

Background: Pyruvate transport into mitochondria is a key step in energy metabolism. Zaprinast is a well known phosphodiesterase inhibitor.
Results: Zaprinast has a strong influence on pyruvate transport into mitochondria.
Conclusion: Inhibition of the mitochondrial pyruvate carrier by Zaprinast causes accumulation of aspartate at the expense of glutamate.
Significance: Maintenance of normal amino acid levels in the retina relies on pyruvate transport into mitochondria.
Keywords: Aspartate, Glutamate, Metabolism, Phosphodiesterases, Pyruvate, Retina
Abstract
Transport of pyruvate into mitochondria by the mitochondrial pyruvate carrier is crucial for complete oxidation of glucose and for biosynthesis of amino acids and lipids. Zaprinast is a well known phosphodiesterase inhibitor and lead compound for sildenafil. We found Zaprinast alters the metabolomic profile of mitochondrial intermediates and amino acids in retina and brain. This metabolic effect of Zaprinast does not depend on inhibition of phosphodiesterase activity. By providing 13C-labeled glucose and glutamine as fuels, we found that the metabolic profile of the Zaprinast effect is nearly identical to that of inhibitors of the mitochondrial pyruvate carrier. Both stimulate oxidation of glutamate and massive accumulation of aspartate. Moreover, Zaprinast inhibits pyruvate-driven O2 consumption in brain mitochondria and blocks mitochondrial pyruvate carrier in liver mitochondria. Inactivation of the aspartate glutamate carrier in retina does not attenuate the metabolic effect of Zaprinast. Our results show that Zaprinast is a potent inhibitor of mitochondrial pyruvate carrier activity, and this action causes aspartate to accumulate at the expense of glutamate. Our findings show that Zaprinast is a specific mitochondrial pyruvate carrier (MPC) inhibitor and may help to elucidate the roles of MPC in amino acid metabolism and hypoglycemia.
Introduction
Pyruvate is a critical metabolite that links glycolysis with the mitochondrial tricarboxylic acid (TCA) cycle, and it is a hub for synthesis of amino acids, carbohydrates, and fatty acids. Pyruvate enters mitochondria through a recently identified mitochondrial pyruvate carrier (MPC),3 a 150-kDa complex of MPC1 and MPC2 located on the inner mitochondrial membrane (1, 2). Knock-out of MPC1 in Drosophila elevates pyruvate and decreases mitochondrial TCA intermediates (1). Children with mutations in a conserved region of human MPC1 have symptoms of lactic acidosis and hyperpyruvatemia (1, 3). MPC inhibitors have been identified in the past 40 years including the classical α-cyanocinnamate analogs, UK5099, α-cyano-4-hydroxycinnamic acid (4), and insulin sensitizer thiazolidinedione (5–7).
Zaprinast, a lead compound used for the development of sildenafil (Viagra), is a well established inhibitor of cGMP-specific phosphodiesterase (PDE). PDEs hydrolyze the cyclic phosphate bond in cAMP and cGMP, and they function to inactivate cyclic nucleotide signaling pathways. Zaprinast has been used as a tool to study PDE5 and PDE6 (8–11). PDE5, PDE6, and PDE9 are cGMP-specific, PDE4, PDE7, and PDE8 are cAMP-specific, and the rest of the members of the PDE family have dual specificity (12). Zaprinast is the only drug that inhibits PDE6 more potently than PDE5 (13). PDE6 is primarily expressed in retinal photoreceptors where it is responsible for light-dependent signal transduction. Mutations in the Pde6a, b, or c genes all cause retinal degeneration in humans (14–16) and in mouse models (17, 18). We performed a study intended to use Zaprinast to simulate the effects of PDE6 dysfunction in an ex vivo retinal degeneration model. We treated cultured mouse retinal explants with Zaprinast, but we found unexpectedly that Zaprinast is a potent inhibitor of mitochondrial pyruvate transport. This led to a novel and important finding, the focus of this report, that inhibition of mitochondrial pyruvate transport triggers dramatic metabolomic changes in neuronal tissues that severely alter the concentrations of glutamate and aspartate.
EXPERIMENTAL PROCEDURES
Reagents
Zaprinast was obtained from EMD Millipore Corp (Billerica, MA). [13C6]Glucose was obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA). [2-14C]Pyruvate was purchased from PerkinElmer Life Sciences. Other 13C tracers and reagents were purchased from Sigma unless otherwise specified.
Animals
C57BL/6 mice (6–8 weeks old) and C3Sn.BLiA-Pde6b+/DnJ (PDE6b+) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). C3H retinal degeneration (rd1) mice, which bear a Pde6b mutation, were provided by Dr. Thomas A Reh, University of Washington. Pde6b+ mice were used as the background control for rd1 mice. Cyclic nucleotide gated channel β1 knock-out (Cngb1−/−) mice on C57BL/6 background were obtained from the Institute for Ophthalmic Research, Tuebingen, Germany (19, 20). Aralar/AGC1+/− mice (21) were crossed to produce Aralar/AGC1−/− and Aralar/AGC1+/+ control littermates in Sv129/C57BL6 background at Dr. Jorgina Satrústegui's laboratory (Madrid, Spain). Experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) recommendations at the University of Washington guidelines after IACUC approval and with procedures approved in the Directive 86/609/EEC of the European Union and the Ethics Committee of the Universidad Autónoma de Madrid.
Retina Isolation and Culture
Mice were euthanized using CO2 and cervical dislocation. The eyes were removed, and retinas were separated from retinal pigment epithelium in cold HBSS. The retina was cultured in Krebs-Ringer/HEPES/bicarbonate buffer (22) with glucose (5 mm) or other 13C-labeled substrate (5 mm) in a 37 °C 5% CO2 incubator.
cGMP Assay
Two mouse retinas were homogenized in 300 μl of 0.1 n HCL. After centrifugation, 100 μl of the supernatant was assayed for cGMP by using a Direct cGMP ELISA kit from Enzo Life Sciences, Inc. (Farmingdale, NY).
Gas Chromatography/Mass Spectrometry (GC/MS) Analysis of Metabolites
Individual mouse retina was rinsed in cold 0.9% NaCl and snap-frozen in liquid nitrogen. The retina was homogenized in a 700:200:50 cold mixture of methanol/chloroform/water with 5 nmol of internal standard methyl succinate. After centrifugation, the supernatant was dried under vacuum, derivatized, and analyzed by GC-MS (Agilent 7890/5975C) (22). The peaks were analyzed using Agilent data analysis software. The measured distribution of mass isotopomers was corrected for natural abundance of 13C metabolite intensity defined with standards and verified by mass after each experiment. Enrichment was calculated by dividing the labeled ions with total ion intensity. The natural abundance from the tracers and derivatization reagents were corrected using IsoCor software (23, 24).
O2 Consumption Measurements of Isolated Mitochondria
Brain mitochondria were isolated as previously reported (22). O2 consumption was measured in an Oroboros Oxygraph-2K. The respiration buffer consisted of 150 mm KCl, 10 mm KH2PO4, 1 mm MgCl2, pH 7.4. 25 μl of 5 mg/ml mitochondria was injected into the chamber for each assay. Respiration was measured in response to serial additions of pyruvate/malate, ADP, drug (DMSO, Zaprinast, or UK5099), glutamate/malate, and succinate. The final concentrations of substrates were 100 μm EGTA, 50 μm CaCl2, 1.5 mm pyruvate, 0.5 mm malate, 2.5 mm ADP, 1 mm glutamate/0.5 mm malate, and 5 mm succinate. After the addition of each component, the O2 consumption rate was allowed to stabilize for 4–5 min before the slope was quantified.
