

Background: CO dehydrogenase oxidizes CO to CO2 under aerobic conditions.
Results: CO dehydrogenase also oxidizes H2, eliciting a new EPR signal.
Conclusion: CO dehydrogenase is an effective hydrogenase, and the EPR signal observed provides new mechanistic information.
Significance: We have gained a deeper understanding of the chemistry catalyzed by this enzyme and new information concerning the mechanism of H2 oxidation.
Keywords: Carbon Monoxide, Copper, Enzyme Kinetics, Hydrogenase, Kinetics, Molybdenum, CO Dehydrogenase
Abstract
The reaction of the air-tolerant CO dehydrogenase from Oligotropha carboxidovorans with H2 has been examined. Like the Ni-Fe CO dehydrogenase, the enzyme can be reduced by H2 with a limiting rate constant of 5.3 s−1 and a dissociation constant Kd of 525 μm; both kred and kred/Kd, reflecting the breakdown of the Michaelis complex and the reaction of free enzyme with free substrate in the low [S] regime, respectively, are largely pH-independent. During the reaction with H2, a new EPR signal arising from the Mo/Cu-containing active site of the enzyme is observed which is distinct from the signal seen when the enzyme is reduced by CO, with greater g anisotropy and larger hyperfine coupling to the active site 63,65Cu. The signal also exhibits hyperfine coupling to at least two solvent-exchangeable protons of bound substrate that are rapidly exchanged with solvent. Proton coupling is also evident in the EPR signal seen with the dithionite-reduced native enzyme, and this coupling is lost in the presence of bicarbonate. We attribute the coupled protons in the dithionite-reduced enzyme to coordinated water at the copper site in the native enzyme and conclude that bicarbonate is able to displace this water from the copper coordination sphere. On the basis of our results, a mechanism for H2 oxidation is proposed which involves initial binding of H2 to the copper of the binuclear center, displacing the bound water, followed by sequential deprotonation through a copper-hydride intermediate to reduce the binuclear center.
Introduction
Oligotropha carboxidovorans expresses an O2-tolerant molybdenum- and copper-containing carbon monoxide dehydrogenase when grown on CO as sole source of both carbon and energy. The enzyme catalyzes the oxidation of carbon monoxide to carbon dioxide according to Reaction 1 (1, 2), with the reducing equivalents obtained from the oxidation of CO passed to the quinone pool (5). A portion of the carbon dioxide thus generated is subsequently fixed nonphotosythetically by the reductive pentose phosphate pathway (3–5). This metabolism is environmentally extremely important as carboxydotrophic bacteria such as O. carboxidovorans remove some 2 × 108 metric tons of CO from the atmosphere annually (6).
 |
The Mo- and Cu-containing CO dehydrogenase from O. carboxidovorans is both mechanistically and structurally distinct from the extremely O2-sensitive Ni- and Fe-containing CO dehydrogenase from organisms such as Moorella thermoacetica or Methanosarcina barkerii (7). The enzyme, encoded by the megaplasmid pHCG3, is an (αβγ)2 hexamer with a large subunit (coxL, 88.7 kDa) containing the binuclear active site, a medium subunit (coxM, 30.2 kDa) with FAD, and a small subunit (coxS, 30.2 kDa) containing two spinach ferredoxin-like [2Fe-2S] clusters (8). Oxidation of carbon monoxide occurs at the binuclear center with reducing equivalents passed from the redox-active molybdenum to the proximal Fe-S cluster I to the distal Fe-S cluster II and finally to the FAD cofactor. On the basis of overall architecture and sequence homology, the Mo/Cu CO dehydrogenase belongs to the xanthine oxidase family of enzymes but is unique among members of this large and broadly distributed family in several regards: the reaction catalyzed is not formally a hydroxylation reaction involving hydride abstraction from substrate; the enzyme utilizes ubiquinone as the oxidizing substrate rather than O2 or NAD+ as oxidizing substrate (5); and, most significantly, its unique binuclear active site contains copper as well as molybdenum, whose structure is shown in Fig. 1 (9, 10). The overall configuration of the binuclear center is LMoVIO2-μS-CuI-Cys388, with L representing a bidentate pyranopterin cofactor common to all molybdenum enzymes other than nitrogenase (in this case present as the cytosine dinucleotide). The molybdenum has a square pyramidal coordination geometry with an apical Mo=O; the equatorial plane consists of the two sulfurs from the pyranopterin cofactor, an equatorial Mo=O and a μ-sulfido sulfur that bridges to the copper, which is also coordinated by Cys388 of the protein (9, 10). Although not commonly referred to in mechanistic discussions of CO dehydrogenase, the copper is also coordinated by a HO−/H2O ligand at a distance of 2.4 Å. Computational studies of the reaction mechanism suggest that this ligand is displaced by CO at the outset of catalysis (11, 12).
FIGURE 1.
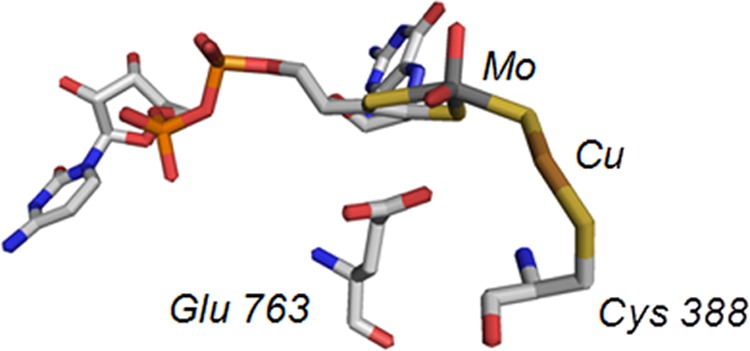
Active site of the enzyme CO dehydrogenase (Protein Data Bank ID code 1N5W). Atom colors are CPK, with the molybdenum atom rendered in teal and the copper in copper color. Cys-388, which coordinates the copper, is shown at far right, and pterin cytosine dinucleotide coordinating the molybdenum is at left.
Santiago and Meyer have also reported that the Mo/Cu CO dehydrogenase also exhibits hydrogenase activity, oxidizing H2 to protons with the reducing equivalents passed on to an artificial oxidant (13); the anaerobic Ni/Fe CO dehydrogenases also catalyze this reaction as well as H2 evolution, with rates of ∼ 3 s−1 for H2 evolution and a rate 0.1 for H2 oxidation (14, 15). In the present work we have characterized the reaction of the Mo/Cu CO dehydrogenase by H2 and found that the enzyme is indeed effectively reduced by H2 over a broad range of pH values. In the course of the reaction, a new EPR signal attributable to the binuclear center is observed which provides important mechanistic clues as to the manner in which H2 is oxidized. A second new signal, exhibited by dithionite-reduced enzyme in the presence of bicarbonate (the hydrated from of the product CO2), provides further insight into the nature of the binuclear cluster.
