystal structure of BirA complexed with its operator is available, and therefore the role of the wing in DNA binding is based only on in vitro studies that show mutual protection of the wing and operator DNA from chemical attack (5, 7). We report that deletion of the wing or its substitution with the wing of an unrelated transcription factor results in loss of DNA binding. Strikingly, the wing deletion protein is deficient in ligase activity, but ligase function is largely restored by insertion of the foreign wing. Moreover, a super-repressor mutation in the BirA wing suppresses the phenotypes of mutations in the biotin-binding loop of the ligase catalytic site. These and other data indicate that the wing organizes the ligase active site in the absence of DNA binding.
EXPERIMENTAL PROCEDURES
Chemicals and Culture Media
The medium used in the physiological experiments was LB or 2XYT as the rich medium, whereas the defined medium was M9 salts supplemented with 0.4% glucose (or another carbon source as stated) and 0.1% vitamin-free casamino acids (Difco) (25). The antibiotics were used at the following concentrations: 100 μg/ml sodium ampicillin, 50 μg/ml kanamycin sulfate, 25 μg/ml chloramphenicol, 12 μg/ml tetracycline HCl, and 50 μg/ml spectinomycin sulfate. The 15:1 mixture of ticarcillin disodium salt and potassium clavulanate (Research Products International) was used at 25 μg/ml. Integrated DNA Technologies provided oligonucleotides. PCR amplification was performed using Taq polymerase (New England BioLabs) and Pfu polymerase (Stratagene) according to the manufacturer's specifications. DNA constructs were sequenced by ACGT, Inc. All reagents and biochemicals were obtained from Sigma-Aldrich and Fisher, unless otherwise noted. New England BioLabs supplied restriction enzymes and T4 DNA ligase, NaF, and Na3VO4. Invitrogen provided SYBER Green I nucleic acid gel stain and the 6% DNA retardation Novex TBE gels. PerkinElmer Life Sciences provided [α-32P]ATP (3,000 Ci/mmol). Analtech TLC uniplates of microcrystalline cellulose matrix were provided by Sigma-Aldrich.
Bacterial Strains and Plasmids
All bacterial strains were derivatives of E. coli K-12 (supplemental Table S1). Bacterial cultures were grown in shake flasks at 37 °C, and growth was measured by absorbance at 600 nm using a Beckman DU600 spectrophotometer unless otherwise indicated. The E. coli B strain, BL21(λDE3) strain was used for protein expression. Strain VC926 was constructed by transducing the birA::km deletion mutant from strain VC156 by P1 phage transduction into BL21(λDE3) carrying the birA plasmid pVC33. The strain was maintained on LB medium supplemented with 400 nm biotin. Other DNA manipulations were carried out by standard procedures.
Plasmid Constructions
The various N-terminal deletions were constructed using naturally occurring restriction sites plus adaptor oligonucleotide cassettes (supplemental Table S2). The Beckett deletion (Δ2–65) was constructed from the plasmid of Xu and Beckett (11). The truncated BirA coding sequence was moved into plasmid pBR329 using the EcoRI and HindIII sites of the donor and vector. A cassette made by annealing oligonucleotides dBeck-T and dBeck-B (supplemental Table S2) was then ligated between the EcoRI and PvuII sites to give an initiation codon PciI site and inactivate the EcoRI site. This modified coding sequence was then moved into pCY374 as described below. The Maeda deletion (Δ2–63) was made by the same manipulations using dMaed-T and dMaed-B to give a BspHI site at the initiation codon. The other deletions (Fig. 2A) were constructed in a plasmid made for another purpose by first inserting the BspHI-BssHII fragment of pBA11 into pMTL22P cut with NcoI and MluI. This plasmid was digested with XhoI (a unique site located upstream of the initiation codon) and AlwNI and ligated to an oligonucleotide cassette that preserved the wild type amino acid sequence plus the XhoI and AlwnI sites, restored the BspHI initiation codon site, and introduced a KpnI site overlapping codons 26 and 27 to give pCY771. To make the Δ3–25 deletion, plasmid CY771 was digested with XhoI and KpnI and ligated to an annealed cassette of birA-KT and birA-KB that inactivated the XhoI site and preserved the KpnI and BspHI sites. The Δ3–60 deletion was made by the same manipulations except that the downstream site was the naturally occurring Bpu10I site, and the cassette consisted of birA-BT and birA-BB. The Δ3–40 deletion was made by the same manipulations except that the downstream site was the naturally occurring AlwNI site, and the cassette consisted of birA-AT and birA-AB. Finally, the Δwing deletion (Δ48–61) was made by ligating an annealed ΔWT and ΔWB cassette between the AlwNI and Bpu10I sites of an intermediate birA plasmid chosen for the lack of complicating vector restriction sites. Each of the deleted birA genes was inserted into pCY374 digested with BspHI and NgoMIV and ligated to the BspHI (or PciI depending on the construct) plus NgoMIV fragments encoding the various deletion alleles (which replaced the wild type birA gene of pCY374). Plasmid pCY374 is a derivative of pBA11 (19) that lacks the BspHI site downstream of the birA gene caused by digestion with NgoMIV and ligation under dilute conditions to delete a 528-bp fragment derived from the remains of tetracycline resistance gene of vector pACYC184.
FIGURE 2.
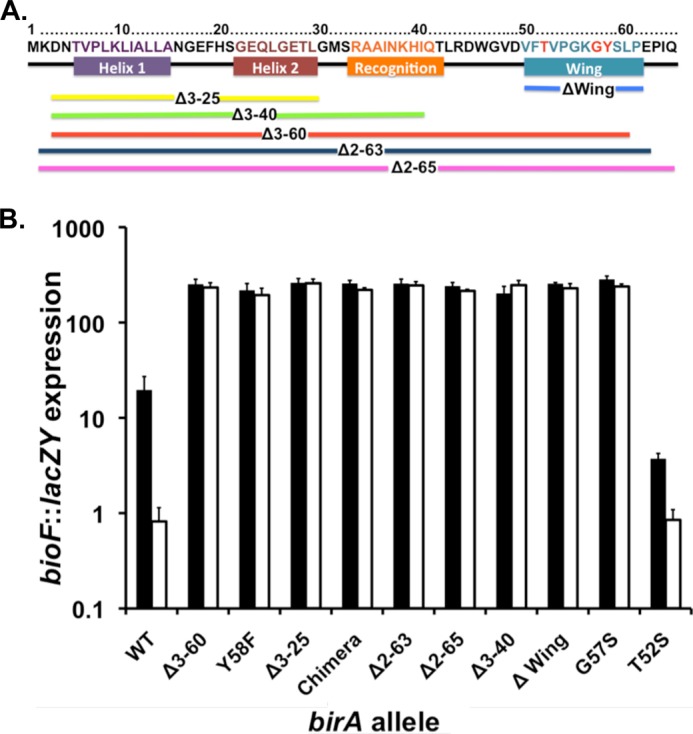
Properties of the BirA N-terminal deletion and point mutant proteins. A, location of the mutants studied. The colored bars show the extents of the various deletions. The BirA residues have the same color code as the helix (the recognition helix is helix 3) except for the wing structure where the residues altered by point mutations are given in red type. B, expression of the bio operon in cultures grown with 10 nm biotin (derepression conditions; solid bars) or 10 μm (repression conditions; open bars) biotin assayed by use of β-galactosidase encoded by a chromosomal bioF::lacZY fusion. The strains used were derivatives of strain VC618 cured of plasmid pVC18 (8). Single colonies were streaked on defined medium plates supplemented with 10 nm biotin and X-gal for phenotypic confirmation. Liquid cultures grown in the same medium lacking X-gal and were then assayed for bio operon transcription by β-galactosidase activity (“Experimental Procedures”). Note that the data are plotted on the y axis as a log scale.
