

Malina et al. readapted the CRISPR/Cas9 genome-editing system for targeted gene disruption positive selection assays. They generated “all-in-one” lentiviral and retroviral delivery vectors heterologously expressing both a codon-optimized Cas9 and its synthetic guide RNA (sgRNA) and linked Cas9 expression to GFP fluorescence for in vivo applications. These results establish Cas9 genome editing as a powerful and practical approach for positive in situ genetic screens.
Keywords: Cas9, CRISPR, genome editing, functional screening, p53
Abstract
RNAi combined with next-generation sequencing has proven to be a powerful and cost-effective genetic screening platform in mammalian cells. Still, this technology has its limitations and is incompatible with in situ mutagenesis screens on a genome-wide scale. Using p53 as a proof-of-principle target, we readapted the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR associated 9) genome-editing system to demonstrate the feasibility of this methodology for targeted gene disruption positive selection assays. By using novel “all-in-one” lentiviral and retroviral delivery vectors heterologously expressing both a codon-optimized Cas9 and its synthetic guide RNA (sgRNA), we show robust selection for the CRISPR-modified Trp53 locus following drug treatment. Furthermore, by linking Cas9 expression to GFP fluorescence, we use an “all-in-one” system to track disrupted Trp53 in chemoresistant lymphomas in the Eμ-myc mouse model. Deep sequencing analysis of the tumor-derived endogenous Cas9-modified Trp53 locus revealed a wide spectrum of mutants that were enriched with seemingly limited off-target effects. Taken together, these results establish Cas9 genome editing as a powerful and practical approach for positive in situ genetic screens.
Modern high-throughput DNA sequencing technologies and their corresponding genome-wide analysis tools have led to an unprecedented amount of publicly available genomic information. However, technologies that are able to capture and translate such a wealth of genetic knowledge and gene regulation into predictive biological models and systems have been lagging (Dow and Lowe 2012). The discovery and exploitation of RNAi has helped close this gap (Chang et al. 2006) and has proven to be a powerful mammalian genetic testing and screening tool but is limited to targeting the exome, and often such mRNA-specific knockdown is only partial or incomplete. Moreover, understanding the biological relevance of gene-specific gain-of-function mutations in cell and animal models is currently incompatible with features of RNAi. The development of modern genome engineering technologies based on modular DNA-binding proteins coupled to nucleases, such as zinc finger nucleases (ZFNs) or transcription activator-like effector nucleases (TALENs), has shown tremendous promise for achieving these goals (Gaj et al. 2013). However, in their present technological states, both are cumbersomely large to use and labor-intensive to produce and rely on prior extensive empirical testing to identify appropriate gene targeting pairs, making either technology impractical and not cost-effective for scaling up in genome-wide studies.
There has been much enthusiasm of late in the development of components of CRISPR (clustered regularly interspaced short palindromic repeats) as an alternative genome-editing system. The CRISPR locus constitutes an ancient and pervasive bacterial and archeal acquired immune system, with its defining characteristic being the use of short complementary CRISPR RNA sequences (crRNAs) that bind to and direct cleavage of foreign invading DNA (and sometimes RNA) molecules in a sequence-specific manner (Sorek et al. 2013). These short crRNA strands originate from a processed longer pre-crRNA transcript that consists of tandem units of palindromic repeat sequences separating short distinct protospacer fragments of foreign DNA sequence that have been integrated into the locus from prior infection events. Several different classes of CRISPR operons have been characterized to date, and while all share these basic hallmarks and steps (foreign DNA integration, crRNA maturation, and crRNA-directed cleavage), they distinguish themselves through sequence divergence among their repeat stretches and also by the set of Cas (CRISPR associated)-encoded proteins that perform all of the system's basic functions. Of these, the type II CRISPR system is perhaps one of the simplest, requiring only three components to execute its DNA interference: one Cas protein (Cas9) that acts as both endonuclease and RNA chaperone and two auxiliary RNAs, the requisite processed crRNA molecule and a Cas9-bridging trans-activating crRNA molecule (tracrRNA) to which the crRNA hybridizes (Chylinski et al. 2013). All three components (specifically from the bacteria Streptococcus pyogenes) were shown to be sufficient to direct DNA cleavage when reconstituted in in vitro reactions, with even further simplification achieved through the fusion of the crRNA to the tracrRNA using a simple hairpin linker region (termed a synthetic guide RNA [sgRNA]) (Jinek et al. 2012). Most notably, Cas9-directed site-specific cleavage can be manipulated by engineering a complementary sequence that is 20 nucleotides (nt) in length (referred to here as the “trigger”) into the sgRNA, as long as an 5′NGG3′ PAM (protospacer-adjacent motif) is present 3′ to the genomically encoded target complement sequence. The latter serves to distinguish self from non-self DNA sequences in the CRISPR system.