EXPERIMENTAL PROCEDURES
Materials
CO and H2 gas were obtained from Airgas. Isotopically enriched D2 (99.6%) and D2O (99.9%) were purchased from Cambridge Isotope Laboratories. All other chemicals and reagents were obtained at the highest quality and purity commercially available and used without additional purification.
Purification/Enzyme Assays
O. carboxidovorans cells (ATCC 49405) were grown at 30 °C, pH 7.0, in a 19-liter Fermentor (BioFlo 415) containing minimal media and CO as the sole carbon and energy source, as described previously (1, 16). CO was introduced in a 20/80 mixture of CO and air, with a total gas flow rate of 2.0 liters/min. Cells were harvested in the late log phase (A436 >6), washed with HEPES, pH 7.2, and stored at −80 °C until needed. CO dehydrogenase was purified according to the procedure described by Zhang et al. (16), using a combination of Q-Sepharose and Sephacryl S-300 FPLC. Enzyme was quantified by the absorbance at 450 nm (ϵ450 = 36 mm−1 cm−1 per active site), and the purity determined by absorbance ratios A280/450 ∼ 5.5, A450/420 ∼1, and A450/550 ∼ 2.9.
Routine enzyme assays were performed at 25 °C using methylene blue (ϵ615 = 37.11 mm−1 cm−1) or 1,4-benzoquinone (Δϵ247 = 20.2 mm−1 cm−1) as oxidizing substrate, as described previously (5, 16). All enzyme preparations were reconstituted with sulfur and copper prior to use following the protocol described by Resch et al. (17) as modified by Wilcoxen et al. (5). Silver-substituted enzyme was prepared as described previously by removing the copper and bridging sulfur followed by incubation with sulfide then AgI-thiourea (18). Enzyme following reactivation was ∼40% functional as determined by the fractional change in absorbance at 450 nm on reduction of enzyme with CO (which reduces only the fully functional enzyme) compared with the absorbance seen with dithionite (which reduces any remaining enzyme) (17). Unless otherwise stated, enzyme concentrations used are those of the functional enzyme, corrected for the amount of nonfunctional enzyme.
Steady-state Kinetics
Steady-state kinetics using H2 as reducing substrate were performed by monitoring the bleaching of 1,4-benzoquinone at 247 nm (Δϵ247 = 20.2 mm−1 cm−1) using a stopped-flow spectrophotometer (Applied Photophysics) at 25 °C. One syringe of 50 μm 1,4-benzoquinone in 50 mm HEPES, pH 7.2, was made anaerobic by bubbling argon gas through a rubber septum into the solution for 15 min; to this solution ∼50 nm CO dehydrogenase was added so the final concentration after mixing was ∼25 nm. A second syringe was prepared by bubbling argon gas through a rubber septum into the solution for 15 min to make the solution anaerobic followed by bubbling with H2 for 1 h. Dilutions of H2-saturated buffer (780 μm) were made with anaerobic 50 mm HEPES, pH 7.2, to give solutions ranging from 20 to 780 μm [H2]. Saturated H2 concentrations were calculated from a Henry's law constant of 0.00078 mol/kg·bar in water (20). Initial rates were measured in triplicate at each concentration of H2 (12–780 μm), averaged, and plotted against H2 concentration. Hyperbolic fits were obtained in SigmaPlot (Systat Software) using the Michaelis-Menten equation to obtain kcat and Km values.
Rapid Reaction Kinetics
The reaction of CO dehydrogenase with H2 under anaerobic conditions was carried out by monitoring the loss of enzyme absorbance at 450 nm and 550 nm upon mixing enzyme with varying concentrations of H2 in a stopped-flow spectrophotometer at 25 °C. The stopped-flow apparatus was made anaerobic by flushing with argon-saturated buffer. Enzyme concentrations in the range of 2.5–5 μm (after mixing) were made anaerobic in a glass tonometer by alternating evacuation an flushing with argon in 10–15-min cycles over the course of a minimum of 1 h. A second syringe containing H2 (12–390 μm H2 after mixing) at the desired concentration was prepared as described above. A pH range from 6.0 to 10.0 was used to examine the reductive half-reaction kinetics using 50 mm phosphate buffer at pH 6.0, 50 mm HEPES at pH 7.2, 50 mm Tris-HCl at pH 8.0 and pH 9.0, and 50 mm CAPS2 at pH 10.0. The activation energy of the reaction with H2 was determined in 50 mm HEPES, pH 7.2, over the temperature range 5–30 °C. Primary and solvent kinetic isotope effects were determined by performing the experiment using D2 as substrate or in D2O as solvent, respectively. In preparing solutions in D2O, the differing ionization constant of D2O relative to H2O was taken into account using the formula pD = (pH meter reading) + 0.4.
Kinetic transients were fit to single exponentials using the stopped-flow manufacturer's ProDataViewer software package to obtain an observed rate constant, kobs. The rate constants thus obtained were then averaged and plotted against substrate concentrations and fit with a hyperbolic fit according to Equation 1 to obtain a reductive half-reaction rate constant, kred, and substrate dissociation equilibrium constant, Kd.
Electron Paramagnetic Resonance Spectroscopy
EPR spectra were recorded using a Brüker Instruments ER 300 spectrophotometer equipped with an ER 035 M gaussmeter and an HP 5352B microwave frequency counter. The temperature was maintained at 150 K with a Brüker ER 4111VT liquid N2 cryostat. Samples were prepared with anaerobic 40–80 μm CO dehydrogenase in 50 mm HEPES, pH 7.2, and added to a sealed argon-flushed EPR tube and incubated with H2 gas for 2–5 s before freezing in a ethanol/dry ice bath. The EPR spectrum in D2O was obtained by exchanging CO dehydrogenase (native or silver-substituted) into a HEPES-D2O buffer, pD 7.2, through a Sephedex G-25 column equilibrated with D2O buffer. Once in the D2O buffer the samples were made anaerobic and reacted with H2 as described above. Dithionite-reduced samples were prepared by making the enzyme (native or silver-substituted) anaerobic in 50 mm HEPES, pH 7.2, 50 mm HEPES-D2O, pD 7.2, or 400 mm bicarbonate, pH 7.0, and titrated to partial reduction with dithionite in 50 mm Tris-HCl, pH 7.0, monitoring the absorbance at 450 and 550 nm. Once partially reduced the samples were frozen as before. Spectra were recorded at 9.45-GHz microwave frequency with 10-milliwatt power and 5-Gauss modulation amplitude at 150 K. Simulations of the collected spectra were done using the EasySpin software package (developed by Dr. Stefan Stoll, Department of Chemistry, University of Washington) (21).
RESULTS
Steady-state Kinetics
Steady-state kinetics were carried out anaerobically as described under “Experimental Procedures,” examining the dependence of catalytic velocity on [H2] at 25 °C, pH 7.2, from 20 to 780 μm, using 1,4-benzoquinone (5) as the oxidizing substrate. The observed rates were plotted against H2 concentration, as shown in Fig. 2 (top), and the data were fit to a hyperbolic function using the Michaelis-Menten kinetics equation, yielding a kcat of 5.1 s−1 and a Km of 283 μm.