MPC Activity Assay
Mouse liver mitochondria were prepared as described before (25). Mitochondrial pyruvate transport was measured as reported (4, 26) with modifications. The liver mitochondria were suspended in medium containing sucrose (250 mm), Tris-HCl (5 mm), and EGTA (2 mm) with pH 7.6. (∼5 mg of mitochondrial protein/ml of medium). An aliquot of the mitochondrial suspension (50 μl or 0.5 mg of mitochondrial protein) was added to 150 μl of medium containing KCl (125 mm) and Tris-HCl (20 mm), pH 6.8, with or without Zaprinast. After incubation for 5 min at room temperature, [2-14C]pyruvate (15 μm or 0.0225 μCi) was spiked into the 200-μl solution to start the assay. After 1 min, the mitochondria-associated radioactivity was separated from the free medium by transferring the mitochondrial suspension to a 0.4-ml centrifuge tube containing a layer of oil consisting of 1:37.5 n-dodecane:bromododecane (Sigma) and spinning at 12,535 × g for 8 s. The portion of the tube containing the mitochondrial pellet was cut off with a razor blade, placed into a scintillation vial, and then counted for radioactivity (26).
Measurement of Mitochondrial Metabolites
Freshly isolated mitochondria had intact mitochondrial membranes. To disrupt them we treated mitochondria with 0.1% Triton and freeze-thawed 5 times. Both membrane-intact and -disrupted mitochondria from separate isolations were incubated with 1 mm [13C3]pyruvate in the buffer used for respiration studies on isolated mitochondria at 30 °C for 10 min. The metabolites were analyzed by GC/MS. To test shuttling of metabolites into and out of mitochondria, we incubated freshly isolated mitochondria with 100 μm EGTA, 1 mm glutamate, 0.5 mm malate, 2.5 mm ADP, 50 μm CaCl2, and 1 mm [13C3]pyruvate in respiration buffer at 30 °C for 10 min. After centrifugation, the mitochondrial pellet and incubation medium were collected for GC/MS analysis.
Glucose Concentration Assay
Glucose in the culture medium was measured using an Amplex® Red Glucose/Glucose Oxidase Assay kit (Invitrogen).
Pyruvate Dehydrogenase Activity (PDH) Activity Assay
PDH activity was measured using an NADH cycling assay in the presence of diaphorase to reduce the generated NADH and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was as an electron acceptor (27). Purified PDH protein from pig heart was incubated with 1 mm MgCl2, 5 mm CaCl2, 5 mm l-carnitine, 0.2 mm thiamine pyrophosphate, 0.1 mm CoA, 2.5 mm NAD+, 1 mm MTT, and 1 unit/ml diaphorase in 50 mm HEPES/KOH buffer, pH 7.5, at 30 °C. 5 mm pyruvate was added into the mixture to start the reaction, and the plate was read at 570 nm every 10 s for 10 min. The Δ570-nm with maximum linear rate was used for calculating the enzyme activity.
Statistical Analysis
Data are expressed as the mean ± S.E. The significance of differences between means was determined by unpaired two-tailed t tests or analysis of variance with an appropriate post hoc test. A p value < 0.05 was considered to be significant.
RESULTS
Zaprinast Causes Depletion of Glutamate and Accumulation of Aspartate
We began the experiments with the intention of identifying the metabolomic signature of PDE6 inhibition. We isolated mouse retinas and incubated them with or without Zaprinast in the presence of 5 mm glucose and then extracted metabolites for GC/MS analysis. We then determined the concentration of Zaprinast required to inhibit PDE activity in mouse retinas. The concentration of Zaprinast required to cause accumulation of cGMP in the retina is in the range of 20∼200 μm (supplemental Fig. 1A). We also found large changes in several metabolite levels at 200 μm (Fig. 1A). Glutamate and aspartate had the largest changes: about a 5-fold decrease and increase, respectively. The effects on both glutamate and aspartate were first detectable at 30 min after the addition of Zaprinast, became more apparent at 60 min, and remained constant from 2 to 6 h (supplemental Fig. 1, B and C). We reasoned that the change of these two metabolites might be primarily responsible for the overall change of metabolic profile as aspartate-derived metabolites, malate, methionine, cysteine, and N-acetyl-aspartate increased, whereas glutamate-derived metabolites, glutamine and α-ketoglutarate (αKG), decreased. We considered that glutamate in the retina might decrease because it is released into the medium. However, both glutamate and aspartate were undetectable in the incubation medium. Pyruvate increased by about 2–3-fold in response to Zaprinast in both retina and medium, but lactate did not change (Fig. 1, A and B). Glucose deprivation (22, 28) causes similar changes of glutamate and aspartate levels in retina and brain slices. Therefore, we tested the effect of Zaprinast on glucose consumption. The retina consumed about 1.2 μmol/h/retina. Zaprinast did not affect the rate of glucose consumption (Fig. 1C).
FIGURE 1.

Zaprinast significantly changes the metabolomic profile in the retina. A, Zaprinast changes metabolites in mouse retina. The retina was cultured for 1 h with 200 μm Zaprinast or with DMSO as a control. Metabolites were extracted, quantified by GC-MS, and normalized to the DMSO alone controls (n = 10). 3-PG, 3-phosphoglyceric acid; PEP, phosphoenolpyruvate; AKG, α-ketoglutarate; GABA, γ-aminobutyric acid; 2-HG, 2-hydroxyglutarate, NAA, N-acetyl aspartate. B, Zaprinast increases pyruvate release from the retina. The medium from A was assayed for metabolites. The data are -fold changes over DMSO control. C, Zaprinast does not change glucose consumption. The medium from A was tested for glucose concentration. NS, no significant difference. * indicates p < 0.05 versus DMSO-treated.
The Effect of Zaprinast on Glutamate and Aspartate Is Independent of PDE Inhibition
To investigate the relationship between PDE6 inhibition and the metabolic effects of Zaprinast, we compared the concentrations of Zaprinast required to inhibit PDE in the retina with the concentration required to cause glutamate to decrease. We found that Zaprinast increases cGMP at 50 and 100 μm and increases both cGMP and cAMP at 200 μm. This indicates that Zaprinast inhibits PDE6 specifically between 50 and 100 μm, but it inhibits other PDEs at 200 μm (Fig. 2A). Surprisingly, Zaprinast decreased glutamate and increased aspartate at much lower concentrations beginning at 2 μm (Fig. 2B).
FIGURE 2.

PDE6 is not involved in the effect of Zaprinast on glutamate and aspartate. A, Zaprinast increases cGMP at 50 μm and cAMP at 200 μm (n = 6). B, Zaprinast dose-dependently decreases glutamate and increases aspartate. Glutamate is shown as white bars, and aspartate is shown as black bars in this and in the next two panels. The retina was treated for 1 h. The decrease of glutamate starts at concentrations of Zaprinast as low as 2 μm (n = 5). C, Cngb1 deficiency does not block the effect of 200 μm Zaprinast (Zap) on glutamate and aspartate. The retina was incubated for 1 h (n = 5). D, Zaprinast at 200 μm decreases glutamate and increases aspartate in rd1 mice. Retinas from 3-month-old PDE6b+ and rd1 mice were incubated with Zaprinast for 1 h (n = 4). E, sildenafil increases cGMP in retinas (n = 3). F, glutamate and aspartate do not change in retinas treated with sildenafil (n = 3). * indicates p < 0.05 versus DMSO-treated.