Building on this system, several groups have shown that Cas9-mediated genome editing can be achieved in a wide variety of organisms: bacteria (Jiang et al. 2013a), yeast (DiCarlo et al. 2013), Caenorhabditis elegans (Dickinson et al. 2013; Friedland et al. 2013; Katic and Grosshans 2013), Drosophila (Bassett et al. 2013; Gratz et al. 2013), zebrafish (Chang et al. 2013; Hwang et al. 2013a,b; Jao et al. 2013), plants (Feng et al. 2013; Jiang et al. 2013b; Li et al. 2013a; Miao et al. 2013; Nekrasov et al. 2013; Shan et al. 2013), and rats (Li et al. 2013b) and mouse and human cells (Cho et al. 2013; Cong et al. 2013; Ding et al. 2013; Jinek et al. 2013; Mali et al. 2013; Shen et al. 2013; Yang et al. 2013). In addition, sgRNAs can be multiplexed to simultaneously achieve genome-editing outcomes at as many as five independent loci in a single cell (Wang et al. 2013). Here, we sought to further extend the utility of the Cas9 genome-editing platform by developing a framework for its use in positive selection screening approaches both in vitro and in vivo using p53 as a model genetic target.
Results
In vitro validation of a single-vector lentiviral-derived Cas9 genome-editing construct
Based on the work by Jinek et al. (2012) and in the same vein as other groups (Cong et al. 2013; Gilbert et al. 2013; Hsu et al. 2013; Mali et al. 2013; Qi et al. 2013), we chose to adapt S. pyogenes Cas9 for genome editing in mammalian cells. It is worth noting those features of our Cas9/sgRNA-based construct that distinguish it and are shared with those that are currently publicly available (Figs. 1A,B; Supplemental Fig. 1): (1) The coding sequence of Cas9 from S. pyogenes was extensively altered for optimal mammalian codon usage bias, with several restriction sites eliminated (Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013). (2) We fused a 3xFlag epitope to the N terminus as an affinity tag flanked by two NLS (nuclear localization signal) tags (one derived from SV40 large T antigen, and the other derived from nucleoplasmin) for efficient nuclear import. (3) We based our hybrid crRNA:tracrRNA sgRNA on what had been described previously (Jiang et al. 2013a) but instead used a longer, more complete tracrRNA sequence that has been subsequently shown to promote more efficient cleavage on a greater number of target sites by Cas9 (Cong et al. 2013; Hsu et al. 2013; Jinek et al. 2013; Mali et al. 2013; Wang et al. 2013). Most importantly, we chose to express both the sgRNA and Cas9 gene in a single vector format embedded within a second-generation self-inactivating (SIN) lentiviral or retroviral (see below) backbone. This design not only allows for flexibility in cell type delivery but is also amenable to the generation of stable single-vector integrants (Cong et al. 2013; Gilbert et al. 2013), a requirement for pooled multiplex screening approaches.
Figure 1.
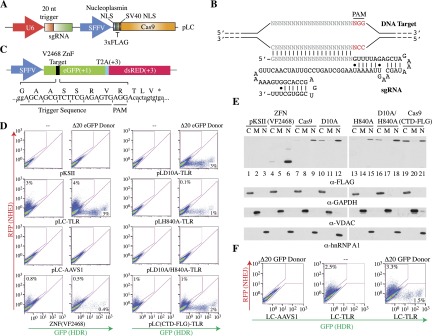
Genome editing of a TLR locus in 293T cells using an engineered all-in-one type II CRISPR system. (A) Schematic diagram of LeGO-based lentivirus (pLC) constructs driving expression of Cas9 and sgRNAs. (B) Predicted secondary structure (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) of sgRNA showing alignment of trigger sequence with target and PAM. The first nucleotide of the trigger sequence is forcibly a G, since the sgRNA is expressed from the murine U6 promoter. (C) Schematic of TLR with the position and nucleotide sequence of the TLR trigger, PAM, and stop codon shown. (D) A genomically integrated TLR is efficiently targeted by pLC-TLR. Quantitation of 293T TLR cells transfected with the indicated Cas9/sgRNA expression constructs and, where indicated, in combination with Δ20 eGFP. (E) Immunoblot showing expression and subcellular localization of Cas9 from the experiment presented in D. (C) Cytoplasmic fraction; (M) membrane fraction; (N) nuclear fraction. Blots were probed with the antibodies indicated below each panel. (F) Lentiviral-mediated NHEJ and HDR in 293T TLR cells. Cells were infected with lentivirus expressing Cas9 and the corresponding sgRNA and analyzed by flow cytometry 6 d later. The Δ20 eGFP donor plasmid was introduced by transfection 1 d prior to transduction with the Cas9/sgRNA lentiviral construct.
To ensure the compatibility of our design with its intended function as a broad-based genome-editing platform, we used the previously described “traffic light reporter” (TLR) assay, which measures simultaneously the frequency of both nonhomologous end-joining (NHEJ) repair and homology-directed repair (HDR) (Fig. 1C; see the Materials and Methods; Certo et al. 2011). We measured the genome-editing efficiencies of several different Cas9 constructs (with or without donor eGFP repair plasmid Δ20 donor) following transient transfection in a 293T cell line stably expressing a single-copy TLR and scoring for both green and red fluorescent cells by flow cytometry (Fig. 1D). Fairly consistent transfection efficiencies across all samples (between ∼40% and 60% of all cells) were achieved, as measured by BFP fluorescence that is coexpressed from the Δ20 eGFP donor repair plasmid (data not shown). Transfection of an empty control vector (pKSII) with or without donor eGFP plasmid yielded virtually no background fluorescent protein expression (Fig. 1D, top two left panels). Transfection of Cas9-TLR (pLC-TLR) harboring an sgRNA targeting the TLR eGFP region promoted 3%–4% of cells to be repaired by NHEJ and, in the presence of the Δ20 eGFP donor plasmid, 4% of cells to be repaired by HDR. We note that our estimates of NHEJ are underreported, since RFP fluorescence scores for only one-third of all editing events. Importantly, this effect was specific for the TLR sgRNA sequence, as no fluorescence events were measured when the sgRNA corresponded to one that targets the AAVS1 genomic region.