FIGURE 2.

Steady-state and rapid reaction kinetics of CO dehydrogenase with H2 as substrate. Top, steady-state H2 concentration dependence of CO dehydrogenase using 50 μm 1,4-benzoquinone as the electron acceptor in 50 mm HEPES, pH 7.2, at 25 °C. Plots of the initial rates, following 1,4-benzoquinone reduction at 247 nm after mixing a final concentration of 25 nm CO dehydrogenase with H2 dissolved in 1,4-benzoquinone, versus [H2] were fit with the Michaelis-Menten equation using Sigma Plot (R2 = 0.995) to give a kcat of 5.1 s−1 and Km of 283 μm. Bottom, rapid reaction kinetic plots of the rate kobs following the reduction of 5 μm CO dehydrogenase at 450 nm versus [H2], from 12 μm to 390 μm, at (50 mm phosphate, pH 6.0, 50 mm HEPES, pH 7.2, 50 mm Tris-HCl, pH 8.0 and pH 9.0, and 50 mm CAPS, pH 10.0), 25 °C. Plots were fit using Equation 1 to yield a rate of reduction and substrate dissociation constant, kred and Kd. The optimum pH, 7.2, gave kred of 5.3 s−1 and Kd of 525 μm (R2 = 0.999).
Rapid Reaction Kinetics
To examine further the reaction of CO dehydrogenase with H2, rapid reaction kinetic experiments were performed, monitoring the bleaching of enzyme at 450 nm (due to accumulation of reducing equivalents in the iron-sulfur and FAD sites) upon reduction by H2 under anaerobic conditions in 50 mm HEPES, pH 7.2, 25 °C. Kinetic transients at 450 nm were well behaved and fit to single exponentials to obtain kobs for each H2 concentration examined. Fig. 2 (bottom) shows a plot of kobs versus [H2], which exhibits the expected hyperbolic shape. When fitted with a hyperbolic equation (“Experimental Procedures,” Equation 1) a limiting rate constant for reduction at infinite [H2], kred, and dissociation constant for H2, KdH2, could be determined to be 5.3 s−1 and 525 μm, respectively. The observed kred is in good agreement with the steady-state kcat of 5.1 s−1, indicating that the rate-limiting step in overall catalysis resides in the reductive half-reaction, as seen with CO as substrate (16). Rate constants obtained at 550 nm, monitoring reduction of the Fe-S clusters alone, were in good agreement with those obtained at 450 nm, with no evidence of the accumulation of long wavelength-absorbing intermediates. This behavior is entirely comparable with that exhibited the closely related and well studied enzyme xanthine oxidase, where intramolecular electron transfer among the several redox-active centers of the enzyme is very fast compared with catalysis (16, 22, 23). The reductive half-reaction was examined over the pH range 6 to 10 (50 mm phosphate (pH 6.0), 50 mm HEPES, pH 7.2, 50 mm Tris-HCl, pH 8.0, 50 mm Tris-HCl, pH 9.0, and 50 mm CAPS, pH 10.0, and in each case hyperbolic plots of kobs versus [H2] were observed (Fig. 2, bottom). The kinetic parameters thus obtained are summarized in Table 1; kred, KdH2, and kred/KdH2 are found to vary only modestly (by no more than a factor of three) over the pH range. We conclude that no reversible substrate- or enzyme-based ionizations are involved in the rate-limiting chemistry of H2 oxidation.
TABLE 1.
Kinetic parameters for CO dehydrogenase as a function of pH
| kred | Kd | kred/Kd | kcat | Km | |
|---|---|---|---|---|---|
| s−1 | μm | s−1 | μm | ||
| pH 6.0 | 5.4 | 750 | 7.2 × 103 | ||
| pH 7.2 | 5.3 | 525 | 1.0 × 104 | 5.1 | 283 |
| pH 8.0 | 5.4 | 700 | 7.7 × 103 | ||
| pH 9.0 | 5.8 | 812 | 7.1 × 103 | ||
| pH 10.0 | 8 | 1562 | 5.1 × 103 | ||
| pD 7.2 | 4.95 | 633 | 7.8 × 103 | ||
| pH 7.2 with D2 | 2.8 | 809 | 3.5 × 103 |
The copper of the binuclear center can be replaced with silver, with retention of activity (albeit at a 6-fold reduced rate compared with native enzyme, 8 s−1 compared with 51 s−1, respectively) (18). Kinetic studies with the silver-substituted enzyme with H2 were also attempted, but, as reflected in the failure to observe any enzyme bleaching under an atmosphere of H2, this form of the enzyme was not reduced by H2 under any reaction conditions examined (data not shown).
Solvent and primary kinetic isotope experiments were next carried out in 50 mm HEPES, pD 7.2 or pH 7.2, respectively, over the concentration range 12–390 μm at 25 °C. Hyperbolic curves were again obtained. In the case of the solvent isotope experiment with H2 in D2O, fits (Table 1) yielded a kred of 5.0 s−1 and KdH2 of 633 μm, with a resulting kinetic isotope effect (Hkred/Dkred) of 1.07 (i.e. unity within the limits of error). The value for kred, 5.3 s−1, is approximately 50-fold greater than seen with the Ni/Fe CO dehydrogenases (14, 15). However, we were unable to observe the reverse reaction, reduction of protons to H2, a reaction that is effectively catalyzed by the Ni/Fe enzyme. In our hands, the fully reduced Mo/Cu CO dehydrogenase was stable indefinitely in aqueous solution, even at low pH (data not shown). The absence of a solvent kinetic isotope effect on kred with the Mo/Cu enzyme is consistent with the lack of pH dependence of the reductive half-reaction. By contrast, similar treatment of the data obtained in the primary isotope effect experiment (D2 in H2O buffer) yielded a kred of 2.8 s−1 and Kd of 809.9 μm, indicating a modest primary kinetic isotope effect of 1.89 (Table 1).
Finally, to obtain the activation parameters for the reaction, the temperature dependence of the rate constant for enzyme reduction over the range 5–30 °C was determined in 50 mm HEPES, pH 7.2, using 780 μm H2 (before mixing). Rate constants obtained from exponential fits to the kinetic transients were averaged, and an Arrhenius plot (Fig. 3, top) of log(kobs) versus 1/T, to obtain an effective activation energy (Ea) of 30.6 kJ/mol an Eyring plot (Fig. 3, bottom) of the same data yielded an enthalpy and entropy of activation (ΔH‡ and ΔS‡) of the transition state yielded values of 28.2 kJ/mol and −146 J/K·mol, respectively. The value for ΔH‡ is in excellent agreement with the value for Ea of 30.6 kJ/mol, given that RT = 1.7 kJ/mol at room temperature) The calculated Gibbs free energy of activation (ΔG‡) of 71.6 kJ/mol, compares with a theoretical activation barrier of 15.7 kJ/mol for the Ni-Fe hydrogenase and a value of 63.4 and 68.4 kJ/mol for the carbon monoxide oxidation reaction with native and silver-substituted CO dehydrogenase, respectively (16, 24). The activation parameters are summarized in Table 2.