To test whether cyclic nucleotide-gated channels and Ca2+ are involved in the decrease of glutamate, we used the retina from Cngb1−/− mice in which the rising cGMP could not activate cyclic nucleotide gated channels to cause depolarization and elevation of intracellular free Ca2+ (19, 29). Remarkably, Zaprinast decreased glutamate and increased aspartate in Cngb1−/− retina (Fig. 2C). Furthermore, neither of the cell-permeable cGMP analogues 8-Br-cGMP and 8-(4-chlorophenylthio)-cGMP nor inhibitors of Protein kinase G and PKA changed the levels of glutamate and aspartate in the retina (supplemental Fig. 2). These findings suggested that Zaprinast increases aspartate and lowers glutamate by a different mechanism than we expected, i.e. in a PDE6-independent fashion.
As further confirmation that the metabolic effects of Zaprinast occur independently of PDE6 inhibition, we applied Zaprinast to the retina from rd1 mice that were 2 months old. The rd1 mutation causes almost all PDE6-expressing photoreceptors to die and disappear by 1 month of age and thus abolishes PDE6 activity. We found that glutamate and aspartate levels in rd1 retina are lower than normal, consistent with photoreceptors being a major source of these amino acids in normal retinas. We also found that Zaprinast potently decreases glutamate and increases aspartate in rd1 retina, suggesting that Zaprinast acted at a site distinct from PDE6 (Fig. 2D). As further confirmation that the effect of Zaprinast on glutamate and aspartate was not through PDE, we evaluated the effect of sildenafil, another PDE5/6 inhibitor. Sildenafil between 0.1–50 μm robustly increased cGMP in WT retina but did not have a significant effect on glutamate or aspartate (Fig. 2, E and F). Taken altogether, these results show unambiguously that the effect of Zaprinast on glutamate and aspartate occurs independently of PDE inhibition.
The Effect of Zaprinast on Glutamate and Aspartate Occurs Independently of GPR35 Activation
Zaprinast also has been identified as a potent agonist of G protein-coupled receptor 35 (GPR35) (30, 31). GPR35 is expressed predominantly in spleen, immune cells, and gastrointestinal tissues, but expression in eye also has been reported (32, 33). To test whether GPR35 contributes to the metabolic effect of Zaprinast, we preincubated the retina with a GPR35 antagonist, CID 2745687, for 30 min followed by an additional 30 min of Zaprinast treatment. CID 2745687 did not block the effect of Zaprinast on glutamate and aspartate. Furthermore, the GPR35 agonist, pamoic acid, did not change glutamate or aspartate levels (supplemental Fig. 3). These results show that GPR35 was not responsible for the observed Zaprinast effects.
Zaprinast Blocks Entry of Glucose-derived Pyruvate into the TCA Cycle
To identify the mechanism behind the metabolic effect of Zaprinast, we incubated the retina with [13C6]glucose for 15 min with/without Zaprinast and then analyzed the isotopomer distributions of the intermediates. Under normal conditions, pyruvate and lactate become fully labeled by glycolysis of [13C6]glucose. Pyruvate loses 1 13C to synthesize acetyl-CoA, and the two-labeled carbons appear in most TCA intermediates in the first round of the TCA cycle (Fig. 3A). In the second cycle citrate gains a total of four labeled carbons, and the rest of the intermediates have either three or four carbons labeled due to the loss of CO2 at one of two different positions (supplemental Fig. 4A). Zaprinast did not change the 13C enrichment in pyruvate and lactate. However, the enrichment of glutamate, aspartate, and TCA intermediates was much lower after Zaprinast treatment (Fig. 3B). Zaprinast increased the concentration of M3 (three labeled carbons) pyruvate but not lactate, and it significantly decreased both enrichment and concentration of the M2 isotopomer of citrate (the form with two labeled carbons from the first cycle), M4 (four labeled carbons from the second cycle), and M5/M6 (after three or more cycles) (Fig. 3, C–E; supplemental Fig. 4B). These results suggest that Zaprinast inhibits the formation of citrate from pyruvate. Accordingly, the downstream intermediates αKG and glutamate had fewer M2 and M3/4 isotopomers in Zaprinast-treated retinas. Zaprinast decreased M2 in both succinate and malate but increased both M2 and M3 in aspartate (Fig. 3, F--J). Zaprinast increased the total amount of labeled aspartate, but it decreased the overall enrichment of aspartate because Zaprinast causes a very large increase in the amount of unlabeled aspartate. Overall, these results indicate that Zaprinast inhibits the incorporation of pyruvate-derived carbons into the TCA cycle. This causes accumulation of oxaloacetate and aspartate. We also noted that the excess oxaloacetate “spills over” into the M3 isotopomers of malate and succinate by the reversible malate dehydrogenase and fumarate hydratase reactions.
FIGURE 3.

Zaprinast blocks synthesis of citrate, αKG, and glutamate from glucose-derived pyruvate. A, schematic of carbon labeling (black circles for 13C) from [13C6]glucose after the first TCA cycle. Pyruvate and lactate are fully labeled on all three carbons. After PDH removes one labeled carbon, the remaining two labeled carbons in acetyl-CoA incorporate into mitochondrial intermediates in the first cycle. OAA, oxaloacetate. B, Zaprinast decreased the enrichment of TCA cycle intermediates from [13C6]glucose. The retina was incubated with 5 mm [13C6]glucose for 15 min. C–J, mass isotopomer distributions of intermediates from B. The fraction of each isotopomer was multiplied by the total metabolite concentration and normalized by protein concentration (pmol or nmol/mg of protein). Zaprinast increases the total incorporation of labeled carbon into pyruvate, but it decreases labeling of TCA intermediates. * indicates p < 0.05 versus DMSO-treated. (n = 3).
Oxidized Glutamate Is the Source of the Aspartate That Accumulates in Response to Zaprinast Treatment
The decrease of glutamate that occurs with Zaprinast treatment always occurs in parallel with an increase of aspartate. We hypothesized that the increased carbons of aspartate come from oxidation of glutamate through αKG dehydrogenase. To test this hypothesis, we labeled the retina with [13C5]glutamine for 5 min and then replaced it with unlabeled glucose for 10 min (Fig. 4). The short time pulse makes it possible to trace the fate of glutamate as TCA intermediates are labeled in the first cycle and then start to lose the labeled carbons in the second cycle. During the first round of the TCA cycle, glutamate and αKG are fully labeled as M5, and the subsequent intermediates have four 13C carbons. A small amount of M5 citrate also accumulates. This must result from the reversal of the isocitrate dehydrogenase reaction (34) (Fig. 4A). For the second cycle, αKG and glutamate begin with three labeled carbons. After oxidative decarboxylation of αKG, the downstream intermediates have two labeled carbons, and citrate ends up having either two or three labeled carbons (supplemental Fig. 5A). Enrichment of citrate decreased with Zaprinast treatment, whereas enrichment of malate and aspartate increased (Fig. 4B). In the first TCA cycle, Zaprinast decreases the labeled citrate (M4) but significantly increases labeled succinate, malate, and especially aspartate (Fig. 4, C–I). These results show that Zaprinast prevents glutamate-derived carbons from passing through citrate to the second cycle. Consistent with this, M2 citrate, M3 glutamate, and M3 αKG decreased in response to Zaprinast. Zaprinast blocked citrate formation and thereby caused accumulation of oxaloacetate and aspartate produced from oxidized glutamate. To confirm this we incubated the retina with and without glutamine for 2 h. Both glutamate and aspartate increased ∼5-fold in response to glutamine (relative to glucose alone), and Zaprinast shifted the glutamate-to-aspartate ratio by decreasing glutamate about 5-fold and increasing aspartate about 10-fold (supplemental Fig. 5B).