Strikingly, when comparing the pLC-TLR construct with that of a well-characterized ZFN pair (VF2468) (Maeder et al. 2008) that targets a region proximal to the sgRNA trigger site, the VF2468 ZFN produced a repair efficiency that was approximately eightfold less despite showing higher expression levels (Fig. 1D,E). We also compared the cleavage efficiencies of two mutant Cas9 constructs (D10A and H840A), which eliminate strand-specific endonuclease activity and behave as nickases (Supplemental Fig. 1A; Jinek et al. 2012; Cong et al. 2013; Mali et al. 2013). As expected, both the D10A mutant and (to a lesser extent) the H840A mutant produced comparable amounts of HDR in the presence of donor plasmid with almost no detectable mutagenic NHEJ, while the Cas9 double mutant D10A/H840A had no detectable double-stranded cutting ability (Fig. 1D). Furthermore, we tested a C-terminal NLS fusion of Cas9 using this system (Supplemental Fig. 2), since initial reports (Cong et al. 2013) indicated reduced activity from N-terminal fusions. We found this not to be the case when engineered into the lentiviral plasmid pLeGO (lentiviral “gene ontology”) (Weber et al. 2008), as the C-terminal domain (CTD)-Flag construct consistently produced lower levels of both NHEJ and HDR (Fig. 1D). All N-terminal domain (NTD)-tagged Cas9 constructs as well as the VF2468 ZNF pair showed preferential nuclear localization (Fig. 1E, lanes 4–18), whereas CTD-tagged Cas9 showed a similar distribution between the cytoplasm and the nucleus (Fig. 1E, lanes 19–21), perhaps explaining the differences seen in repair efficiencies. Finally, our Cas9 lentiviral constructs produced comparable levels of NHEJ and HDR following transduction of virus into the TLR 293T cell line, enabling one to perform genome editing in a broader range of cell types (Fig. 1F). In sum, our combined Cas9–sgRNA-expressing single-vector lentiviral construct offers a convenient and adaptable template for in situ genome modification in cells.
Endogenous gene targeting
Having functionally validated our Cas9 system using an exogenously synthetic reporter locus, we next sought to target an endogenous gene. We chose to target exon 7 of Trp53 as a suitable candidate for mutagenesis in a positive selection assay, as this region is frequently mutated in a wide range of tumor types (commonly referred to as a p53 “hot spot” region) and can confer resistance to DNA damage-inducing chemotherapy (Supplemental Fig. 3; Oren and Rotter 2010). Because we ultimately wanted to track Cas9-mediated mutagenesis in a live mouse model, we linked Cas9 to eGFP as a facile means to monitor its expression and as an indirect indicator for potential genome-editing events (Fig. 2A). We also transferred our expression cassette into a SIN-based MMLV-based vector backbone, which, in our hands, has proven to be a more reliable means of transducing lymphocytic mouse cells (data not shown). We chose to test for Cas9-driven targeted mutagenesis on the endogenous Trp53 gene in mouse embryonic fibroblasts (MEFs), a cell line in which the biology of p53 has been thoroughly characterized and where the gene is structurally intact. Both Arf−/− MEFs (p53-proficient) and p53−/− MEFs were transduced with retroviruses expressing an sgRNA/Cas9 construct targeting either Rosa26 (which served as a neutral endogenous target) or Trp53, which we termed pQCiG-Rosa and pQCiG-p53, respectively. A previously validated short hairpin construct against p53 (p53.1224) that also coexpresses GFP served as a positive control (Dickins et al. 2005). Seventy-two hours post-transduction, cells were cultured in the presence or absence of Nutlin-3a, a specific inhibitor of the MDM2–p53 interaction (Tovar et al. 2006). Cells were scored for the fraction of GFP+ cells after 4 d of Nutlin-3a exposure using flow cytometry (Fig. 2B). Nutlin-3a potently activates a p53-dependent anti-proliferative cellular response, which strongly and specifically selects for any cells with disrupted Trp53 function (Efeyan et al. 2007). Much like MLP-p53.1224 infected cells, the pQCiG-p53 GFP+ cells were rapidly enriched for in the presence of Nutlin-3a, suggesting that CRISPR-mediated gene disruption had initially occurred in at least a proportion of the initial Cas9-expressing cells. This is in contrast to pQCiG-Rosa-infected cells whose GFP+ proportion remained constant throughout the assay. Crucially, all of the effects from exposure to Nutlin-3a were completely eliminated when performed in p53−/− MEFs, suggesting that the selected GFP+ cell population originated from a pool of cells with a Cas9-induced p53 gene disruption and not due to any off-target effects (Fig. 2B). Further reinforcing this notion, alternate trigger sequences targeting exons 5, 6, and 8 of Trp53 (Supplemental Fig. 3B) also promoted the strong selection for GFP+ cells in the presence of Nutlin-3a (Supplemental Fig. 4A), making it unlikely that separate altered genomic regions contributed to this phenotype. Moreover, it is worth pointing out that in all of these experiments, Cas9-linked GFP expression was stable over time, indicating that continuous Cas9 expression is not toxic to cells (Fig. 2B; see below).