For construction of the plasmid encoding BirA with the wing chimera, first the pVC26 fragment encoding the chimera was obtained using the DraIII and BstXI and ligated to plasmid pCY216 (26) digested with the same enzymes to give plasmid pVC27. Using primers VCC65 and VCC66 (supplemental Table S2), the region encoding the chimera wing was PCR-amplified to give a 598-bp DNA fragment. This fragment was then digested with PvuI and DraIII, gel-purified, and ligated to plasmid pBA11 digested with the same enzymes to give pVC28. Plasmids encoding the C-terminal hexahistidine-tagged derivatives of the chimera, Δ2–65 BirA, and Δwing BirA proteins were constructed by amplifying the BirA coding sequences from the pBA11 derivatives using primers VCC67 (or primer VCC60 for Δ2–65 BirA) and VCB17. The fragments were gel-purified, digested with BspHI (or PciI for Δ2–65 BirA) and BamHI, and ligated to pET19b cut with NcoI and BamHI. The BirAG57S and T52S derivatives were made using plasmid pVC10 as template and primers VCC23-VCC24 and VCB77-VCB78, respectively. All plasmids constructed were verified by restriction digestion followed by DNA sequencing.
The 10b strain is a BirA super-repressor mutant strain isolated in the selection and screening approach previously reported (8). The initial isolate encoded a protein with three amino acid substitutions (T52S, G115S, and Q75R). The effects of these mutations were deconvoluted by construction of plasmids carrying each mutation separately plus all pairwise double mutant derivatives by site-directed mutagenesis (supplemental Fig. S2). To construct the expression plasmid encoding a C-terminal hexahistidine-tagged version of the strain, 10b-derived BirA mutant protein was amplified from the plasmid pVC46 using primers VCB16 and VCB17 (8), which added NcoI and BamHI sites as well as the purification tag. The digested products were ligated into the same sites of plasmid pET19b, to give plasmid pVC48. Plasmid VC48 was then digested with NcoI, treated with mung bean nuclease to generate blunt ends, and ligated to give plasmid pVC49. Plasmid VC47 was constructed by DraIII and PvuI digestion of pVC46. The 574-bp fragment encoding the mutations T52S, G115S, and Q75R was then inserted into plasmid pBA11 digested with the same enzymes. All of the plasmid constructs were confirmed by sequence analysis
Plasmids VC37, 38, and 39 were constructed by site-directed mutagenesis (Stratagene QuikChange II protocol) using the following primer pairs (supplemental Table S2): VCB79-VCB80, VCC75-VCC76, and VCC77-VCC78, respectively, and pVC50 as the template. The template plasmids were digested with DpnI prior to transformation of strain DH5α. All the mutations were confirmed by sequence analysis. The plasmids encoding the T52S mutant and the additional mutation at the biotin-binding loop were then digested with PvuI and DraIII and ligated into pBA11 digested with the same enzyme. The plasmids encoding BirA having single amino acid mutations were generated using site-directed mutagenesis using the primers in supplemental Table S2 and pBA11 as the template.
In Vivo Complementation
Strain VC618 was transformed with the plasmid pBA11, pVC28, pVC29, pCY910, pCY895, pCY911, pCY897, pCY896, and pCY909, and the transformants were selected for on LB plates supplemented with ampicillin and chloramphenicol at 30 °C. The temperature-sensitive plasmid pVC18 was cured from these strains by growth at 42 °C on defined media supplemented with chloramphenicol. The loss of the temperature-sensitive plasmid was confirmed by ampicillin sensitivity. These strains were then used to carry out growth curve analyses as previously described (8).
β-Galactosidase Assays
The cultures were grown overnight and then diluted 1:400 into fresh medium of the same composition and grown to the early to mid-log phase before assays were performed. β-Galactosidase activity was determined as described (25) following disruption of the cells by sodium dodecyl sulfate-chloroform treatment. The data collected were from a minimum of three independent experiments.
Protein Purification
Expression, purification, and quantitation of the truncated AccB (AccB-87) apo acceptor protein was performed as previously described (8). The BirA plasmids encoding the wild type, BirA10b mutant, BirA Δ2–65, chimera, G57S, and T52S proteins were purified from 1-liter LB cultures. The BL21 λDE3 strains containing plasmids encoding the wild type BirA, the 10b mutant protein, chimera, G57S, or T52S BirA were grown to an A600 0.8 at 37 °C, and T7 RNA polymerase expression was induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside for 4 h at 30 °C. For the strain carrying the Δ2–65 BirA plasmid, the culture was grown at 30 °C, and T7 RNA polymerase expression was induced by adding 100 μm isopropyl β-d-thiogalactopyranoside and growing the culture for 4 h at 16 °C. The cells were harvested by centrifugation and stored at −80 °C. Cell lysis and protein purification were performed by the previously described protocols (8) with the following modification. Following the nickel affinity chromatography, the pooled fractions containing the BirA protein were dialyzed overnight in 50 mm sodium phosphate buffer (pH 8) containing 10% glycerol (Buffer A). The dialyzed sample was subjected to anion exchange chromatography using a Vivapure IEX Q Maxi H spin column. The column was equilibrated with buffer A prior to loading the dialyzed sample. The column was successively washed with the above buffer, and the eluate was collected. The BirA proteins eluted in the flow through and the first wash steps, whereas the impurities bound to the column and only eluted with sodium phosphate buffer containing 2 m NaCl. The eluates were dialyzed against storage buffer (50 mm Tris-HCl, pH 8.0, containing 150 mm KCl, 10% glycerol, and 0.1 mm DTT) overnight, concentrated using Millipore concentrators, flash frozen, and stored at −80 °C. All the protein samples were found to be 95% pure except for Δ2–65 BirA, which was found to be 80% pure by using 12% SDS-PAGE gels.