FIGURE 4.

Carbons used to increase aspartate come from oxidation of glutamate. A, schematic of carbon labeling (black circles represent 13C) from [13C5]glutamine after the first TCA cycle. Most of the 13C from [13C5]glutamine labels five carbons in αKG and four carbons for the rest of the intermediates in the first cycle. However, a small fraction (<10%) of the [13C5]glutamine also labels five carbons in citrate by reversal of the isocitrate dehydrogenase reaction. B, Zaprinast increases enrichment of aspartate and malate from [13C5]glutamine. The retina was pulsed with 5 mm [13C5]glutamine for 5 min and then chased with unlabeled glucose for 10 min. C–I, mass isotopomer distribution of intermediates from B. The metabolite concentration was normalized by protein concentration (pmol or nmol/mg protein). Zaprinast increases M4 succinate, malate, and aspartate from [13C5]glutamine. * indicates p < 0.05 versus DMSO treated (n = 3).
Zaprinast Inhibits O2 Consumption in Intact Retina
To test the effects of Zaprinast and UK5099 on mitochondrial function in ex vivo retina, we measured O2 consumption from small pieces of intact mouse retina. Zaprinast caused a rapid increase followed by sustained inhibition of O2 consumption. This biphasic effect can be explained by a mixed effect of Zaprinast on PDE and MPC activities. PDE inhibition increases O2 consumption by stimulating energy consumption whereas MPC inhibition decreases availability of fuel to mitochondria. Consistent with this we found that sildenafil increases O2 consumption whereas UK5099 inhibits it (Supplemental Fig. 6). UK5099 causes a slow but strong inhibition, probably due to its ability to inhibit complex II (Results not shown). Consistent with this we found that UK5099 at 100 μm but not 10 nm inhibits succinate-driven O2 consumption.
Zaprinast Inhibits Pyruvate-driven O2 Consumption in Brain Mitochondria
Pyruvate enters via the MPC into the mitochondrial matrix where PDH transforms it into acetyl-CoA. PDH and the dehydrogenases of the TCA cycle produce NADH that provides reducing power for O2 consumption. To test the hypothesis that Zaprinast inhibits pyruvate oxidation, we isolated brain mitochondria, and measured rates of O2 consumption fueled either by pyruvate, glutamate, or succinate. Zaprinast almost completely blocked pyruvate-driven O2 consumption (Fig. 5, A--D). However, Zaprinast did not block O2 consumption fueled by glutamate or succinate. This is consistent with our isotopic labeling experiment in which Zaprinast did not inhibit glutamate oxidation. We next tested the hypothesis that Zaprinast blocks pyruvate transport by inhibiting MPC. Similar to Zaprinast, MPC inhibitor UK5099 blocked pyruvate-driven but increased glutamate and succinate-driven oxygen consumption (Fig. 5, E and F, supplemental Fig. 7, A and B). There was no significant difference in the rate of oxygen consumption between Zaprinast and UK5099 (supplemental Fig. 7, C and D).
FIGURE 5.

Zaprinast inhibits pyruvate-driven oxygen consumption. Zaprinast inhibits O2 consumption in brain mitochondria when fueled by pyruvate but not when fueled by glutamate or succinate (Suc) in conditions starting with either glutamate/malate (Glu/Mal, A and B) or pyruvate/malate (Pyr/Mal, C and D). The large dip upon the addition of both Zaprinast (200 μm) and DMSO is an artifact caused by differences in O2 solubility between water and DMSO. The substrate was added at the time point indicated by arrows. B and D are increments (ΔOCR) over the previous rate by subtracting OCR initiated by the substrate added before. OCR, oxygen consumption rate (n = 3). E and F, MPC inhibitor UK5099 inhibits pyruvate-driven oxygen consumption. Like Zaprinast, UK5099 at 10 nm inhibits oxygen consumption fueled by pyruvate but not glutamate and succinate. E is a representative trace from F, the normalized data (n = 3). * indicates p < 0.05 versus DMSO treated. Drug indicates the adding of DMSO, Zaprinast, or UK5099. * indicates p < 0.05 versus DMSO treated (n = 3).
Zaprinast Inhibits MPC but Not PDH
To confirm that Zaprinast targets the MPC, we measured MPC activity directly using isolated mouse liver mitochondria. The MPC kinetics we measured are similar to previous reports (2, 4). We measured flux of [2-14C]pyruvate transport into isolated liver mitochondria. We confirmed the specificity of the assay by showing that 10 mm unlabeled pyruvate inhibits uptake of the labeled pyruvate (supplemental Fig. 8, A and B). Zaprinast inhibits pyruvate uptake (Fig. 6A) at a dose (Fig. 6B) that is similar to the dose required for its effect on glutamate and aspartate in the retina (Fig. 2A).
FIGURE 6.

Zaprinast inhibits transport of pyruvate into mitochondria but does not inhibit PDH. A and B, Zaprinast (Zap) inhibits pyruvate influx into liver mitochondria. A, isolated liver mitochondria was preincubated with DMSO or Zaprinast at 100 μm for 5 min before adding 15 μm [2-14C]pyruvate for 1 min. As a positive control, 10 mm unlabeled pyruvate (Cold Pyr) was added into mitochondria instantly after [2-14C]pyruvate addition (n = 5). B, Zaprinast dose-dependently inhibits mitochondrial pyruvate transport (n = 5). C, Zaprinast does not affect PDH activity. PDH protein was incubated with either Zaprinast at different concentrations or PDH inhibitor 3-fluropyruvate (3-FP) at 5 mm for 10 min. PDH activity was measured by cycling assay (n = 4). D–E, both Zaprinast and UK5099 inhibit citrate and glutamate synthesis from [13C3]pyruvate in brain mitochondria with intact membranes but not when mitochondrial membranes are disrupted. Membrane intact and disrupted mitochondria were incubated with 1 mm [13C3]pyruvate and Zaprinast (100 μm) or UK5099 (UK, 100 μm) for 10 min at 30 °C. The 13C citrate and glutamate were measured by GC-MS. Mem, membrane (n = 3). F–G, MPC inhibitors increase pyruvate and aspartate but decrease glutamate in the retina. Retinas were treated with α-cyano-4-hydroxycinnamic acid (4CIN, 100 μm), UK5099 (100 μm), and Zaprinast (100 μm) for 1 h. (n = 3). * indicates p < 0.05 versus DMSO treated.
To exclude the possibility that Zaprinast might inhibit PDH, we incubated pure PDH enzyme with Zaprinast or the known PDH inhibitor 3-fluropyruvate. Zaprinast did not affect the activity of PDH at any concentration tested. In contrast, 3-fluropyruvate effectively inhibited PDH activities (Fig. 6C).