Figure 2.

Cas9-mediated editing of Trp53 in Arf−/− MEFs leads to Nutlin-3a resistance. (A) Schematic diagram of the pQ-based retroviral constructs driving expression of Cas9, GFP, and sgRNAs (pQCiG). (B) Flow cytometric analysis of Arf−/− and p53−/− MEFs transduced with QCiG-Rosa, QCiG-p53, or MLP-p53.1224 retroviruses, cultured 3 d later in the presence of vehicle or 10 μM Nutlin-3a for 24 h, and then allowed to recover for 4 d. (C) Colony formation assay of infected Arf−/− and p53−/− MEFs with QCiG-Rosa, QCiG-p53, or MLP-p53.1224. Five-thousand cells were seeded, exposed to 10 μM Nutlin-3a for 24 h, and allowed to recover for 12 d in the absence of drug, at which point they were stained with crystal violet. (D) SURVEYOR assay of DNA isolated from QCiG-p53- and QCiG-Rosa-infected Arf−/− MEFs exposed to 10 μM Nutlin-3a for 24 h and allowed to recover for 4 d. The arrowhead denotes the expected SURVEYOR cleavage products. (E) Immunoblot documenting Cas9 and p53 expression in QCiG- and MLP-infected MEFs. The asterisk denotes the position of a prominent p53 truncated product.
Similar results were obtained in a separate colony formation assay, where low numbers of transduced cells were allowed to proliferate over the course of 10 d in the presence or absence of Nutlin-3a (Fig. 2C; Supplemental Fig. 4B). Here, in the presence of Nutlin-3a, the pQCiG-p53-infected population formed ample size colonies, much like their MLP-p53.1224 counterparts, while the proliferative defects of pQCiG-Rosa-infected cells became quite apparent (Fig. 2C). At the molecular level, we detected an insertion/deletion (indel) frequency that ranged from 4% to 36% at the Trp53 locus in the pQCiG-p53-infected cells (Fig. 2D; Supplemental Fig. 4C), while no mutated Trp53 alleles could be distinguished among pQCiG-Rosa-infected cells (Fig. 2D). By immunoblotting, we could also detect the presence of truncated p53 protein products from extracts of Arf−/−-infected pQCiG-p53 samples, consistent with a Trp53 reading frameshift following NHEJ repair (Fig. 2E, lane 4, denoted by the asterisk). Moreover, our ability to specifically disrupt an endogenous Trp53 locus was not simply limited to mouse primary cells—we were also able to successfully mutagenize p53 in U2OS, an osteosarcoma cell line with an intact TP53 locus (Supplemental Fig. 5). Here, too, we could readily select for cells with a modified TP53 locus in the presence of Nutlin-3a, as shown for cells transduced with LC-hp53 in a similarly designed colony formation assay (Supplemental Fig. 5A). Multiple truncated p53 protein products were detected in extracts from pLC-hp53-infected U2OS cells and, in this case, an overall reduction in total p53 levels (Supplemental Fig. 5B, cf. lanes 3 and 2). Thus, we can stably and easily transduce both mouse and human cell lines in culture with our Cas9 targeting vector as a means to alter various regions of an endogenous genetic locus and select for that specific genome-editing event with no apparent long-term toxic effects.
Positive in vivo selection of targeted Cas9-induced p53 gene disruption
Having established the simplicity of our Cas9 setup to target and select for an altered Trp53 locus in cell culture, we next wanted to know whether we could adapt this for use in a mouse model of cancer. We chose to use the Eμ-myc mouse as a candidate model, in part due to the ease with which ex vivo retroviral transduction of B-cell lymphomas can be accomplished, the fact that multiple cohorts of syngeneic mice can be rapidly generated, and the reliable and predictable p53 tumor response that can be elicited from a clinically relevant therapeutic intervention. Moreover, very low numbers of injectable cells are required for propagating Eμ-myc-driven lymphomagenesis in mice, and thus almost every cell can individually contribute to tumor formation following transplantation (Kelly et al. 2007). This mouse model is therefore uniquely suitable for performing in vivo screens. As before, we first tested the ability to select for Cas9-modified Trp53 in Arf−/−Eμ-myc lymphomas in vitro using a fitness assay (Supplemental Fig. 6). Cells were partially transduced with QCiG-p53 or QCiG-Rosa Cas9 targeting vectors along with MLP-p53.1224 as a positive control; treated with either Nutlin-3a, doxorubicin (DXR), or vehicle; and allowed to divide over the course of 3 d. No toxicity was observed from expression of either Cas9 construct, as the initial GFP expression remained constant over time in the presence of vehicle treatment (Supplemental Fig. 6). When compared with vehicle, there was an obvious and substantial shift in the fluorescence peak following Nutlin-3a or DXR treatment that was only apparent for QCiG-p53-infected cells but not for the QCiG-Rosa-infected cells (Supplemental Fig. 6). These effects also appeared to be entirely reliant on the p53-dependent DNA damage-induced apoptotic response, since enrichment for GFP observed in the presence of drug was not apparent in p53−/−Eμ-myc lymphomas or Eμ-myc lymphomas overexpressing Bcl-2 for either QCiG-p53 or MLP-p53.1224 (Supplemental Fig. 6).