To purify the BirA Δwing protein strain, VC926 lacking the chromosomal birA gene was used. The protein was purified as above with some modifications. The induction medium and temperature were optimized to increase solubility of the protein. Rather than growing cultures in grown in LB with induction at 37 °C, which caused the protein to form inclusion bodies, the strain was grown in 1 liter of 2XYT medium buffered with M9 salts and supplemented with 400 nm biotin plus the ticarcillin-clavulanate mixture. The cultures were grown at 30 °C to an A600 of 1.4, at which point T7 RNA polymerase expression was induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside for 3 h at 16 °C. The cells were then harvested and stored at −80 °C. The lysate and protein purification was achieved using the protocol mentioned above. After nickel affinity purification, the protein was found to be 70% pure, judged by running the sample on a 12% SDS-PAGE gels. The protein was dialyzed into storage buffer, flash frozen, and stored at −80 °C. Attempts to further purify the protein using chromatography on ion exchange, Blue Sepharose affinity, HiTrap phenyl-HP hydrophobic, or size exclusion columns were fruitless. The Δwing BirA protein was found to be quite unstable in that it either precipitated on all the columns tested or eluted together with impurities (as in the case of the size exclusion chromatography).
Bio-5′-AMP Synthesis Reactions
The assay for BirA-catalyzed in vitro protein biotinylation activity was performed as described previously (8) with some modifications. Protein concentrations were determined using the extinction coefficients calculated from the protein sequence using ExPASY Tools website. The assays contained 50 mm Tris-HCl (pH 8), 5 mm tris-(2-carboxyethyl) phosphine, 5 mm MgCl2, 20 μm biotin, 5 μm ATP plus 16.5 nm [α-32P] ATP, 100 mm KCl, and 1 μm BirA protein. Each of the reaction mixtures were incubated at 37 °C for 30 min. For each BirA protein tested, two identical tubes were used, and at the end of 30 min, AccB-87 (50 μm) was added to one of each pair of tubes, whereas the other tube was left untreated. The tubes were incubated for an additional 15 min at 37 °C. One μl of each reaction mixture was applied to a cellulose thin layer chromatography plate of microcrystalline cellulose, and the plates were developed in isobutyric acid-NH4OH-water (66:1:33) (27). The thin layer chromatograms were dried for 10 h, exposed to a phosphorimaging plate, and visualized using a Fujifilm FLA-3000 Phosphor Imager and Fujifilm Image Gauge software (version 3.4 for Mac OS). To preclude the possibility of contaminating ATPase activity, different concentrations of sodium fluoride (50, 25, 10, or 5 mm) or Na3VO4 (10, 5, 2.5 or 1 mm) were added either separately or together to the biotinylation reaction mixtures containing the wild type BirA or Δwing BirA proteins.
Electrophoretic Mobility Shift Assays
To assay the DNA binding ability of the mutant BirA proteins and the wild type protein, the method used is similar to the previously published protocol (8). A 112-bp DNA containing the biotin operator was PCR-amplified using primers VCB36 and VCB37 and Taq DNA polymerase. The resulting PCR product was checked for size and quality on a 2% agarose gel. The DNA was then extracted from the gel using the Qiagen gel extraction kit. The DNA was quantitated using a Nanodrop spectrophotometer (Thermo Pierce). The DNA probe (40 nm) was incubated with varying concentrations of the BirA protein in binding buffer (50 mm Tris HCl, pH 8.0, 50 mm NaCl, and 10% glycerol) together with 50 μm biotin, 1 mm ATP, 5 mm MgCl2, 100 mm KCl, and 5 mm tris-(2-carboxyethyl) phosphine at 37 °C for 30 min. The products were then separated on a 6% DNA retardation gel and run for 1 h at 100 V and 20 milliamps. The gel was then stained in 1× SYBR Green I nucleic acid gel stain solution made according to the manufacturer's protocol. The gel was then visualized using Bio-Rad Chemi Doc XRS and Quantity One software.
Bioinformatics and Secondary Structure Analyses
The structural data for the proteins comparison were entered into Chimera program (28) along with the N-terminal region of BirA (Protein Data Bank code 1HXD). Using the Match Align tool, the alignment data were collected based on the secondary structure. The amino acid sequences for the aligned proteins were then trimmed based on the secondary structure alignment. Each sequence was then entered into Clustal Omega program. The Clustal data were then entered into the ESPRIPT 2.2 program. The structural data from IHXD was used as the reference structure, and similarity settings were set so as to calculate the percentage of equivalent residues calculated per columns, considering their physicochemical properties.
RESULTS
N-terminal Deletion Mutants of BirA
The original N-terminal deletion protein, Δ2–65, had the expected defect in DNA binding (11) and retained in vitro catalytic activity, although it was severely defective in binding both biotin and biotinoyl-AMP (11). However, another group subsequently reported a similar N-terminal deletion, Δ2–63, and found the truncated protein to be functional in vivo (12). To address this apparent contradiction and because the Δ2–65 had not been tested in vivo, we constructed genes encoding these two mutant proteins and several proteins having smaller N-terminal deletions, Δ3–25, Δ3–40, and Δ3–60 with various end points within the wHTH motif (Fig. 2A). In the smallest of these deletions (called Δwing), only the 14-residue wing sequence of the wHTH motif was deleted. Each of these constructs was used to replace the birA gene in the medium copy number plasmid pBA11 (or a pBA11 derivative lacking an irrelevant vector sequence), in which a constitutive promoter drives birA expression.
To determine the effects of these deletions on the regulation of the biotin operon and the in vivo ligation abilities of the mutant BirA proteins, each of the plasmids encoding an N-terminally truncated protein was transformed into strain VC618 (ΔbirA::kan bioF::lacZY ΔlacZY/pVC18) at 30 °C. Expression of LacZ (β-galactosidase) provides an assay for transcription of the bioBFCD operon (16). The strain is a biotin auxotroph caused by the lacZY insertion into the bio operon and lacks birA because of deletion of the entire gene. Because birA is essential for growth, this strain survives because of expression of the yeast Bpl1 ligase (29) from a plasmid (pVC18) that is temperature-sensitive for replication (8). Following transformation with the plasmids to be tested, the transformants were cured of the Bpl1 ligase plasmid by growth at 42 °C. The cured strains producing the various mutant proteins were plated on MacConkey indicator plates that contained a repressing biotin concentration (115 nm biotin) or lacked biotin supplementation (the medium contains about 15 nm biotin). A strain expressing wild type BirA gives white colonies on 115 nm biotin and red colonies on 15 nm biotin (red colonies are due to high level lactose utilization of the acidic products, which react with indicator dyes in the medium). In contrast, the strains expressing the N-terminal deletion mutants gave red colonies on both the 115 and 15 nm biotin plates, indicating that the mutant proteins were deficient in bio operon regulation. That is, a normally repressing biotin concentration failed to repress. To quantify these results, β-galactosidase levels were assayed on the strains after growth in minimal media supplemented with biotin concentrations that were either strongly repressing (10 μm) or depressing (10 nm) (Fig. 2B). The strain expressing wild type BirA was strongly repressed at the higher biotin concentration and showed an ∼14-fold greater activity at the lower biotin concentration, whereas the strains expressing the N-terminal deletion proteins had essentially the same activities at both biotin concentrations. Moreover, when grown with 10 μm biotin, the deletion mutant strains had activities 100-fold greater than the strain expressing wild type BirA, indicating that the mutant proteins were completely unable to repress bio operon expression.