To confirm that the metabolic effect of Zaprinast is caused by MPC inhibition and not by inhibition of PDH activity, we bypassed MPC by adding [13C3]pyruvate to mitochondria with disrupted membranes (see “Experimental Procedures”). In this experiment pyruvate is accessible directly to PDH. Both Zaprinast and UK5099 decrease the amount of labeled citrate produced by mitochondria with intact membranes, but they have no effect on citrate formation when mitochondrial membranes are disrupted (Fig. 6D). As expected, disruption of the mitochondrial membrane blocks the ability of either Zaprinast or UK5099 to stimulate depletion of glutamate (Fig. 6D).
Known Inhibitors of MPC Replicate the Effects of Zaprinast
We then asked whether other known MPC inhibitors cause metabolic effects similar to those of Zaprinast. Two known MPC inhibitors, α-cyano-4-hydroxycinnamic acid and UK5099, produce effects nearly identical to the effects of Zaprinast; they are increased aspartate and decreased glutamate, increased pyruvate, and suppressed pyruvate-driven O2 consumption (Fig. 6, F–G). Taken together, these results are consistent with our other data showing that Zaprinast blocks pyruvate transport into mitochondria.
Inactivation of Aspartate Glutamate Carrier (AGC1) Does Not Prevent the Decrease of Glutamate Caused by Zaprinast
Aspartate normally is transported out of mitochondria via AGC1 in exchange for glutamate from cytosol (Fig. 7A). AGC1 allows the carbons from aspartate to bypass the conventional “complete” version of the TCA cycle. In the absence of AGC1, the carbons that would normally be used to make aspartate are forced into the complete TCA cycle, thus decreasing the levels of the amino acid in AGC1−/− retina. In the absence of AGC1, the malate aspartate shuttle becomes impaired, and pyruvate is diverted to produce lactate and not acetyl-CoA. Consistently, the retina from AGC1−/− mice had lower levels of aspartate and pyruvate than WT mice (Fig. 7B) (35).
FIGURE 7.

Disruption of AGC1 activity does not prevent Zaprinast from stimulating oxidation of glutamate into aspartate. A, schematic of malate-aspartate shuttle. OAA, oxaloacetate; OGC, 2-oxoglutarate carrier. B, the retina from AGC1−/− mice has lower aspartate and pyruvate, but Zaprinast (Zap) induces similar metabolic changes. The retina was incubated with DMSO or Zaprinast for 1 h in the presence of glucose. C–F, pyruvate, citrate, glutamate, and aspartate labeled from [13C6]glucose. Retinas from WT, AGC1+/−, and AGC1−/− mice were incubated with 5 mm [13C6]glucose for 15 min. Data were expressed as -fold increase of 13C label over the DMSO control. * indicates p < 0.05 versus DMSO treated within groups, and # indicates versus DMSO or Zaprinast treated in the WT group. (n = 5).
We investigated whether AGC1 plays a role in the Zaprinast effect and found that it does not (Fig. 7B). When incubated with [13C6]glucose, AGC1+/− or AGC1−/− retinas have 13C enrichment similar to WT except that the overall levels of pyruvate and aspartate are lower than normal (supplemental Fig. 9). AGC1 deficiency impaired production of pyruvate, citrate, glutamate, and especially aspartate, consistent with a previous study of AGC1-deficient neurons (35). Nevertheless, Zaprinast still decreased labeling of glutamate from glucose and increased labeling of aspartate and pyruvate in both AGC1+/− and AGC1−/− retinas (Fig. 7, C–F).
Zaprinast Causes Accumulation of Aspartate in Mitochondria and Prevents Generation of Glutamate from Aspartate
To examine the distribution of accumulated aspartate in or outside of mitochondria, we incubated isolated intact brain mitochondria with EGTA, malate, glutamate, Ca2+, ADP, and [13C3]pyruvate for 10 min and then measured the metabolites from the mitochondria and from the incubation medium. Consistent with previous experiments, Zaprinast blocked incorporation of 13C from pyruvate into citrate both in and outside of mitochondria. Similarly, Zaprinast also inhibited incorporation of 13C into aspartate. At the same time, Zaprinast stimulated the accumulation of unlabeled aspartate in mitochondria (Fig. 8, A–C). Some glutamate may enter the mitochondria by a glutamate carrier (36), contributing to aspartate increase in mitochondria, but not the medium, within 10 min of incubation. Each of these findings is consistent with inhibition by Zaprinast of MPC-catalyzed uptake of pyruvate into mitochondria.
FIGURE 8.

Zaprinast causes accumulation of aspartate inside mitochondria and decreases aspartate utilization. A–C, most of the aspartate increase induced by Zaprinast (Zap) does not come from [13C3]pyruvate. Brain mitochondria were incubated with 1 mm [13C3]pyruvate and 100 μm Zaprinast for 10 min supplemented with EGTA 100 μm, 1 mm glutamate, 0.5 mm malate, 2.5 mm ADP, and 50 μm Ca2+. Metabolites from both mitochondria and medium were analyzed (n = 3). OAA, oxaloacetate. D, schematic of carbon labeling (black circles represent 13C) from [13C4]aspartate. E, [13C4]aspartate increased glutamate enrichment and TCA cycle intermediates. The retina was incubated with 250 μm [13C4]aspartate and unlabeled glucose for 1 h (n = 3). F–H, M4 aspartate increased, but M4 citrate and M3 glutamate decreased in response to Zaprinast (100 μm). I, Zaprinast decreased the lactate/pyruvate ratio (n = 3). J, Zaprinast decreased the level of NADH in the retina. Retinas were treated with Zaprinast for 1 h. * indicates p < 0.05 versus DMSO treated or without Zaprinast (n = 3).
Unlabeled aspartate accumulates in mitochondria during Zaprinast treatment (Fig. 8, A–C). To confirm that Zaprinast decreases utilization of aspartate, we incubated the retina with labeled aspartate in the presence of glucose. During 1 h without Zaprinast, aspartate contributed 13C to ∼30% of glutamate and other TCA cycle intermediates (Fig. 8, D–E). However, Zaprinast blocked the incorporation of 13C from aspartate into citrate and glutamate (Fig. 8, F–H). This reflects diminished acetyl-CoA synthesis in mitochondria, consistent with inhibition of pyruvate carrier activity by Zaprinast. In addition, the decreased lactate/pyruvate ratio (Fig. 8I) might lower cytosolic reducing power, which causes accumulation of aspartate by reversing the malate dehydrogenase reaction in the cytoplasm (Fig. 9). Consistent with this, we found that Zaprinast decreases NADH levels in the retina (Fig. 8J).
FIGURE 9.