We next sought to corroborate these results in vivo in mice. Cohorts of mice were injected with equivalent amounts of the same ex vivo sgRNA–Cas9-transduced Arf−/−Eμ-myc lymphoma cells as described above. Once lymphomas had developed and were palpable, mice were separated into two groups and received a single injection of either DXR or vehicle (Fig. 3A). The Arf−/−Eμ-myc tumors infected with QCiG-Rosa readily responded to DXR therapy, with regression occurring almost immediately followed by a tumor-free remission lasting a median of 8 d (Fig. 3B). In contrast, all other tumor types showed complete resistance to DXR treatment (Fig. 3B). Dissection of moribund QCiG-p53 mice revealed a pronounced GFP fluorescence from various lymphoid tissues and lymphomas (Fig. 3C). Extraction and disaggregation followed by FACS analysis of cells and tissues at various stages of lymphomagenesis (preinjection, post-injection [pre-DXR], and post-DXR treatment) confirmed that GFP fluorescence was due to progressive enrichment of the QCiG-p53-expressing lymphoma cells (much like what was seen for MLP-p53.1224) (Fig. 3D). In contrast, QCiG-Rosa-expressing lymphoma cells were not enriched (Fig. 3D). Importantly, this result was dependent on a prior intact p53 gene, as the entire selective pressure for QCiG-p53 expression was eliminated in p53−/−Eμ-myc lymphomas following DXR treatment, implying that exon 7 of Trp53 in Arf−/−Eμ-myc cells had been altered by Cas9. Clear evidence of mutation was confirmed using the SURVEYOR assay, showing an indel frequency of 19% (Fig. 3E). Furthermore, immunoblots comparing protein extracts from dissected lymphomas pre- and post-DXR treatment revealed a strong preference for elevated Cas9 expression in the DXR-treated QCiG-p53 samples relative to QCiG-Rosa controls, which correlated with the increase in the proportion of GFP+ cells as seen by flow cytometry (Fig. 3F). Strikingly, in addition to the accumulation of lower truncated p53 protein products in the QCiG-p53 samples following DXR treatment, there was also a marked increase in the total levels of p53 expression, a likely result of a dominant-negative mutant interaction (see the Discussion). Taken together, these results demonstrate the feasibility and practicality of using CRISPR as a tool for positive selective screening approaches in mouse models of cancer.
Figure 3.
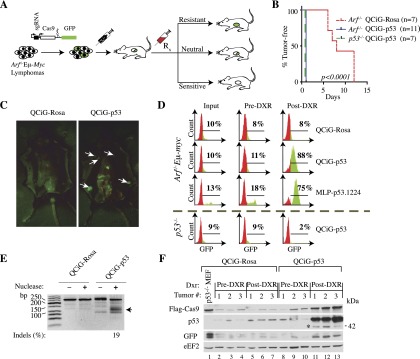
Cas9-mediated editing of Trp53 in Arf−/−Eμ-myc lymphomas is positively selected for following DXR treatment in vivo. (A) Schematic diagram of in vivo fitness assay. (B) Kaplan-Meier analysis of tumor-free survival of mice injected with Rosa26 or Trp53 Cas9 targeted Arf−/−Eμ-myc and p53−/−Eμ-myc lymphomas following treatment with DXR. (C) Detection of GFP in tumors arising from QCiG-p53-infected Arf−/−Eμ-myc lymphomas following exposure to DXR and analyzed 3 d later. White arrows denote GFP fluorescence in lymph nodes originating from the presence of QCiG-p53 in the resulting tumors. (D) FACS analysis of the indicated Cas9 targeted Eμ-myc lymphomas analyzed before injection into mice (input), from tumors arising in vivo (pre-DXR), and from tumors for which the host had received DXR treatment (post-DXR). (E) SURVEYOR assay of DNA from QCiG-p53- and QCiG-Rosa-infected Arf−/−Eμ-myc lymphomas isolated from mice prior to DXR treatment. (F) Immunoblot showing long-term Cas9, p53, and GFP expression in QCiG-Rosa and QCiG-p53 Arf−/−Eμ-Myc lymphomas in vivo. Samples are from three separate tumors isolated prior to (pre-DXR) or following (post-DXR) DXR treatment. In the case of post-DXR samples for QCiG-Rosa Arf−/−Eμ-myc lymphomas, tumors were harvested after relapse (∼10 d after post-DXR treatment). The asterisk highlights a truncated p53 protein arising in the Cas9-edited samples.