The finding that the strains expressing the N-terminal deletion proteins survived loss of the yeast Bpl1 plasmid argued that they retained at least a minimal level of ligase activity. To test this indication, we grew the strains in minimal media containing various levels of biotin (Fig. 3). All of the mutant strains grew normally on 40 nm or 4 μm biotin but grew more slowly than the wild type strain on 4 nm biotin, and growth was especially slow with 1.6 nm biotin. Therefore, the defects in biotin and biotinoyl-AMP demonstrated in vitro for the Δ2–65 protein by Xu and Beckett (11) can be overcome by high extracellular biotin concentrations, and the protein biotinylation proficiency of the Δ2–63 protein reported by Maeda and co-workers (12) can be ascribed to high level biotin production by their host strain and the high biotin concentration used in their in vitro experiments (10 μm). Hence, there seems to be no contradiction between the two reports.
FIGURE 3.

Complementation of the E. coli ΔbirA bioF::lacZY birA strain by expression of WT BirA, BirA T52S mutant protein, and BirA N-terminal deletion mutant proteins. The strains were assayed for growth in defined medium liquid cultures supplemented with various concentrations of biotin as given by the colored symbols of the third row as described under “Experimental Procedures.” Each of the panels is the host strain expressing a different birA allele. Each growth assay was carried out in triplicate, and this plot shows the averages of the triplicate cultures. Growth was measured by optical density at 600 nm. Note that the data are plotted on the y axis as a log scale. All the panels share the same color code. Expression was driven by the constitutive promoter of a medium copy number plasmid.
Insertion of a Nearly Isosteric Foreign Wing Largely Restores in Vivo Biotinylation Ability to the Δwing BirA
The wing structures of wHTH proteins often aid HTH DNA binding by making contacts in the DNA minor groove (13); therefore it was not surprising that the Δwing BirA protein was defective in regulation of bio operon expression. However, an unexpected finding was that the small 14-residue deletion protein was as compromised in the ability to support growth at low biotin concentrations, as were the proteins with much larger deletions (Fig. 3). These results argued that the wing is required for full biotin ligase activity and might play a structural role or contribute residue side chains involved in catalysis. To test these possibilities, we searched the protein structure databases for wHTH wings that closely resembled the BirA wing (Fig. 4C). The BirA wing is small, which limited the possibilities, but one protein, OmpR, has a wing that is a very close match to the BirA wing (Fig. 4, A and B). Unlike BirA, the wHTH structure is located at the C terminus of OmpR, a transcriptional regulator of E. coli outer membrane permeability. Given the close match of the two structures, we substituted the OmpR wing for that of BirA and found that expression of this chimera protein allowed almost normal growth at the low biotin concentrations where the Δwing protein functioned poorly (Fig. 3). Hence, the wing structure rather than its exact sequence is required for normal ligase activity.
FIGURE 4.

Sequences and structural alignments of the DNA-binding domains of several wHTH regulatory proteins. A, alignment of the C-terminal DNA-binding domain of OmpR with the N-terminal BirA DNA-binding domain. B, superimposition of the OmpR DNA-binding domain on the BirA N-terminal domain. The model was made using the Match Align Tool of Chimera (28) with Protein Data Bank files 1HXD and 1OPC. C, alignments of the wing motifs for several wHTH regulatory proteins of known structure were generated using the method described under “Experimental Procedures.” The Protein Data Bank files used were: BirA, 1HXD; OmpR, 1OPC; Rap30, 1BBY; CRP, 1CGP; FadR, 1E2X; PhoB, 1QQI; LexA, 1JHF; HNF-3γ, 1VTN; RFX1, 1DP7; and PcoR, 2JZY. Identical residues are denoted by white letters on a red background, similar residues are denoted by red letters on a white background, variable residues are in black letters, and dots represent gaps. In A and C, the alignment was generated using the BirA secondary structure as the reference structure. In A and C, the secondary structures of the proteins are shown at the top of the panel: β, β sheets; coils, β-turns; α, α-helices. In B, H1, H2, and H3 denote helices 1, 2, and 3, respectively. W denotes the wing structure, whereas α denotes the OmpR loop that interacts with RNA polymerase subunit α. Helix 3 is the recognition helix.
The chimera protein remained defective in bio operon regulation, indicating that the role of the BirA wing in DNA binding is specific. However, five of the ten wing residues are conserved between the BirA and OmpR wings (Fig. 4A). To test whether one of these residues played an important role in wing function, we altered each of these BirA residues in turn, often to residues that were reported to alter DNA binding by OmpR (Table 1) (14). All of these single residue substitutions within the wing resulted in normal growth on 1.6 nm biotin as seen for the T52S mutant protein (Fig. 3), and thus it seemed likely that the structure rather than the sequence of the wing is the important factor in ligase activity. Moreover, only those BirA proteins having substitutions of BirA residues Gly-57 and Tyr-58 (G57S and Y58F, respectively), both of which are conserved in the OmpR wing, were defective in transcriptional repression (Fig. 2B). In the latter case, the tyrosine hydroxyl group seems to be the important functional group because the Y58T BirA had bio operon repressor function (data not shown). We also studied a wing mutant, T52S, that was serendipitously isolated in our prior search for super-repressor BirA mutants (8) and found it showed a 5-fold increase in repression at 10 nm biotin relative to wild type BirA, whereas at a higher biotin concentration (10 μm), its repression ability was indistinguishable from that of wild type BirA (Fig. 2B). In OmpR, it was reported that substitution of isoleucine for the OmpR Thr-224, a wing residue analogous to Thr-52 of BirA, resulted in a protein defective in DNA binding (14), and thus we made the same substitution in BirA and found the T52I BirA protein had normal repression ability (data not shown). Hence, although the OmpR and BirA wings show strong structural conservation (Fig. 4B), the residues conserved between the two proteins have different functions.
TABLE 1.
N-terminal deletion and point mutant proteins of BirA plus OmpR protein mutants
| Repression | Biotin requirementa | Mutation siteb | Secondary structurec | OmpR mutantd | Reference |
|---|---|---|---|---|---|
| nm | |||||
| + | 1.6 | None | Ref. 16 | ||
| ++ | 1.6 | T52S | Wing | This study | |
| + | 1.6 | T52I | Wing | T224I | This study |
| + | 1.6 | V53A | Wing | This study | |
| + | 1.6 | V53M | Wing | This study | |
| + | 1.6 | G55S | Wing | This study | |
| − | 1.6 | G57S | Wing | G229S | This study |
| − | 1.6 | Y58F | Wing | This study | |
| + | 1.6 | Y58T | Wing | This study | |
| − | 41–4000 | Δ2–65 | wHTH | Ref. 11 | |
| − | 41–4000 | Δ2–63 | wHTH | Ref. 12 | |
| − | 41–4000 | Δ3–60 | wHTH | This study | |
| − | 41–4000 | Δ3–40 | Helix I, II, III | This study | |
| − | 41–4000 | Δ3–25 | Helix I, II | This study | |
| − | 41–4000 | Δ48–61 | ΔWing | This study | |
| − | 1.6 | 50–62 substitution | Chimera | This study | |
| − | 4000 | G115S | BBLe | Refs. 15 and 19 | |
| − | 41–4000 | R118G | BBL | Ref. 16 | |
| − | 4 | R119W | BBL | Ref. 16 |
a The minimal biotin requirements of the deletion and the point mutants constructed in this study. The data for the N-terminal mutants are directly from Refs. 15, 16, and 19.
b Positions are amino acid residue positions.
c Locations of the mutations in the secondary structure of BirA protein.
d Wing residue substitutions having regulatory effects in the OmpR system.
e BBL, biotin binding loop.