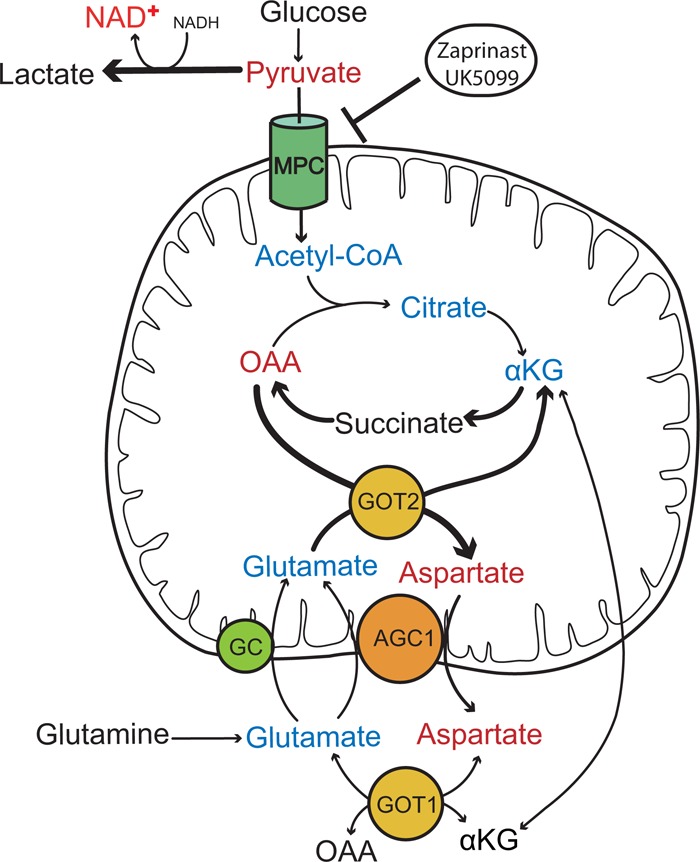
MPC inhibition causes accumulation of aspartate at the expense of glutamate. Inhibition of MPC by Zaprinast or UK5099 blocks entry of pyruvate into mitochondria, which decreases production of acetyl-CoA, citrate, αKG, glutamate, and NADH and increases oxaloacetate (OAA) and aspartate. Aspartate exits mitochondria in exchange for glutamate via AGC1 to oxidize more glutamate into aspartate. In the cytosol the glutamate may be transaminated into αKG, which exchanges with malate or enters the mitochondria through either AGC1 or glutamate carrier (GC). Metabolites with red represent increases, blue represents decrease, and black represents unchanged or untested. Thick lines represent more flux in that direction. GOT1, cytosolic glutamate oxoglutarate transaminase; GOT2, mitochondrial glutamate oxoglutarate transaminase.
DISCUSSION
We demonstrated that the well known PDE5/6 inhibitor Zaprinast blocks pyruvate transport into mitochondria by a mechanism that is independent of PDE inhibition. Inhibition of pyruvate transport either by Zaprinast or by known MPC inhibitors decreases de novo synthesis of glutamate from glucose and aspartate, and it increases the net oxidation of glutamate into aspartate. The result is severe depletion of glutamate and severalfold accumulation of aspartate. These findings are significant because glutamate is important as a fuel for energy production, as a neurotransmitter, and as a substrate for glutathione synthesis.
The Effect of Zaprinast on Glutamate and Aspartate Occurs Independently of Its Previously Known Effects on PDE
The ability of Zaprinast to inhibit PDE has been characterized (9, 10). Here we report four lines of evidence that Zaprinast also has an independent activity that causes severe depletion of glutamate and accumulation of aspartate. 1) The concentration at which Zaprinast decreases glutamate is about 20 times lower than that required to inhibit PDE. 2) Other PDE inhibitors, sildenafil or cell-permeable cGMP analogues, do not influence glutamate or aspartate. 3) Inactivation of cyclic nucleotide gated channels and inhibition of PKG or PKA do not rescue Zaprinast-induced glutamate depletion. 4) Zaprinast causes depletion of glutamate and accumulation of aspartate even in the retina, which has no PDE6.
Previous studies found that depletion of glutamate correlates with cell death in the retina (22, 28). Interestingly, in long term organotypic retinal explant cultures, at concentrations of up to 200 μm, Zaprinast induces selective photoreceptor death without affecting other retinal neurons (37). Why does Zaprinast not cause generalized cell death and instead appears to affect only PDE6 under culture conditions (37–40)? Tissue culture medium, such as R16 (41), used for organotypic retinal culture contains both fatty acids and glutamine. This provides two opportunities to bypass the consequences of MPC inhibition, either via direct acetyl-CoA uptake from fatty acid oxidation or via glutamine to glutamate conversion. Hence, the composition of the culture medium may help to discriminate between PDE6 and MPC effects of Zaprinast in cultured retina.
Zaprinast Inhibits Transport of Pyruvate into Mitochondria
MPC transports pyruvate into mitochondria. UK5099 or α-cyano-4-hydroxycinnamic acid are known inhibitors of MPC activity (1, 4). In this study we showed that MPC can also be inhibited by Zaprinast. Inhibition or knockdown of MPC prevents pyruvate-dependent acetyl-CoA formation and O2 consumption (5, 6, 42) and decreases the lactate/pyruvate ratio (43). Our study shows that Zaprinast and UK5099 not only block pyruvate-dependent mitochondrial O2 consumption and citrate synthesis in brain mitochondria but also cause increases in pyruvate and aspartate and decrease in glutamate in brain mitochondria and the retina.
We confirmed by direct measurements of pyruvate uptake that Zaprinast dose-dependently inhibits MPC activity in isolated mitochondria. MPC is an integral membrane protein in the mitochondrial inner membrane. To rule out any effect on PDH activity we measured the effect of Zaprinast on mitochondria whose membrane structure was disrupted by detergent. There was no effect of Zaprinast on these preparations. This shows that Zaprinast does not target PDH or citrate synthase. We also confirmed directly that Zaprinast does not affect the activity of purified PDH. In our MPC activity assay some labeled pyruvate may have been metabolized. We did not correct for this or for labeled pyruvate that may have been at the surface of the pellets. This may explain why the pyruvate uptake continued for more than 1 min and neither cold pyruvate nor Zaprinast completely eliminated the labeled pyruvate in mitochondrial pellet. Future studies should address the structural basis for Zaprinast inhibition of MPC and whether Zaprinast directly binds MPC.
Zaprinast Causes Oxidation of Glutamate and Accumulation of Aspartate
Hypoglycemia or inhibition of glycolysis leads to decreased glutamate and cellular accumulation or release of aspartate in brain (44–46), synaptosomes (47), and the retina (22, 28). During glucose deprivation, neurons turn to glutamine and glutamate for energy (48). Zaprinast does not affect glucose uptake or glycolysis (as assessed by lactate production), but its block of pyruvate transport mimics hypoglycemic conditions because less glucose-derived pyruvate is available to mitochondria. The lack of two carbons from pyruvate causes oxaloacetate to accumulate at the expense of glutamate. Some pyruvate may enter mitochondria before Zaprinast takes effect or there might be some residual MPC activity, so some citrate, αKG, glutamate, and succinate can be labeled by 13C glucose. However, once these labeled carbons are incorporated into oxaloacetate, there is so little acetyl-CoA that they cannot enter a second turn of TCA cycle. The result is similar to what happens during glucose deprivation. Glutamine starts to replenish the TCA cycle to rescue the mitochondrial energy crisis, but it can only lead to accumulation of more oxaloacetate. By Le Chatelier's principle, the increased oxaloacetate will shift aspartate transaminase toward producing more aspartate and more αKG from glutamate.
In the retina, >60% of glutamate de novo synthesis depends on transaminases such as alanine transaminase, aspartate transaminase, and branched amino acids transaminase (49). However, only aspartate and not alanine or branched amino acids increase with Zaprinast. Glutamate may also decarboxylate into GABA, but Zaprinast does not increase GABA content in the retina. Therefore, the amino group of aspartate most likely comes from glutamate transamination (49).