The spectrum of mutations derived from CRISPR-mediated Trp53 targeted cleavage
One of the unique features of CRISPR-driven mutagenesis is the initial mixture of mutational events that arises in a pool of cells from NHEJ repair. Genetic mutations conferring unique growth advantages should be selected for, potentially yielding novel biological insight into the target gene's cellular role. Spurred by our previous results, we decided to look in more detail at the dynamics of the mutational spectrum over the course of drug treatment that arose following repair of the double-strand breaks induced by Cas9 targeted cleavage in Trp53 exon 7. Deep sequencing was performed on DNA isolated from Arf−/− MEFs and Arf−/−Eμ-myc lymphoma cells both pre- and post-drug treatment (Fig. 4). As expected from previous studies, a majority of the indels that occur after NHEJ repair centered on the predicted Cas9 cleavage site (∼4 nt 5′ to the PAM site) (Fig. 4A). The total number of mutations increased following exposure to DXR in tumor cells, from an initial average NHEJ rate of ∼10% (among three independent tumors) to a final average NHEJ rate of ∼55% (a fivefold relative increase) (Fig. 4B). We detected a significant rise in the percentage of mutant Trp53 exon 7 reads in cells infected with QCiG-p53 following either Nutlin-3a treatment in vitro or DXR treatment in vivo (about fourfold relative increase) (Fig. 4B), correlating very tightly with the fold enrichment in GFP-expressing cells (see Supplemental Fig. 5). Importantly, baseline mutant reads (<1%) remained constant in all QCiG-Rosa samples even following drug treatment (Fig. 4B). To an extent similar to Arf−/−Eμ-myc lymphoma cells, the number of mutant reads in exon 7 of Trp53 increased by about threefold in QCiG-p53 transduced Arf−/− MEFs following Nutlin-3a treatment but not in their QCiG-Rosa counterparts, indicating that these effects were not limited to transformed cells (Supplemental Fig. 7A). While there was a wide range of nucleotide mismatches in the p53 target site that arose following drug treatment, closer inspection of the mutant reads revealed rank order preferences that were enriched, mostly resulting in frameshift mutations (Supplemental Fig. 7B,C).
Figure 4.
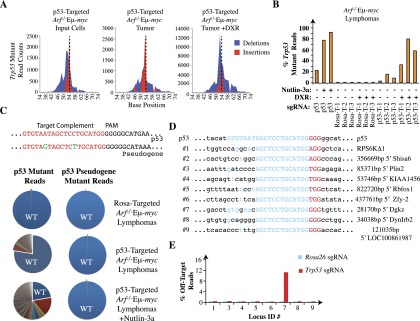
Analysis of indels at the Trp53 locus and at predicted off-target sites in Arf−/−MEFs and Arf−/−Eμ-myc tumors edited with Rosa26 and Trp53 sgRNAs. (A) Total count and location of insertions and deletions in exon 7 of Trp53 in Arf−/−Eμ-myc cells prior to injection, post-implantation, and post-DXR treatment, respectively. The vertical dashed line represents the predicted Cas9 cleavage site. (B) Frequency of mutant reads obtained following sequencing of Trp53 exon 7 from the indicated cells and tumors. T-1, T-2, and T-3 represent three independent tumors. (C, top panel) Sequence alignment of the trigger site in the Trp53 and Trp53 pseudogene. Differences are highlighted in green. (Bottom panel) Pie charts illustrating the proportion of mutated sequence reads at Trp53 (left) and the Trp53 pseudogene (right) relative to wild-type sequences (wt; blue). DNA was isolated from samples of Arf−/−Eμ-myc lymphoma cells infected with QCiG-Rosa-infected (top), QCiG-p53-infected (middle), or QCiG-p53-infected cells that were exposed to 10 μM Nutlin-3a for 3 d followed by a 10-d recovery period (bottom). (D) Prediction of genomic sequences showing sequences complementary to the first 13 perfectly matched nucleotides 5′ to the PAM of the Trp53 trigger sequence with all possible combinations of PAM. The trigger sequence is shown in blue, PAM is in red, and flanking nucleotides are in black. The genomic location is shown at the right. (E) Percent mutant reads at the indicated genomic locus in Rosa26- and Trp53-modified Arf−/− MEFs. The total read count for each amplified region ranged from ∼11,000 to 15,000 (sample #8), ∼18,000 to 23,000 (sample #7), and ∼20,000 to 53,000 (all others). Read counts for locus #2 are absent, since the barcode that had been used in the preparation of that sample could not be deciphered from the output of reads.
One interesting result of the deep sequencing analysis was the almost complete lack of sequencing reads that contained mutations in the homologous region of the Trp53 pseudogene (Fig. 4C). Only two mismatches in the sgRNA target region distinguish Trp53 from the pseudogene. Mutant reads for the Trp53 pseudogene remained consistently at baseline levels across all samples analyzed, unlike the p53 mutant reads, indicating the absence of off-target cutting by Cas9 at this locus (Fig. 4C). We further analyzed our tumor samples for other predicted off-target modifications (Fig. 4D). From both the SURVEYOR assay and deep sequencing reads, only one (locus #7) of nine potential off-target sites showed consistent Cas9-mediated off-target mutagenesis, as only QCiG-p53 but not QCiG-Rosa transduced cells demonstrated this effect (Fig. 4D,E; Supplemental Fig. 7D). Incidentally, locus #7 had the least number of total mismatches (two) among all predicted off-target regions, suggesting that Cas9 cleavage can tolerate a small number of mismatches in the 5′ end of the trigger sequence, in line with previously reported data (see the Discussion; Semenova et al. 2011; Jinek et al. 2012). Thus, the repair of Cas9-induced double-strand breaks in Trp53 enriched for a wide variety of indels covering a large range of sequence diversity with seemingly minimal off-target effects, underscoring the versatility of CRISPR-mediated in situ genome editing.