FIGURE 3.
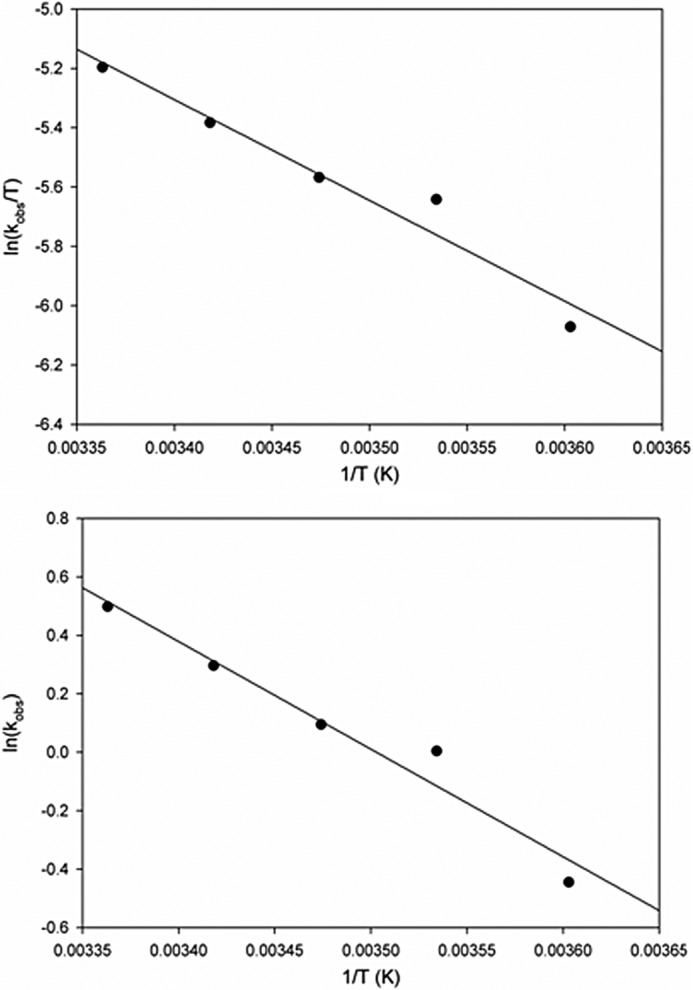
Temperature dependence of CO dehydrogenase reduction by H2. Rates were obtained from 5 to 30 °C using 5 μm CO dehydrogenase reduced with 390 μm H2. Observed rates were plotted in Arrhenius (top) and Erying (bottom) plots for the determination of activation energy of CO dehydrogenase reduction.
TABLE 2.
Activation parameters for CO dehydrogenase
Electron Paramagnetic Resonance Spectroscopy
We have previously described the EPR spectrum of the binuclear center of CO dehydrogenase that has been partially reduced by CO, and found g1,2,3 = 2.0010, 1.9604, 1.9549, with extremely large hyperfine coupling to the 63,65Cu nucleus (I = 3/2) of A1,2,3 = 117, 164, 132 MHz but no coupling to protons (16); this spectrum is reproduced in Fig. 4A. The oxidized enzyme has Mo(VI) and Cu(I), and with Cu(I) being a closed shell d10 system. The one-electron reduced, EPR-active state of the binuclear center is formally Mo(V) and Cu(I), but a recent investigation of a model compound with a bridging Mo(V)-μS-Cu(I) core that exhibits 63,65Cu hyperfine coupling comparable to that seen with the enzyme has shown that the redox-active orbital of the system is extensively delocalized over the three core atoms, with a composition that is 44% Mo dxy, 25% S p, and 21% Cu d (dxz, and dz2, as well as some undefined amount of Cus) character (25). It is the highly delocalized nature of the redox-active orbital, singly occupied in the EPR-active state, that accounts for the extremely large copper hyperfine observed in the signal. Enzyme that has been partially reduced with H2 exhibits a new EPR spectrum that is quite distinct from that seen with the CO-reduced enzyme, as shown in Fig. 4B. In an attempt to simplify this complex spectrum, the sample was prepared in buffer containing D2O. As shown in Fig. 4C (black line), the observed spectrum was indeed considerably simplified, with fits (Fig. 4C, red line), yielding g1,2,3 = 2.0127, 1.9676, 1.9594, with approximately isotropic hyperfine coupling A1,2,3(63,65Cu) = 169, 200, and 170 MHz. The quality of the fit (Fig. 4C) clearly indicates that the signal arises from a single species. These values are quite different from those exhibited by the CO-reduced enzyme, with substantially larger gav and greater hyperfine coupling to the copper. With the g tensor and 63,65Cu A tensor thus defined, the signal seen with H2-reduced spectrum in H2O could be adequately fit with only proton hyperfine coupling constants as variables. The parameters that yielded the best fits reflected coupling to two approximately equivalent solvent-exchangeable protons with A1,2,3(1H) = 80, 20, 130 MHz, in addition to coupling for the copper of A1,2,3(63,65Cu) = 170, 200, 170 MHz (and g1,2,3 = 2.0127, 1.9676, 1.9594), as shown in Fig. 4B (red line). Although the quality of the fit is considered acceptable, particularly given the number of variables that are involved, we cannot exclude the possibility that additional, more weakly coupled protons are present. When the enzyme is reduced with D2, the spectrum is identical to the spectrum of the H2-reduced enzyme in H2O, indicating that if the coupled protons originate from substrate, they exchange very rapidly with solvent under turnover conditions. As with the CO-reduced enzyme previously reported, the H2-reduced enzyme was not stable, as evidenced by the accumulation of CuII in the sample; the inactivation of the enzyme was in fact much faster with H2 than with CO (4, 16).
FIGURE 4.