In Vitro Studies of the BirA Point Mutant, Δwing, and Chimera Proteins
Our in vitro studies omitted the proteins having large N-terminal deletions because their in vivo properties were very similar to those of the Δ2–65 deletion protein of Xu and Beckett (11), which have been thoroughly studied in vitro. We purified the C-terminal hexahistidine-tagged BirA chimera, BirA Δwing, BirA Δ2–65, BirAG57S, BirAT52S, and wild type proteins all to apparent homogeneity with the exception of the BirA Δwing and BirA Δ2–65 proteins, which were recalcitrant to purification of about 80% purity (Fig. 5A). We first tested the DNA binding abilities of the purified BirAs proteins by electromobility shift assays using a 112-bp biotin operator.
FIGURE 5.
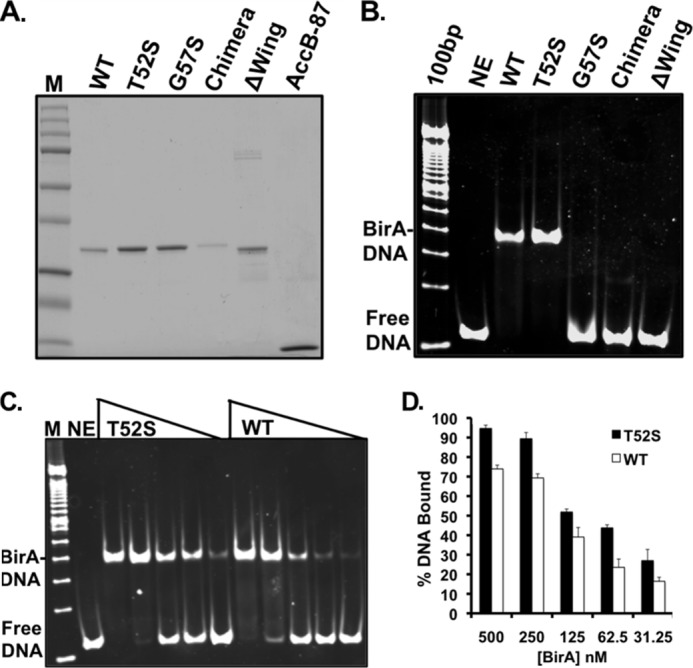
Purification and operator binding of purified BirA proteins. A, the BirA and AccB-87 proteins were subjected to electrophoresis in a 12% polyacrylamide gel. The Precision Plus protein standards from Bio-Rad are shown in lane M, and AccB-87 is in the far right-hand lane. The proteins were purified as described under “Experimental Procedures” and dialyzed in storage buffer prior to storage at −80 °C. In B, at 1 μm, the BirA mutant derivatives and wild type BirA purified as described under “Experimental Procedures” were assayed for binding to a 112-bp bioO duplex DNA prepared as described under “Experimental Procedures.” The reaction products were separated by native gel electrophoresis, and the gels were stained with SYBR Green I nucleic acid gel stain and visualized using the Bio-Rad Chemi Doc XRS. In C, the point mutant T52S and wild type BirA proteins (0–500 nm) were subjected to the same analysis. In D, the results from three independent experiments were quantified with the Quantity One program. % DNA bound represents the percentage of DNA bound to BirA protein versus the percentage of remaining unbound calculated for each lane using the no enzyme (NE) lane as control for the amount of DNA loaded. The results are presented as the averages and standard errors of triplicate determinations. The open bars represent the wild type BirA, and the closed bars represent the T52S mutant protein. The identities of the BirA proteins tested are given at the tops of A, B, and C. The standards in B and C are the 100-bp DNA ladder (Life Technologies).
The Δwing and the G57S BirA proteins showed no DNA binding even at the highest protein concentrations tested (Fig. 5B). High concentrations of the Δwing protein could not be tested because of its precipitation and clogging of the wells. The chimera protein gave a weak but detectable shift (Fig. 5B). However, the shift differed from that seen with the wild type protein. To test whether this shift represented a nonspecific interaction, we carried out a similar mobility shift experiment using a 117-bp XmnI-ScaI DNA fragment of vector pUC19 in place of the operator sequence. The chimera protein shifted this fragment, and thus the protein seems able to bind DNA, but without sequence specificity (data not shown).
The T52S BirA showed increased ability to bind the bio operator (Fig. 5, C and D), consistent with its in vivo behavior (Fig. 2B). At BirA concentrations of 250 nm, the T52S protein bound 90% of the operator DNA compared with 75% binding by the wild type BirA protein (Fig. 5D). Moreover, significant DNA binding was observed at 62.5 nm BirA for T52S BirA, whereas wild type protein showed only barely detectable binding (Fig. 5, C and D). In past work, we have seen a significant variation in binding by wild type BirA (8), and therefore we conducted these studies over a range of concentrations and assayed the mutant protein in parallel with the wild type protein. We found that the T52S mutant invariably exhibited a higher extent of DNA binding than the wild type protein (Fig. 5D).
We also tested the abilities of the purified proteins to synthesize Bio-5′-AMP in the first ligase partial reaction and to transfer the biotin moiety to the AccB-87 acceptor protein in the second partial reaction (Fig. 6). Because Bio-5′-AMP is required for DNA binding, an inability to synthesize the ligand would be manifested as a defect in operator binding. We tested the purified BirA proteins for the ability to synthesize Bio-5′-AMP from biotin and [α-32P]ATP. All of the mutant proteins synthesized Bio-5′-AMP (Fig. 6), and thus the ligand required for DNA binding was present in the gel shift experiments. However, a striking result was seen in the reactions containing the Δwing protein. This protein converted ATP to ADP, an off target reaction in which the incorrect phosphoric anhydride linkage was cleaved. This off target product can be attributed to loss of the wing because it was not seen in reactions containing the chimera protein or any other protein. However, it remained possible that the Δwing protein preparations were contaminated with a co-purifying ATPase activity. To test this possibility, we added two general inhibitors of ATPase activity, 25 mm NaF and 5 mm Na3VO4, either separately or together, to reactions containing the Δwing BirA and found no significant decrease in ADP production in the presence of the inhibitors (supplemental Fig. S1). Hence, ADP production is a property of the Δwing protein.