Discussion
Versatility and limitations
With the recent emergence and development of sequence-specific endonucleases, the prospect of targeting and editing any genetic element in an organism's genome in situ appears within reach. Methodologies and techniques based on ZFNs and TALENs have broadened our capability of manipulating the genome to the point where their application as gene therapy is undergoing early-phase clinical trials (NCT00842634, NCT01044654, and NCT01252641). Nevertheless, the widespread adoption of these technologies has been hampered somewhat by their intrinsic design constraints, which involves the de novo construction of pairs of lengthy modular DNA-binding domains for each and every DNA target region and iterative empirical testing for target modification efficiency and general cellular toxicity. Thus, in their current states, converting them for use in genome-wide studies and screens is hindered by their lack of flexibility and scalability. The simplicity of the Cas9-based genome-editing procedure, which uses a single monomeric endonuclease that is guided to the DNA cleavage site through RNA hybridization, is uniquely suited to resolve many of these adaption barriers and issues. In this study, we exploited these unique traits of Cas9/CRISPR to tailor a version of the Cas9/CRISPR system specifically for broad in vivo use in a relatively simple platform intended for implementation in positive-screening approaches.
Central to the general applicability of a modified type II-based CRISPR system in genome-wide screening approaches (and, for that matter, in any genome-editing application) is the ability to target any homologous DNA sequence through simple alteration of the trigger portion of the sgRNA, requiring only the partially degenerate 3-base 5′NGG3′ PAM sequence to be present in the noncomplementary strand of the targeted region. The frequency of this motif in standard mammalian genomes and the small oligo sgRNA design requirement should allow for the generation of complex pools of targeted whole-genome sgRNA libraries. In its current embodiment, libraries based on Cas9-driven cleavage are only amenable to positive selection screening approaches given the rapid normalization of noncutting or neutral mutagenic events in the population following DNA cleavage and subsequent repair. Nevertheless, when compared with RNAi, the current gold standard in pooled high-throughput whole-genome screening approaches, Cas9-based libraries would offer several advantages. First, unlike shRNA/siRNA libraries, sgRNA-based libraries could have a larger targeting range than shRNAs to include genomic regions responsible for generating noncoding RNAs as well as probing intronic and intergenic regions for functional consequences. Second, there is inherent predictability in Cas9-directed targeted gene mutation in terms of both the potency of its disruption (either monoallelic or biallelic) and the permanency of its alteration, avoiding confusing hypomorphic phenotypes. Third, the confounding interpretation of off-target effects should also be greatly reduced, since the probability of a single sgRNA targeting multiple exons is greatly diluted by the sequence space of a whole genome, while off-target effects arising from shRNA must, out of design, affect another mRNA. In principle, this also greatly reduces the need for high “coverage” of distinct sgRNAs targeting multiple areas of the same transcript, hence reducing overall pool sizes.
A necessary prerequisite for any CRISPR-based large-scale multiplex screen is the use of a surrogate marker for monitoring genome-editing events and ensuring its stability over the course of the assay. In our design, we opted for GFP fluorescence as a convenient means to track genome editing in vivo by linking it to Cas9 expression. While GFP fluorescence cannot be used to score directly for a mutagenic or gene-disruptive event, it does allow for enrichment of cells that have undergone such events (Ding et al. 2013). It is worth noting that when compared with MLP-p53.1224, the GFP fluorescence originating from the pQCiG constructs appears dimmer (mean fluorescence ∼10-fold above baseline), perhaps a consequence of reduced viral titers owing to the larger size of the Cas9 cDNA or due to differential promoter activity between MLP and pQCiG (PGK vs. SV40) (Supplemental Fig. 6).
Unlike what has been observed for ZFNs or TALENs, long-term Cas9 expression (even in complex with its sgRNA) does not appear to be toxic. We observed no overt loss of expression in cells that continuously produced Cas9/sgRNA for up to 18 d in culture as well as 24 d in vivo (Fig. 3D; data not shown). More telling is that the proportion of GFP+ Rosa26 targeted Arf−/−Eμ-myc lymphoma cells remained constant when tumors arose in vivo and did not change in resistant tumors that arose following DXR treatment (Fig. 3D). Thus, despite continuous expression of our control Rosa26 targeting Cas9/sgRNA construct under conditions of strong negative selective pressure in mouse tissue, our Cas9 variant was not depleted, demonstrating both the robustness of our vector design and the neutrality of the CRISPR system.