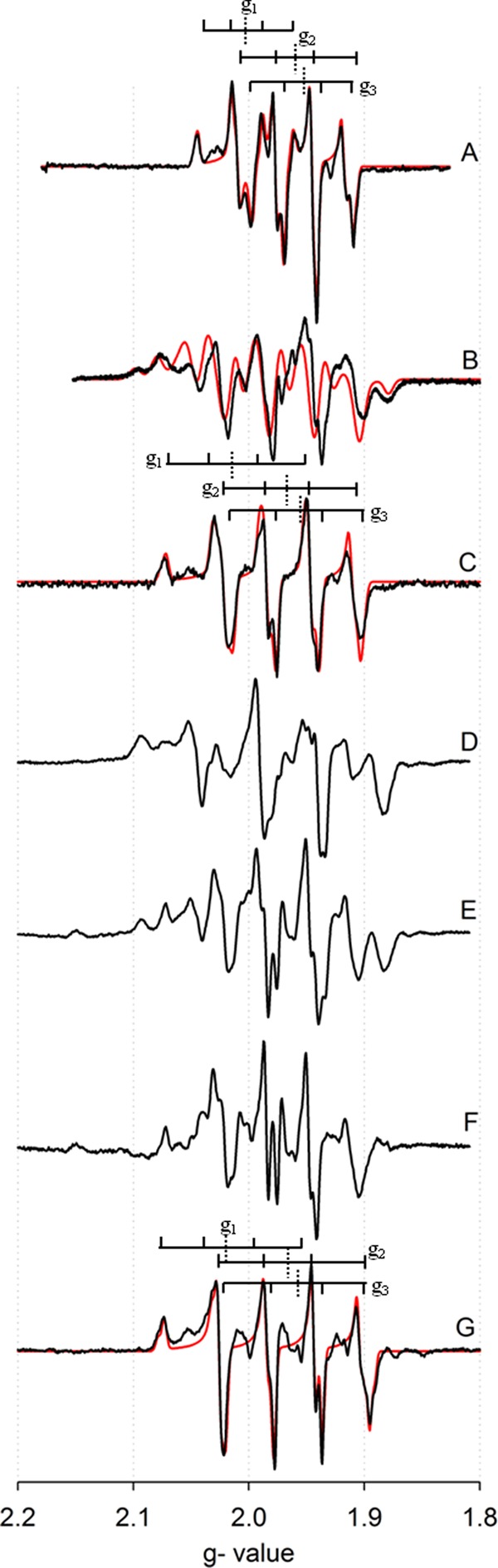
EPR spectra manifested by CO dehydrogenase under various conditions of 100 μm CO dehydrogenase partially reduced by CO, H2, or dithionite. A, CO-reduced CO dehydrogenase in 50 mm HEPES, pH 7.2, with simulation in red. B, H2-reduced CO dehydrogenase in 50 mm HEPES, pH 7.2, with simulation in red. C, H2-reduced CO dehydrogenase in 50 mm HEPES, pD 7.2, with simulation in red. D, dithionite-reduced CO dehydrogenase in 50 mm HEPES, pH 7.2. E, dithionite-reduced CO dehydrogenase in 50 mm HEPES, pD 7.2. F, spectrum E minus spectrum D. G, dithionite-reduced CO dehydrogenase in 400 mm bicarbonate, pH 8.0, with simulation in red. Brackets indicate approximate g1,2,3 values (dashed lines) with copper splittings (solid lines). The EPR instrument settings were: 9.45 GHz microwave frequency; 4 milliwatt microwave power; 5 Gauss modulation amplitude; 150 K.
The EPR signal of H2-reduced CO dehydrogenase resembles, but is distinct from, that seen when enzyme is partially reduced noncatalytically using sodium dithionite (17), as reproduced in Fig. 4D. There is similar splitting from the copper and likely two protons, but with a distinctly different line shape particularly around g = 2.08 and 1.90 (compare line spectra B and D in Fig. 4). Given the presence of the protons in the spectrum, an attempt was made to record the dithionite-reduced enzyme at high pH, pH 9.0 and 10.0, but no difference in the spectrum was observed, indicating that the pKa values for the site(s) were >11. Furthermore, several attempts were made to produce a dithionite-reduced spectrum in D2O. These included extended incubation with the oxidized enzyme in D2O (>36 h) prior to reduction and enzyme reduced by dithionite and incubated in D2O under anaerobic conditions (>30 min). The resulting EPR signal was always a mixture of the protonated and deuterated spectra, as shown in Fig. 4E. When an appropriately weighted fraction of the protonated spectrum (Fig. 4D) is subtracted out from that in Fig. 4E, the resulting spectrum (Fig. 4F) resembles that seen with H2-reduced enzyme in the presence of D2O (Fig. 4C), but with distinct g and A tensors (Table 3). The EPR spectrum of silver-substituted enzyme reduced by dithionite was obtained (Fig. 5C), but in this case the observed spectrum was essentially identical to that of the CO-reduced silver-substituted enzyme in either H2O or D2O (Fig. 5, A and B) and exhibited no proton hyperfine coupling. This strongly suggests that in the former case CO is not bound to silver in the signal-giving species. The lack of proton coupling in the silver-substituted enzyme makes it unlikely that the molybdenum subnucleus is the site of protonation.
TABLE 3.
EPR simulation parameters for CO dehydrogenase
| Cu/CO dehydrogenase CO/H2Oa | Ag/CO dehydrogenase CO/H2O | Cu/CO dehydrogenase H2/H2O | Cu/CO dehydrogenase H2/D2O | Cu/CO dehydrogenase Dt/CO3 | |
|---|---|---|---|---|---|
| g1 | 2.0010 | 2.0043 | 2.0127 | 2.0127 | 2.0020 |
| g2 | 1.9604 | 1.9595 | 1.9676 | 1.9676 | 1.9618 |
| g3 | 1.9549 | 1.9540 | 1.9594 | 1.9594 | 1.9548 |
| gavg | 1.9721 | 1.9726 | 1.9799 | 1.9799 | 1.9729 |
| A1 (MHz) | 117 | 81.98 | 169.13 | 169.13 | 192.87 |
| A2 (MHz) | 164 | 78.85 | 200.28 | 200.28 | 190.93 |
| A3 (MHz) | 132 | 81.89 | 170.23 | 170.23 | 221.34 |
| A1 (MHz) Ha (Hb) | 80 (80) | ||||
| A2 (MHz) Ha (Hb) | 20 (20) | ||||
| A3 (MHz) Ha (Hb) | 130 (120) | ||||
| Euler angle α (radians) | −0.55 | −0.55 | −0.197 | ||
| Euler angle β (radians) | 0.54 | −1.95 | −1.95 | −1.028 | |
| Euler angle γ (radians) | 0.54 | 1.93 | 1.93 | 2.777 | |
| Line width (Gauss) | 0.51 | 0.8 | 0.71 | 0.63 |
a Zhang et al. (16).
FIGURE 5.