FIGURE 6.
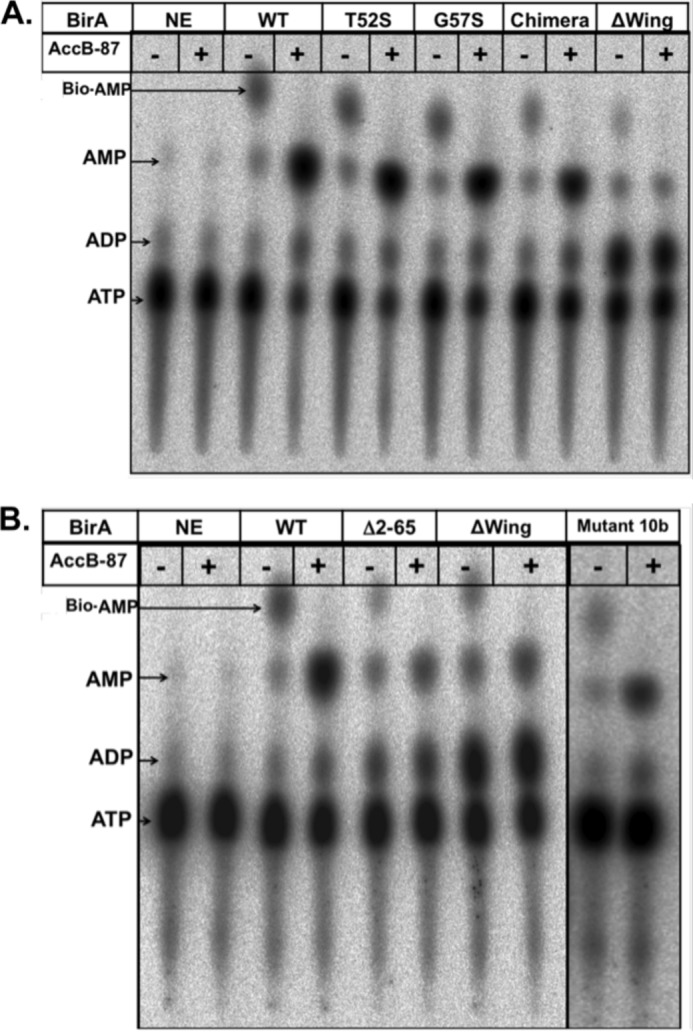
Thin layer chromatographic analysis of various BirA proteins to synthesize Bio-5′-AMP and transfer the biotin moiety to AccB-87. Synthesis of biotinoyl-adenylate in reaction mixtures containing 1 μm BirA, 20 μm biotin, 5 μm ATP, 5 mm MgCl2, 100 mm KCl, 5 mm TCEP, and 16.5 nm [α-32P]ATP is shown. Each reaction was run in duplicate and incubated for 30 min at 37 °C, after which time point plus signs (+) purified apo-AccB-87 (50 μm) was added to one of the duplicate reactions followed by incubation for an additional 15 min. The second duplicate reaction did not receive AccB-87 (minus signs). One μl of each reaction was spotted onto a cellulose thin layer chromatography plate. The right panels with 10b BirA mutant show a separate experiment. Following plate development, the reaction products (indicated by arrows), biotinoyl-5′-AMP (Bio-AMP), ADP, AMP, and the remaining ATP were visualized by autoradiography. NE denotes the control reactions lacking BirA. The proteins assayed are given at the top of the figure, and the identities of the spots are on the left margin. A and B show the products of the BirA reaction in the presence or absence of the AccB-87 acceptor protein. The identity of each protein is given at the top of each pair of lanes. The wild type (WT) and Δwing proteins were included in both analyses as controls.
We tested the activities of these proteins in the second partial reaction of BirA: transfer of the biotin moiety to the AccB-87 acceptor protein. Each of Bio-5′-AMP synthesis reactions was run in duplicate and incubated to allow accumulation of Bio-5′-AMP. AccB-87 was added to one of the duplicate tubes followed by further incubation and chromatography (Fig. 6). Transfer of the biotin moiety to AccB-87 results in disappearance of the Bio-5′-AMP spot and strong augmentation of the AMP spot (8). Each of the mutant proteins showed this behavior, although the Δwing protein appeared somewhat deficient in transfer (Fig. 6).
Genetic Evidence for Direct Interaction of the Wing with the Ligase Active Site
The birA gene was originally defined by a single mutant allele, birA1 (15). This mutation encoded a G115S substitution within a highly conserved BPL motif that resulted in a loss of the ability to repress bio operon expression and an inability to grow at low biotin concentrations (16, 17). This protein was subsequently studied in vitro by Kwon and Beckett (18) and was found to be severely defective in binding both the biotin substrate and the Bio-5′-AMP intermediate, properties that readily explain the physiological deficiencies in repression and growth. This and neighboring residues defined the biotin-binding loop that becomes structured upon binding biotin or Bio-5′-AMP (10).
The T52S wing mutation discussed above was originally isolated in a triply mutant birA strain called 10b (8). In addition to the T52S alteration, the mutant gene of strain 10b encoded the G115S birA1 mutation plus a Q75R mutation (supplemental Fig. S2). Strikingly, the triply mutant protein strongly repressed bio operon transcription (Fig. 7), despite the presence of the G115S mutation, which abolishes repression as the sole mutation (16). We deconvoluted the lesions of the triple mutant gene and found that upon removal of the T52S mutation, the encoded protein showed the lack of repression ability characteristic of the G115S BirA protein (supplemental Fig. S2). Elimination of the G115S mutation had little or no effect on the proteins that retained the T52S mutation; both the T52S and T52S Q75R proteins repressed much more strongly than wild type BirA (Fig. 7 and supplemental Fig. S2). Finally, elimination of the Q75R mutation, leaving the T52S and G115S mutations intact, resulted in a protein that repressed bio operon expression as strongly as the original super-repressor strain, indicating that the Q75R mutation is silent (Fig. 7 and supplemental Fig. S2). We also carried out in vitro biotinylation assays with the purified BirA10b mutant protein and found that the protein synthesized the Bio-5′-AMP ligase intermediate and transferred the biotin moiety to the AccB-87 acceptor protein (Fig. 6B). Hence, the T52S BirA not only binds operator DNA more tightly than the wild type protein but also overcomes the ligation defect engendered by the G115S mutation.
FIGURE 7.