In conclusion, independently of its known effects on PDE, Zaprinast potently inhibits pyruvate transport into mitochondria, impairing glucose oxidation and mitochondrial function. Inhibition of pyruvate transport mimics the effect of hypoglycemia: accumulation of aspartate, depletion of glutamate and glutamine, and mitochondrial dysfunction. Our findings suggest a new route for pharmacological application and development of Zaprinast, they highlight the physiological influence of MPC on metabolism, and they contribute to understanding of the basic metabolic reaction to hypoglycemia.
Acknowledgments
The Cngb1−/− mice were kindly provided by Stylianos Michalakis and Martin Biel, Center for Integrated Protein Science CIPS-M and Department of Pharmacy, Ludwig-Maximilians Universität München. Oxygen consumption and pyruvate uptake experiments were carried out by Diabetes Research Center Cell Function Analysis Core (National Institutes of Health Grant P30DK017047).
This work was supported, in whole or in part, by National Institutes of Health Grants EY06641 and EY017863 (to J. B. H.). This work was also supported by grants from the Deutsche Forschungsgemeinschaft (Pa1751/4-1), EU (DRUGSFORD: HEALTH-F2-2012-304963), and the Kerstan Foundation (to F. P.-D.) and the Ministerio de Economíay Competitividad (BFU2011-30456-C02-01/BMC) and Coumunidad Autónoma de Madrid (S2010/BMD-2402) (to J. S.). This work was also funded by the CIBERER, an initiative from the Instituto de Salud Carlos III, and an institutional grant from the Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa.

This article contains supplemental Methods and Figs. 1–9.
- MPC
- mitochondrial pyruvate carrier
- PDE
- phosphodiesterase
- PDH
- pyruvate dehydrogenase activity
- αKG
- α-ketoglutarate
- GPR35
- G protein-coupled receptor 35
- AGC1
- aspartate glutamate carrier.
REFERENCES
- 1. Bricker D. K., Taylor E. B., Schell J. C., Orsak T., Boutron A., Chen Y. C., Cox J. E., Cardon C. M., Van Vranken J. G., Dephoure N., Redin C., Boudina S., Gygi S. P., Brivet M., Thummel C. S., Rutter J. (2012) A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Herzig S., Raemy E., Montessuit S., Veuthey J. L., Zamboni N., Westermann B., Kunji E. R., Martinou J. C. (2012) Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96 [DOI] [PubMed] [Google Scholar]
- 3. Brivet M., Garcia-Cazorla A., Lyonnet S., Dumez Y., Nassogne M. C., Slama A., Boutron A., Touati G., Legrand A., Saudubray J. M. (2003) Impaired mitochondrial pyruvate importation in a patient and a fetus at risk. Mol. Genet. Metab. 78, 186–192 [DOI] [PubMed] [Google Scholar]
- 4. Halestrap A. P. (1975) The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem. J. 148, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colca J. R., McDonald W. G., Cavey G. S., Cole S. L., Holewa D. D., Brightwell-Conrad A. S., Wolfe C. L., Wheeler J. S., Coulter K. R., Kilkuskie P. M., Gracheva E., Korshunova Y., Trusgnich M., Karr R., Wiley S. E., Divakaruni A. S., Murphy A. N., Vigueira P. A., Finck B. N., Kletzien R. F. (2013) Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT). Relationship to newly identified mitochondrial pyruvate carrier proteins. PloS ONE 8, e61551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Divakaruni A. S., Wiley S. E., Rogers G. W., Andreyev A. Y., Petrosyan S., Loviscach M., Wall E. A., Yadava N., Heuck A. P., Ferrick D. A., Henry R. R., McDonald W. G., Colca J. R., Simon M. I., Ciaraldi T. P., Murphy A. N. (2013) Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. U.S.A. 110, 5422–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hildyard J. C., Ammälä C., Dukes I. D., Thomson S. A., Halestrap A. P. (2005) Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim. Biophys. Acta 1707, 221–230 [DOI] [PubMed] [Google Scholar]
- 8. Gillespie P. G., Beavo J. A. (1989) Inhibition and stimulation of photoreceptor phosphodiesterases by dipyridamole and M&B 22,948. Mol. Pharmacol. 36, 773–781 [PubMed] [Google Scholar]
- 9. Thompson W. J. (1991) Cyclic nucleotide phosphodiesterases. Pharmacology, biochemistry, and function. Pharmacol. Ther. 51, 13–33 [DOI] [PubMed] [Google Scholar]
- 10. Gibson A. (2001) Phosphodiesterase 5 inhibitors and nitrergic transmission-from zaprinast to sildenafil. Eur. J. Pharmacol. 411, 1–10 [DOI] [PubMed] [Google Scholar]
- 11. Turko I. V., Francis S. H., Corbin J. D. (1998) Potential roles of conserved amino acids in the catalytic domain of the cGMP-binding cGMP-specific phosphodiesterase. J. Biol. Chem. 273, 6460–6466 [DOI] [PubMed] [Google Scholar]
- 12. Francis S. H., Blount M. A., Corbin J. D. (2011) Mammalian cyclic nucleotide phosphodiesterases. Molecular mechanisms and physiological functions. Physiol. Rev. 91, 651–690 [DOI] [PubMed] [Google Scholar]
- 13. Zhang X., Feng Q., Cote R. H. (2005) Efficacy and selectivity of phosphodiesterase-targeted drugs in inhibiting photoreceptor phosphodiesterase (PDE6) in retinal photoreceptors. Invest. Ophthalmol. Vis. Sci. 46, 3060–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McLaughlin M. E., Ehrhart T. L., Berson E. L., Dryja T. P. (1995) Mutation spectrum of the gene encoding the β subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 92, 3249–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang B., Grau T., Dangel S., Hurd R., Jurklies B., Sener E. C., Andreasson S., Dollfus H., Baumann B., Bolz S., Artemyev N., Kohl S., Heckenlively J., Wissinger B. (2009) A homologous genetic basis of the murine cpfl1 mutant and human achromatopsia linked to mutations in the PDE6C gene. Proc. Natl. Acad. Sci. U.S.A. 106, 19581–19586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang S. H., Pittler S. J., Huang X., Oliveira L., Berson E. L., Dryja T. P. (1995) Autosomal recessive retinitis pigmentosa caused by mutations in the α subunit of rod cGMP phosphodiesterase. Nat. Genet. 11, 468–471 [DOI] [PubMed] [Google Scholar]
- 17. Bowes C., Li T., Danciger M., Baxter L. C., Applebury M. L., Farber D. B. (1990) Retinal degeneration in the rd mouse is caused by a defect in the β subunit of rod cGMP-phosphodiesterase. Nature 347, 677–680 [DOI] [PubMed] [Google Scholar]
- 18. Chang B., Hawes N. L., Hurd R. E., Davisson M. T., Nusinowitz S., Heckenlively J. R. (2002) Retinal degeneration mutants in the mouse. Vis. Res. 42, 517–525 [DOI] [PubMed] [Google Scholar]
- 19. Paquet-Durand F., Beck S., Michalakis S., Goldmann T., Huber G., Mühlfriedel R., Trifunovi D., Fischer M. D., Fahl E., Duetsch G., Becirovic E., Wolfrum U., van Veen T., Biel M., Tanimoto N., Seeliger M. W. (2011) A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet. 20, 941–947 [DOI] [PubMed] [Google Scholar]