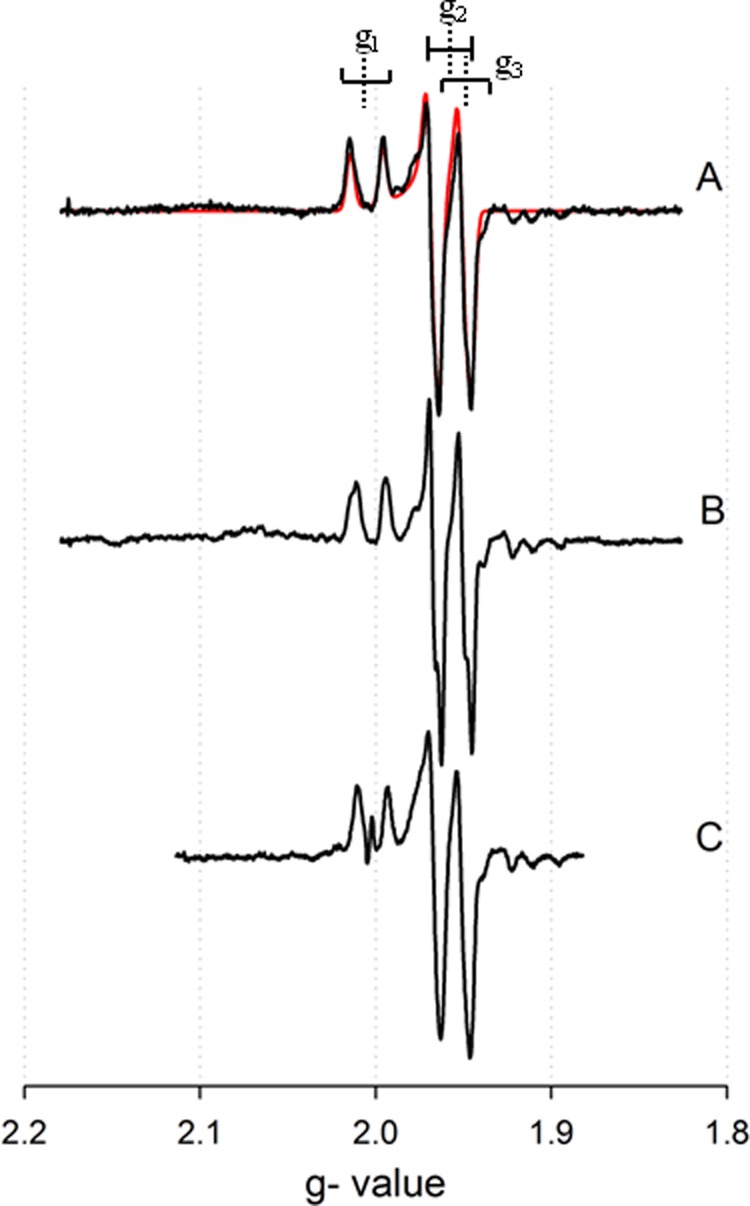
EPR spectra manifested by the silver-substituted CO dehydrogenase under various conditions. A, CO-reduced silver-substituted CO dehydrogenase in 50 mm HEPES, pH 7.2, with simulation in red. B, CO-reduced silver-substituted CO dehydrogenase in 50 mm HEPES, pD 7.2. C, dithionite-reduced silver-substituted CO dehydrogenase in 50 mm HEPES, pH 7.2. Brackets indicate approximate g1,2,3 values (dashed lines) with copper splittings (solid lines). The EPR instrument settings were: 9.45 GHz microwave frequency; 4 milliwatt microwave power; 5 Gauss modulation amplitude; 150 K.
Finally, the effect of bicarbonate (the hydrated form of product CO2 in the reaction) was examined. The resulting EPR spectrum (Fig. 4G) did not exhibit any coupling to protons even in H2O, and in fact resembles that exhibited by H2-reduced enzyme in D2O (Fig. 4C), albeit with somewhat different spectral parameters: g1,2,3 = 2.0020, 1.9618, 1.9548 and A1,2,3(63,65Cu) = 193, 191, 221 MHz. Again, the quality of fit indicates the presence of only a single signal-giving species. That bicarbonate abolishes the proton hyperfine coupling seen in EPR signal of the dithionite-reduced enzyme strongly suggests that it displaces those ligands in the binuclear center that are responsible for the proton hyperfine.
DISCUSSION
We find that the Mo- and Cu-containing CO dehydrogenase from O. carboxidovorans is readily reduced by H2, with a kred, 5.3 s−1 and a KdH2 of 525 μm. kred from the rapid reaction study is in very good agreement with the steady-state derived kcat, 5.1, indicating the rate-limiting step in turnover is in the reductive half-reaction (as is the case with CO as substrate (16)). The lack of a significant pH dependence to the reductive half-reaction over the pH range 6–10 (Table 1) indicates that no substrate- or enzyme-based ionizations in the experimentally accessible pH range are involved in the rate-limiting step of the reaction. We note that the Ni-Fe hydrogenase of Allochromatium vinosum also exhibits no pH dependence in its hydrogenase activity (26).
Partial reduction of enzyme with H2 yields an EPR signal distinctly different from that seen with CO as reductant, with more anisotropic g values and larger coupling to the 63,65Cu nucleus (g1,2,3 = 2.0127, 1.9676, 1.9594 and A1,2,3(63,65Cu) = 169, 200, 170 MHz, Table 3) and, significantly, strong coupling to two protons that is lost on preparation of the EPR sample in D2O. The spectra exhibited by H2-reduced CO dehydrogenase in both H2O and D2O are similar in form to, but with distinct g and A parameters from, those produced when the enzyme is simply partially reduced by sodium dithionite under similar conditions. By contrast, the EPR signal seen with dithionite-reduced silver-substituted enzyme exhibits no proton coupling (bearing in mind that H2 does not reduce the silver-substituted enzyme).
Considering first the structure giving rise to the signal seen with dithionite-reduced enzyme, the likeliest source of the two strongly coupled protons is the HO−/H2O known crystallographically to be bound to the copper at a distance of 2.4 Å (9, 10) Indeed, given the Cu-O distance and the fact that the oxygen is stacked on top of the π cloud of Phe-290, it is most likely likely that the ligand is a water molecule (with two protons) rather than a negatively charged hydroxide. Presumably for steric reasons (given the restricted access to the active site), the water is not readily solvent-exchangeable. This interpretation is consistent with the absence of coupled protons with the dithionite-reduced silver-substituted enzyme, which lacks the coordinated water at the silver. The signal-giving species with native and silver-substituted enzyme can thus be formulated as Mo(V)/Cu(I)·H2O and Mo(V)/Ag(I), respectively, as shown in Fig. 6A.
FIGURE 6.
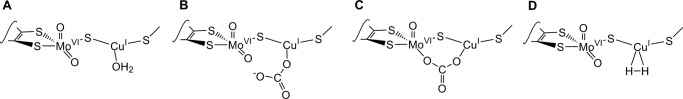
Possible structures for the EPR-active species seen with CO dehydrogenase. Structures A and B represent CO dehydrogenase reduced by dithionite or H2, respectively. Structures C and D are possible structures for the bound bicarbonate species.
Considering next the EPR signal seen with dihtionite-reduced enzyme in the presence of bicarbonate, the absence of proton coupling suggests strongly that bicarbonate has displaced the bound water from the copper of the binuclear center. Two alternate structures can be considered, as shown in Fig. 6 (B and C). The first of these has bicarbonate coordinated in a monodentate fashion to the copper and with the equatorial Mo=O unprotonated (or, alternatively, protonated but in a configuration that leads to very weak coupling). The second possibility has bicarbonate bridging between the Mo and Cu of the binuclear center in a bidentate fashion, displacing both the copper-coordinated water and the equatorial Mo=O. Our present preference on entropic grounds is the second structure, but a definitive answer must await crystallographic analysis of the bicarbonate complex. We consider it unlikely that bicarbonate displaces only the equatorial Mo=O of oxidized enzyme (to give a Mo(V)/Cu(I) species analogous to a presumed Mo(VI)/Cu(I) intermediate proposed on the basis of computational studies (11, 12), as this does not account for the experimentally observed loss of proton coupling in the EPR signal in the presence of bicarbonate.