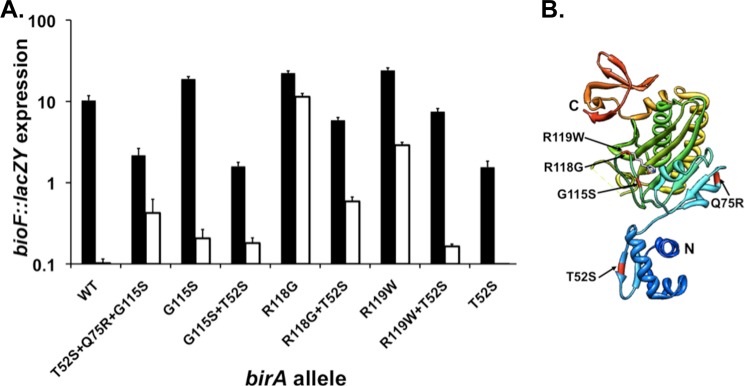
Genetic evidence that the wing plays a direct role in the enzymatic function of BirA. A, transcription of the bio operon in cultures grown with 4 nm biotin (derepression conditions; depicted with solid bars) or 4 μm biotin (repression conditions; depicted by open bars) assayed by β-galactosidase production from a chromosomal bioF::lacZY fusion. The strains used were derivatives of strain VC618 cured of plasmid pVC18 (8). Single colonies were streaked on defined medium plates supplemented with 4 nm biotin and X-gal for phenotypic confirmation. Liquid cultures were grown in the same medium lacking X-gal and were then assayed for bio operon transcription by β-galactosidase activity (“Experimental Procedures”). Note that the data are plotted on the y axis as a log scale. B, three-dimensional structure of monomeric BirA protein. The single residue substitutions tested are highlighted. The model was constructed using Chimera and Protein Data Bank file 1HXD.
Because the G115S mutation alters the BirA biotin-binding loop (6), two other repression-defective biotin-binding loop birA alleles isolated by Barker and Campbell (16), the protein products of which had been studied by Kwon and Beckett (18), were tested to ascertain whether the T52S mutation showed allele specificity. The R118G BirA is similar to the G115S BirA in that it also binds biotin and Bio-5′-AMP poorly, whereas the R119W BirA binds biotin and Bio-5′-AMP with essentially wild type binding constants (18). We constructed plasmids encoding the R118G and R119W proteins and transformed the plasmids into strain VC618. After driving out the Bpl1 plasmid, we found that the strains grew with 4 nm biotin and were defective in repression of biotin operon transcription at 4 μm biotin. As in the case of the G115S protein, introduction of the T52S allele into the genes encoding the R118G and R119W proteins restored repression (Fig. 7). Therefore, the ability of the T52S mutation to restore repression to otherwise repression-defective biotin-binding loop mutants is not restricted to G115S BirA. Note that our ability to grow strains encoding the G115S and R118G proteins in media containing 4 nm biotin is due to overproduction of the proteins, and hence our data do not conflict with those of Barker and Campbell (16), who assayed expression from the chromosomal locus.
DISCUSSION
All of the birA deletion mutations lacked both repression activity and the ability to grow normally at low biotin concentrations (Figs. 2 and 3) and thus were similar in phenotype to the original construct of Xu and Beckett (11), which had not previously been characterized in vivo. The growth defects of the various deletion mutants were only apparent at low biotin concentrations (Fig. 3), whereas the repression defect was seen at all biotin concentrations (Fig. 2). As mentioned above, the multiple BirA structures available (unliganded or liganded with biotinoyl-lysine, biotin, or a Bio-5′-AMP analog) fail to provide an explanation for the loss of ligase activity resulting from deletions within the N-terminal domain.
We were surprised to find that the Δwing BirA was as defective in growth and repression as the proteins with much larger deletions (Figs. 2 and 3). To test the role of the wing in BirA ligase activity and repressor function, we replaced the BirA wing with the foreign isosteric wing of OmpR. This replacement largely restored growth at low biotin concentrations to the Δwing protein (Fig. 3), indicating that the wing structure must stabilize the enzyme active site and that the OmpR wing is a good mimic of the BirA wing. Both the chimera and Δwing BirAs were defective in repressor function, indicating that although half the residues are conserved between the BirA and OmpR wings (Fig. 4A), the operative DNA contacts are not conserved. The chimera protein synthesized Bio-5′-AMP and biotinylated the AccB-87 acceptor protein (Fig. 6). The Δwing BirA has a flawed active site as shown by hydrolysis of much of the ATP substrate to ADP, an off pathway product. The OmpR wing overcomes this problem further, indicating that the wing stabilizes and organizes the active site to allow both ligase partial reactions to proceed efficiently (Fig. 6).
The importance of the BirA wing was strikingly demonstrated by a remarkable fortuitous intragenic epistasis (Fig. 7). The protein carrying both the T52S super-repressor mutation and the G115S biotin-binding loop mutation has the phenotype of a super-repressor mutant rather than displaying the lack of repression and deficient ligase activity characteristic of the G115S BirA (18, 19). Similar results were seen with two other biotin-binding loop mutants. These data strongly argue that when free in the cytosol, the wing contacts the flexible loop (residues 116–124) that becomes ordered in the presence of biotin and Bio-5′-AMP (7, 10, 20) and aids the ordering process. Proteins having amino acid residue substitutions within or adjacent to the loop have poorly ordered loops and therefore bind biotin and biotinoyl-AMP poorly; however, this is overcome when the wing carries the T52S mutation. These data together with the properties of the Δwing and chimera proteins indicate that Bio-5′-AMP synthesis would proceed only when BirA is free in the cytosol where the wing-biotin-binding loop interaction can occur. When BirA is DNA-bound, the wing is removed from the biotin-binding loop region, but this is of no consequence because the loop is fully ordered because of the binding of the Bio-5′-AMP required for dimerization and DNA binding.
To our knowledge, this is the first report that the wing of a wHTH protein determines the activity of an enzyme. This also may be the first case of a successful “wing swap” in a wHTH protein. Most members of the wHTH family use the wing to directly assist DNA binding, as is the case for BirA in its regulatory role. However, in the SOS response LexA repressor, the wing structures fulfill spacer length requirements such that the two DNA-binding domains of the dimeric protein are positioned to make specific contacts within the same minor groove of the SOS regulon operators (21). The wHTH of the plasmid stability protein TubR is involved in DNA binding, but in a very atypical manner. The TubR recognition helices mediate dimerization, thereby precluding canonical HTH-DNA interactions. Instead the two wing structures plus the N termini of the recognition helices mediate DNA binding by insertion into the same major groove (22). Other wHTH proteins have evolved to use the wing for more diverse purposes. For example, in the heat shock transcriptional factor, the wing plays a role in dimerization of the protein (23), whereas in DEAH helicases, the wHTH domain has also been implicated in opening double-stranded DNA and RNA molecules in an ATP-independent manner (24). Therefore, despite the structural conservation of wHTH motifs, no model protein-DNA complex can be chosen to represent a paradigm for wHTH motif-containing proteins. Indeed, we probably have just scratched the surface of the diverse functions that this ancient and highly conserved motif might encompass.
This work was supported, in whole or in part, by National Institutes of Health Grants AI15650.

This article contains supplemental Tables S1 and S2 and Figs. S1 and S2.
- Bio-5′-AMP
- biotinoyl-adenylate
- BPL
- biotin protein ligase
- wHTH
- winged helix-turn-helix.