- 20. Hüttl S., Michalakis S., Seeliger M., Luo D. G., Acar N., Geiger H., Hudl K., Mader R., Haverkamp S., Moser M., Pfeifer A., Gerstner A., Yau K. W., Biel M. (2005) Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci. 25, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jalil M. A., Begum L., Contreras L., Pardo B., Iijima M., Li M. X., Ramos M., Marmol P., Horiuchi M., Shimotsu K., Nakagawa S., Okubo A., Sameshima M., Isashiki Y., Del Arco A., Kobayashi K., Satrústegui J., Saheki T. (2005) Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J. Biol. Chem. 280, 31333–31339 [DOI] [PubMed] [Google Scholar]
- 22. Chertov A. O., Holzhausen L., Kuok I. T., Couron D., Parker E., Linton J. D., Sadilek M., Sweet I. R., Hurley J. B. (2011) Roles of glucose in photoreceptor survival. J. Biol. Chem. 286, 34700–34711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Millard P., Letisse F., Sokol S., Portais J. C. (2012) IsoCor. Correcting MS data in isotope labeling experiments. Bioinformatics 28, 1294–1296 [DOI] [PubMed] [Google Scholar]
- 24. van Winden W. A., Wittmann C., Heinzle E., Heijnen J. J. (2002) Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 80, 477–479 [DOI] [PubMed] [Google Scholar]
- 25. Frezza C., Cipolat S., Scorrano L. (2007) Organelle isolation. Functional mitochondria from mouse liver, muscle, and cultured fibroblasts. Nat. Protoc. 2, 287–295 [DOI] [PubMed] [Google Scholar]
- 26. Sweet I. R., Cook D. L., Lernmark A., Greenbaum C. J., Wallen A. R., Marcum E. S., Stekhova S. A., Krohn K. A. (2004) Systematic screening of potential β-cell imaging agents. Biochem. Biophys. Res. Commun. 314, 976–983 [DOI] [PubMed] [Google Scholar]
- 27. Janke R., Genzel Y., Wahl A., Reichl U. (2010) Measurement of key metabolic enzyme activities in mammalian cells using rapid and sensitive microplate-based assays. Biotechnol. Bioeng. 107, 566–581 [DOI] [PubMed] [Google Scholar]
- 28. Zeevalk G. D., Nicklas W. J. (2000) Lactate prevents the alterations in tissue amino acids, decline in ATP, and cell damage due to aglycemia in retina. J. Neurochem. 75, 1027–1034 [DOI] [PubMed] [Google Scholar]
- 29. Biel M., Michalakis S. (2009) Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 191, 111–136 [DOI] [PubMed] [Google Scholar]
- 30. Taniguchi Y., Tonai-Kachi H., Shinjo K. (2006) Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 580, 5003–5008 [DOI] [PubMed] [Google Scholar]
- 31. Guo J., Williams D. J., Puhl H. L., 3rd, Ikeda S. R. (2008) Inhibition of N-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J. Pharmacol. Exp. Ther. 324, 342–351 [DOI] [PubMed] [Google Scholar]
- 32. Wang J., Simonavicius N., Wu X., Swaminath G., Reagan J., Tian H., Ling L. (2006) Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 281, 22021–22028 [DOI] [PubMed] [Google Scholar]
- 33. Mackenzie A. E., Lappin J. E., Taylor D. L., Nicklin S. A., Milligan G. (2011) GPR35 as a novel therapeutic target. Front. Endocrinol. 2, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., Jewell C. M., Johnson Z. R., Irvine D. J., Guarente L., Kelleher J. K., Vander Heiden M. G., Iliopoulos O., Stephanopoulos G. (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pardo B., Rodrigues T. B., Contreras L., Garzón M., Llorente-Folch I., Kobayashi K., Saheki T., Cerdan S., Satrústegui J. (2011) Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J. Cereb. Blood Flow Metab. 31, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fiermonte G., Palmieri L., Todisco S., Agrimi G., Palmieri F., Walker J. E. (2002) Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 277, 19289–19294 [DOI] [PubMed] [Google Scholar]
- 37. Sahaboglu A., Tanimoto N., Kaur J., Sancho-Pelluz J., Huber G., Fahl E., Arango-Gonzalez B., Zrenner E., Ekström P., Löwenheim H., Seeliger M., Paquet-Durand F. (2010) PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PloS ONE 5, e15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vallazza-Deschamps G., Cia D., Gong J., Jellali A., Duboc A., Forster V., Sahel J. A., Tessier L. H., Picaud S. (2005) Excessive activation of cyclic nucleotide-gated channels contributes to neuronal degeneration of photoreceptors. Eur. J. Neurosci. 22, 1013–1022 [DOI] [PubMed] [Google Scholar]
- 39. Sahaboglu A., Paquet-Durand O., Dietter J., Dengler K., Bernhard-Kurz S., Ekström P. A., Hitzmann B., Ueffing M., Paquet-Durand F. (2013) Retinitis pigmentosa. Rapid neurodegeneration is governed by slow cell death mechanisms. Cell Death Dis. 4, e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martínez-Fernández de la Cámara C., Sequedo M. D., Gómez-Pinedo U., Jaijo T., Aller E., García-Tárraga P., García-Verdugo J. M., Millán J. M., Rodrigo R. (2013) Phosphodiesterase inhibition induces retinal degeneration, oxidative stress, and inflammation in cone-enriched cultures of porcine retina. Exp. Eye Res. 111, 122–133 [DOI] [PubMed] [Google Scholar]
- 41. Romijn H. J. (1988) Development and advantages of serum-free, chemically defined nutrient media for culturing of nerve tissue. Biol. Cell 63, 263–268 [DOI] [PubMed] [Google Scholar]
- 42. Proudlove M. O., Beechey R. B., Moore A. L. (1987) Pyruvate transport by thermogenic-tissue mitochondria. Biochem. J. 247, 441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Halestrap A. P., Denton R. M. (1975) The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by α-cyano-4-hydroxycinnamate and related compounds. Biochem. J. 148, 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Butterworth R. F., Merkel A. D., Landreville F. (1982) Regional amino acid distribution in relation to function in insulin hypoglycaemia. J. Neurochem. 38, 1483–1489 [DOI] [PubMed] [Google Scholar]
- 45. Sandberg M., Nyström B., Hamberger A. (1985) Metabolically derived aspartate. Elevated extracellular levels in vivo in iodoacetate poisoning. J. Neurosci. Res. 13, 489–495 [DOI] [PubMed] [Google Scholar]
- 46. Szerb J. C., O'Regan P. A. (1987) Reversible shifts in the Ca2+-dependent release of aspartate and glutamate from hippocampal slices with changing glucose concentrations. Synapse 1, 265–272 [DOI] [PubMed] [Google Scholar]
- 47. Yudkoff M., Nelson D., Daikhin Y., Erecińska M. (1994) Tricarboxylic acid cycle in rat brain synaptosomes. Fluxes and interactions with aspartate aminotransferase and malate/aspartate shuttle. J. Biol. Chem. 269, 27414–27420 [PubMed] [Google Scholar]
- 48. Peng L., Gu L., Zhang H., Huang X., Hertz E., Hertz L. (2007) Glutamine as an energy substrate in cultured neurons during glucose deprivation. J. Neurosci. Res. 85, 3480–3486 [DOI] [PubMed] [Google Scholar]
- 49. LaNoue K. F., Berkich D. A., Conway M., Barber A. J., Hu L. Y., Taylor C., Hutson S. (2001) Role of specific aminotransferases in de novo glutamate synthesis and redox shuttling in the retina. J. Neurosci. Res. 66, 914–922 [DOI] [PubMed] [Google Scholar]