Finally, with the H2-reduced native enzyme, given the effectiveness with which CO dehydrogenase oxidizes H2, we consider it very likely that that, as has been proposed for CO oxidation where CO initially binds to the copper of the binuclear center, H2 also first binds at the copper of the binuclear center (11, 12), displacing the H2O coordinated to the copper prior to catalysis. We believe that the observed EPR signal (as with CO as substrate) arises from enzyme that has already become partially reduced by reaction with prior turnover under the reaction conditions, such that H2 is bound to a partially reduced Mo(V)/Cu(I) binuclear center. Although the complex cannot progress through the catalytic cycle until the center has become fully oxidized (via intramolecular electron transfer to the other redox-active sites of the enzyme), the signal-giving species in fact reflects a paramagnetic analog of the Michaelis complex. The lack of observed reaction of the silver-substituted enzyme with H2 indicates that the copper is necessary for activity and it is the copper rather than molybdenum is the site of H2 binding. The situation is directly analogous to the long studied “rapid” signals seen with the closely related molybdenum-containing enzyme xanthine oxidase (27) which have been shown to arise from substrate similarly bound to partially reduced enzyme (28). Binding of H2 to metals, and specifically Cu(I), such as is proposed has chemical precedent (29–31) and is known to involve a side-on η2 binding that polarizes substrate, facilitating its ionization to a metal hydride, for which precedent also exists (29–32). (By contrast, no precedent exists that we are aware of for molybdenum hydrides or H2 complexes.) This accounts for the rapid exchange with solvent, but the pH independence of the observed reaction with H2 and the observation of two strongly coupled protons in the EPR signal of H2-reduced enzyme requires that the pKa for this ionization be greater than ∼12. An EPR sample of the enzyme in H2O but reduced with D2 and quickly frozen (within 10 s) showed the same strong coupling to protons seen in the H2-reduced sample, indicating that this exchange is relatively rapid, likely facilitated by the highly conserved glutamate residue in the active site (Glu-763). The rapid protonation/deprotonation of bound H2 is in fact catalytically productive, as the Cu(I)-hydride species formed transiently can itself deprotonate, with (when beginning with the fully oxidized Mo(VI)/Cu(I) binuclear center) the two reducing equivalents simply entering the highly delocalized redox-active orbital of the binuclear center.
Catalysis with H2 as substrate is thus proposed to proceed as shown in Fig. 7, with H2 first binding at the copper of the binuclear center, displacing the bound water, and becoming polarized. Successive ionizations, first of bound H2 and then the resulting hydride, lead to full deprotonation and two-electron reduction of the binuclear center, formally to the Mo(IV)/Cu(I) state. Given the experimentally observed rapid exchange of solvent protons into the bound hydride, at least the first of these prototropic equilibria must be rapid compared with catalysis. Furthermore, given the pH independence of the reaction the pKa for this step must be high, meaning that the equilibrium favors the H2 complex over the hydride. This accounts for the observation of two protons rather than one in the EPR signal and rationalizes the more modest reactivity of enzyme toward H2 compared with CO. The failure to observe any reverse reaction (i.e. reduction of protons to H2), as seen with the Ni-Fe CO dehydrogenases, suggests that deprotonation of the copper hydride intermediate is strongly downhill thermodynamically and functionally irreversible (15). Nevertheless, there are common elements of the reaction mechanism proposed here for reduction by H2 and that proposed previously for reduction by CO (11, 12). These include initial binding of substrate to the copper of the binuclear center in a manner that activates substrate (by back-bonding from copper into a π* orbit in the case of CO, by polarization of the H-H bond in the case of H2) and utilization of the highly delocalized nature of the redox-active orbital of the binuclear center to bring about its reduction via the copper rather than the molybdenum.
FIGURE 7.

The proposed reaction mechanism of CO dehydrogenase with H2. Catalysis proceeds by displacement of H2O from the copper of the binuclear center by H2, followed by successive deprotonations as shown to introduce a pair of reducing equivalents into the highly delocalized redox-active orbital of the binuclear center.
In conclusion, the oxidation of H2 by the Mo/Cu CO dehydrogenase requires a fully constituted active site with bound copper, not silver, and occurs with a pH-independent kred of 5.3 s−1. The observed EPR signal seen upon reduction of enzyme with H2 is distinct from that seen with CO as substrate or with dithionite as noncatalytic reductant. The proton coupling observed in the dithionite-reduced enzyme is lost in the presence of bicarbonate and is proposed to arise from the crystallographically observed water molecule coordinated to the copper of the binuclear center. On the basis of these observation, a reaction mechanism can be proposed in which catalysis is initiated by side-on binding of H2 to the copper of the binuclear center, which predisposes its ionization through a copper hydride intermediate; chemical precedent exists for both such species (19, 30–33). Subsequent ionization of the copper hydride leads to formal reduction of the binuclear center to a Mo(IV)/Cu(I) state, taking advantage of the extremely delocalized redox-active orbital of the center. Glutamate 763 is well positioned in the active site to facilitate these deprotonation events.
This work was supported by Department of Energy Grant 13ER16411 (to R. H.).
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid.