REFERENCES
- 1. Beckett D. (2007) Biotin sensing. Universal influence of biotin status on transcription. Annu. Rev. Genet. 41, 443–464 [DOI] [PubMed] [Google Scholar]
- 2. Cronan J. E., Jr. (1988) Expression of the biotin biosynthetic operon of Escherichia coli is regulated by the rate of protein biotination. J. Biol. Chem. 263, 10332–10336 [PubMed] [Google Scholar]
- 3. Streaker E. D., Gupta A., Beckett D. (2002) The biotin repressor. Thermodynamic coupling of corepressor binding, protein assembly, and sequence-specific DNA binding. Biochemistry 41, 14263–14271 [DOI] [PubMed] [Google Scholar]
- 4. Mukhopadhyay B., Purwantini E., Kreder C. L., Wolfe R. S. (2001) Oxaloacetate synthesis in the methanarchaeon Methanosarcina barkeri. Pyruvate carboxylase genes and a putative Escherichia coli-type bifunctional biotin protein ligase gene (bpl/birA) exhibit a unique organization. J. Bacteriol. 183, 3804–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Streaker E. D., Beckett D. (1998) A map of the biotin repressor-biotin operator interface. Binding of a winged helix-turn-helix protein dimer to a forty base-pair site. J. Mol. Biol. 278, 787–800 [DOI] [PubMed] [Google Scholar]
- 6. Wilson K. P., Shewchuk L. M., Brennan R. G., Otsuka A. J., Matthews B. W. (1992) Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc. Natl. Acad. Sci. U.S.A. 89, 9257–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Streaker E. D., Beckett D. (1999) Ligand-linked structural changes in the Escherichia coli biotin repressor. The significance of surface loops for binding and allostery. J. Mol. Biol. 292, 619–632 [DOI] [PubMed] [Google Scholar]
- 8. Chakravartty V., Cronan J. E. (2012) Altered regulation of Escherichia coli biotin biosynthesis in BirA superrepressor mutant strains. J. Bacteriol. 194, 1113–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weaver L. H., Kwon K., Beckett D., Matthews B. W. (2001) Corepressor-induced organization and assembly of the biotin repressor. A model for allosteric activation of a transcriptional regulator. Proc. Natl. Acad. Sci. U.S.A. 98, 6045–6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wood Z. A., Weaver L. H., Brown P. H., Beckett D., Matthews B. W. (2006) Co-repressor induced order and biotin repressor dimerization. A case for divergent followed by convergent evolution. J. Mol. Biol. 357, 509–523 [DOI] [PubMed] [Google Scholar]
- 11. Xu Y., Beckett D. (1996) Evidence for interdomain interaction in the Escherichia coli repressor of biotin biosynthesis from studies of an N-terminal domain deletion mutant. Biochemistry 35, 1783–1792 [DOI] [PubMed] [Google Scholar]
- 12. Maeda Y., Yoshino T., Matsunaga T. (2010) In vivo biotinylation of bacterial magnetic particles by a truncated form of Escherichia coli biotin ligase and biotin acceptor peptide. Appl. Environ. Microbiol. 76, 5785–5790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brennan R. G., Matthews B. W. (1989) The helix-turn-helix DNA binding motif. J. Biol. Chem. 264, 1903–1906 [PubMed] [Google Scholar]
- 14. Kato N., Tsuzuki M., Aiba H., Mizuno T. (1995) Gene activation by the Escherichia coli positive regulator OmpR. A mutational study of the DNA-binding domain of OmpR. Mol. Gen. Genet. 248, 399–406 [DOI] [PubMed] [Google Scholar]
- 15. Campbell A., Del Campillo-Campbell A., Chang R. (1972) A mutant of Escherichia coli that requires high concentrations of biotin. Proc. Natl. Acad. Sci. U.S.A. 69, 676–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barker D. F., Campbell A. M. (1980) Use of bio-lac fusion strains to study regulation of biotin biosynthesis in Escherichia coli. J. Bacteriol. 143, 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buoncristiani M. R., Howard P. K., Otsuka A. J. (1986) DNA-binding and enzymatic domains of the bifunctional biotin operon repressor (BirA) of Escherichia coli. Gene 44, 255–261 [DOI] [PubMed] [Google Scholar]
- 18. Kwon K., Beckett D. (2000) Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci. 9, 1530–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barker D. F., Campbell A. M. (1981) Genetic and biochemical characterization of the birA gene and its product. Evidence for a direct role of biotin holoenzyme synthetase in repression of the biotin operon in Escherichia coli. J. Mol. Biol. 146, 469–492 [DOI] [PubMed] [Google Scholar]
- 20. Xu Y., Nenortas E., Beckett D. (1995) Evidence for distinct ligand-bound conformational states of the multifunctional Escherichia coli repressor of biotin biosynthesis. Biochemistry 34, 16624–16631 [DOI] [PubMed] [Google Scholar]
- 21. Zhang A. P., Pigli Y. Z., Rice P. A. (2010) Structure of the LexA-DNA complex and implications for SOS box measurement. Nature 466, 883–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ni L., Xu W., Kumaraswami M., Schumacher M. A. (2010) Plasmid protein TubR uses a distinct mode of HTH-DNA binding and recruits the prokaryotic tubulin homolog TubZ to effect DNA partition. Proc. Natl. Acad. Sci. U.S.A. 107, 11763–11768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Littlefield O., Nelson H. C. (1999) A new use for the “wing” of the “winged” helix-turn-helix motif in the HSF-DNA cocrystal. Nat. Struct. Biol. 6, 464–470 [DOI] [PubMed] [Google Scholar]
- 24. Teichmann M., Dumay-Odelot H., Fribourg S. (2012) Structural and functional aspects of winged-helix domains at the core of transcription initiation complexes. Transcription 3, 2–7 [DOI] [PubMed] [Google Scholar]
- 25. Miller J. H. (1972) Experiments in Molecular Genetics, pp. 352–355, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 26. Chapman-Smith A., Turner D. L., Cronan J. E., Jr., Morris T. W., Wallace J. C. (1994) Expression, biotinylation and purification of a biotin-domain peptide from the biotin carboxy carrier protein of Escherichia coli acetyl-CoA carboxylase. Biochem. J. 302, 881–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prakash O., Eisenberg M. A. (1979) Biotinyl 5′-adenylate. Corepressor role in the regulation of the biotin genes of Escherichia coli K-12. Proc. Natl. Acad. Sci. U.S.A. 76, 5592–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 29. Cronan J. E., Jr., Wallace J. C. (1995) The gene encoding the biotin-apoprotein ligase of Saccharomyces cerevisiae. FEMS Microbiol. Lett. 130, 221–229 [DOI] [PubMed] [Google Scholar]
- 30. Solbiati J., Cronan J. E. (2010) The switch regulating transcription of the Escherichia coli biotin operon does not require extensive protein-protein interactions. Chem. Biol. 17, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]