REFERENCES
- 1. Meyer O., Schlegel H. G. (1978) Reisolation of the carbon monoxide utilizing hydrogen bacterium Pseudomonas carboxydovorans (Kistner) comb. nov. Arch. Microbiol. 118, 35–43 [DOI] [PubMed] [Google Scholar]
- 2. Meyer O., Schlegel H. G. (1980) Carbon monoxide:methylene blue oxidoreductase from Pseudomonas carboxydovorans. J. Bacteriol. 141, 74–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cypionka H., Meyer O. (1983) Carbon monoxide-insensitive respiratory chain of Pseudomonas carboxydovorans. J. Bacteriol. 156, 1178–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meyer O., Gremer L., Ferner R., Ferner M., Dobbek H., Gnida M., Meyer-Klaucke W., Huber R. (2000) The role of Se, Mo, and Fe in the structure and function of carbon monoxide dehydrogenase. Biol. Chem. 381, 865–876 [DOI] [PubMed] [Google Scholar]
- 5. Wilcoxen J., Zhang B., Hille R. (2011) Reaction of the molybdenum- and copper-containing carbon monoxide dehydrogenase from Oligotropha carboxydovorans with quinones. Biochemistry 50, 1910–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moersdorf G., Frunzke K., Gadkari D., Meyer O. (1992) Microbial growth on carbon monoxide. Biodegradation 3, 61–82 [Google Scholar]
- 7. Ragsdale S. W., Pierce E. (2008) Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784, 1873–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santiago B., Schübel U., Egelseer C., Meyer O. (1999) Sequence analysis, characterization and CO-specific transcription of the cox gene cluster on the megaplasmid pHCG3 of Oligotropha carboxidovorans. Gene 236, 115–124 [DOI] [PubMed] [Google Scholar]
- 9. Dobbek H., Gremer L., Meyer O., Huber R. (1999) Crystal structure and mechanism of CO dehydrogenase, a molybdo iron-sulfur flavoprotein containing S-selanylcysteine. Proc. Natl. Acad. Sci. U.S.A. 96, 8884–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dobbek H., Gremer L., Kiefersauer R., Huber R., Meyer O. (2002) Catalysis at a dinuclear [CuSMo(=O)OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 99, 15971–15976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hofmann M., Kassube J. K., Graf T. (2005) The mechanism of Mo-/Cu-dependent CO dehydrogenase. J. Biol. Inorg. Chem. 10, 490–495 [DOI] [PubMed] [Google Scholar]
- 12. Siegbahn P. E., Shestakov A. F. (2005) Quantum chemical modeling of CO oxidation by the active site of molybdenum CO dehydrogenase. J. Comput. Chem. 26, 888–898 [DOI] [PubMed] [Google Scholar]
- 13. Santiago B., Meyer O. (1996) Characterization of hydrogenase activities associated with the molybdenum CO dehydrogenase from Oligotropha carboxydovorans. FEMS Microbiol. Lett. 136, 157–162 [Google Scholar]
- 14. Bhatnagar L., Krzycki J. A., Zeikus J. G. (1987) Analysis of hydrogen metabolism in Methanosarcina barkeri: regulation of hydrogenase and role of CO-dehydrogenase in H2 production. FEMS Microbiol. Lett. 41, 337–343 [Google Scholar]
- 15. Menon S., Ragsdale S. W. (1996) Unleashing hydrogenase activity in carbon monoxide dehydrogenase/acetyl-CoA synthase and pyruvate:ferredoxin oxidoreductase. Biochemistry 35, 15814–15821 [DOI] [PubMed] [Google Scholar]
- 16. Zhang B., Hemann C. F., Hille R. (2010) Kinetic and spectroscopic studies of the molybdenum-copper CO dehydrogenase from Oligotropha carboxidovorans. J. Biol. Chem. 285, 12571–12578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Resch M., Dobbek H., Meyer O. (2005) Structural and functional reconstruction in situ of the CuSMoO2 active site of carbon monoxide dehydrogenase from the carbon monoxide oxidizing eubacterium Oligotropha carboxidovorans. J. Biol. Inorg. Chem. 10, 518–528 [DOI] [PubMed] [Google Scholar]
- 18. Wilcoxen J., Snider S., Hille R. (2011) Substitution of silver for copper in the binuclear Mo/Cu center of carbon monoxide dehydrogenase from Oligotropha carboxidovorans. J. Am. Chem. Soc. 133, 12934–12936 [DOI] [PubMed] [Google Scholar]
- 19. Mahoney W. S., Brestensky D. M., Stryker J. M. (1988) Selective hydride-mediated conjugate reduction of α,β-unsaturated carbonyl compounds using [(Ph3P)CuH]6. J. Am. Chem. Soc. 110, 291–293 [Google Scholar]
- 20. Speight J. (2005) Lange's Handbook of Chemistry, 16th Ed., pp. 1.312–1.313, McGraw-Hill, New York [Google Scholar]
- 21. Stoll S., Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 [DOI] [PubMed] [Google Scholar]
- 22. Hille R., Anderson R. F. (2001) Coupled electron/proton transfer in complex flavoproteins: solvent kinetic isotope effect studies of electron transfer in xanthine oxidase and trimethylamine dehydrogenase. J. Biol. Chem. 276, 31193–31201 [DOI] [PubMed] [Google Scholar]
- 23. Olson J. S., Ballou D. P., Palmer G., Massey V. (1974) Mechanism of action of xanthine oxidase. J. Biol. Chem. 249, 4363–4382 [PubMed] [Google Scholar]
- 24. Siegbahn P. E., Tye J. W., Hall M. B. (2007) Computational studies of [NiFe] and [FeFe] hydrogenases. Chem. Rev. 107, 4414–4435 [DOI] [PubMed] [Google Scholar]
- 25. Gourlay C., Nielsen D. J., White J. M., Knottenbelt S. Z., Kirk M. L., Young C. G. (2006) Paramagnetic active site models for the molybdenum-copper carbon monoxide dehydrogenase. J. Am. Chem. Soc. 128, 2164–2165 [DOI] [PubMed] [Google Scholar]
- 26. Léger C., Jones A. K., Roseboom W., Albracht S. P., Armstrong F. A. (2002) Enzyme electrokinetics: hydrogen evolution and oxidation by Allochromatium vinosum [NiFe]-hydrogenase. Biochemistry 41, 15736–15746 [DOI] [PubMed] [Google Scholar]
- 27. Bray R. C., Vänngård T. (1969) “Rapidly appearing” molybdenum electron paramagnetic resonance signals from reduced xanthine oxidase. Biochem. J. 114, 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hille R., Kim J. H., Hemann C. (1993) Reductive half-reaction of xanthine oxidase: mechanistic role of the species giving rise to the “rapid type 1” molybdenum(V) electron paramagnetic resonance signal. Biochemistry 32, 3973–3980 [DOI] [PubMed] [Google Scholar]
- 29. Wang N., Wang M., Chen L., Sun L. (2013) Reactions of [FeFe]-hydrogenase models involving the formation of hydrides related to proton reduction and hydrogen oxidation. Dalton Trans. 42, 12059–12071 [DOI] [PubMed] [Google Scholar]
- 30. Frohman D. J., Grubbs G. S., 2nd, Yu Z., Novick S. E. (2013) Probing the chemical nature of dihydrogen complexation to transition metals, a gas phase case study: H2-CuF. Inorg. Chem. 52, 816–822 [DOI] [PubMed] [Google Scholar]
- 31. Kemper P. R., Weis P., Bowers M. T., Maıtre P. (1998) Origin of bonding interactions in Cu+(H2)n clusters: an experimental and theoretical investigation. J. Am. Chem. Soc. 120, 13494–13502 [Google Scholar]
- 32. Hulley E. B., Welch K. D., Appel A. M., DuBois D. L., Bullock R. M. (2013) Rapid, reversible heterolytic cleavage of bound H2. J. Am. Chem. Soc. 135, 11736–11739 [DOI] [PubMed] [Google Scholar]
- 33. Moser R., Bosković Z. V., Crowe C. S., Lipshutz B. H. (2010) CuH-catalyzed enantioselective 1,2-reductions of a,b-unsaturated ketones. J. Am. Chem. Soc. 132, 7852–7853 [DOI] [PMC free article] [PubMed] [Google Scholar]