Mutational spectrum of Cas9-edited Trp53 following drug selections
Another distinct advantage of CRISPR-based mutagenesis screens is the ability to not only induce permanent and selectable genetic alterations among a diverse set of genes but also select for distinct mutations within a given gene. Thus, one has the potential to infer more than a simple loss-of-function genetic analysis from the output of mutations that are selected for during a positive selection CRISPR-based screen. In fact, following our Cas9-mediated targeted p53 disruption, we observed a proportion of indels in exon 7 that were selected for following drug treatment (Supplemental Fig. 7C). While most of the cancer-linked TP53 mutations are distributed evenly over its coding region, a majority tends to occur in the DNA transactivation domains that span roughly exons 4–9. Specifically on exon 7, several “hot spot” missense mutations (which modify residues G245, R248, and R249) frequently arise in a wide variety of tumor samples (Supplemental Fig. 3; Petitjean et al. 2007; Edlund et al. 2012). Many of these mutations permanently affect p53 transcriptional activity and are destabilizing in general but can also exert dominant-negative effects on wild-type p53 (through sequestration of the unaffected allelic protein in a nonfunctional tetrameric complex) or, more subtly, acquire gain-of-function properties that could confer selective advantages during key stages of the tumorigenic process (Muller and Vousden 2013). The majority of the indels generated through Cas9 cleavage in the Eμ-myc mouse model that were enriched during drug treatment were frameshifts causing premature termination at 27 codons or three codons downstream from the PAM. For both cases, the truncated proteins would have lost much of their DNA transactivation and oligomerization domains (including NLS), which we presume would result in loss-of-function (as opposed to dominant-negative) gene products, of which several of these frameshift mutant alleles are annotated in human tumor TP53 curated databases (Petitjean et al. 2007; Forbes et al. 2011). Nevertheless, we detected strong dominant-negative-like phenotypic increases in total p53 levels via immunoblots in all DXR-resistant tumors (Fig. 3F), which, in the Eμ-myc model, is often the result of allelic dominant-negative inactivating mutations in Trp53 that interrupt the negative feedback loop brought on by p53-dependent transactivation of MDM2 (Eischen et al. 1999). Although we did not confirm this via direct Trp53 transcript sequencing, we speculate that this might be the result of coselection for dominant-negative-conferring mutations in other non-Cas9 targeted exons. One of the top-ranked mutant reads (labeled “A” and comprising ∼6% of total reads averaged across p53-modified Arf−/−Eμ-myc lymphomas) (Supplemental Fig. 7B,C) consists of a single A insertion at the site of Cas9 cleavage shifting the reading frame by +1 and prematurely terminating p53 translation at an out-of-frame stop codon 27 codons downstream. In fact, this mutation results in the deletion of ∼12 kDa from the C terminus and might correspond to the prominent lower band detectable in Western blots that migrates ∼10 kDa lower than native p53 (Figs. 2E, 3F). The high proportion of the single A insertion mutants reads could have arisen from initial offset Cas9 cleavage events on opposite DNA strands (Jinek et al. 2012), with the resulting staggered ends undergoing repair and ligation (Supplemental Fig. 7B), thus biasing the resulting mutagenic outcome and leading to the higher incidence of the single-base insertion that we observed. Other common indels do result in in-frame deletions that should drastically impair transactivation function, but, in either case, further empirical testing will be necessary to fully grasp their true mutational role.
Off-target Cas9 cleavage events
For any large-scale CRISPR-based library, screening efforts controlling for target specificity will be of paramount importance. Despite the degenerate nature of the PAM and Cas9's apparent tolerance for base mismatches 5′ distal to its cleavage site and purported strict seed region, early reports in both lower and higher model organisms suggested minimal cutting at predicted off-target genomic sites in situ (albeit often only circumstantially) (Sapranauskas et al. 2011; Jinek et al. 2012; Cong et al. 2013; Jiang et al. 2013a). While not an unbiased genome-wide analysis, we devised a similar heuristic list of probable p53 off-targets containing sequences that aligned genomically but conformed to 13 nt of an exactly matching sequence 5′ distal to the PAM and found very little off-target cutting. We noted Cas9-driven modification at one predicted site, a site bearing only two mismatches (Fig. 4D, locus 7, positions 14 and 17 relative to PAM). This was in stark contrast to another potential off-target locus, the p53 pseudogene, for which we could not detect any Cas9-driven mutations. Quite fortuitously, the p53 pseudogene also contains exactly two mismatches in its trigger sequence, differing only in the locations of the mismatches (Fig. 4C, positions 8 and 15 relative to PAM), reinforcing the notion that Cas9 cleavage has a much less stringent 5′ end bias for mismatch tolerance. This also underscores the fact that for purposes of sgRNA library construction, much fewer gene-specific (or other genomic-specific) sgRNAs will be needed to control for false positive (or false negative) hits.
More recent work suggests that there is even greater sequence degeneracy of DNA cleavage by Cas9, implying that simple rules for off-target prediction will need to be largely amended and expanded. Some reports suggest substantial off-target mutagenesis (Cradick et al. 2013; Fu et al. 2013), while others emphasized a more nuanced portrait of Cas9 cleavage specificity (Hsu et al. 2013; Pattanayak et al. 2013). One issue highlighted by these reports is the importance in controlling both the amount of Cas9/sgRNA delivered and the stoichiometry of Cas9:sgRNA as determinants to limiting off-target cleavage. This is one key benefit designed into our constructs: The combined amounts of each component (Cas9 and sgRNA) would be fairly consistent upon cellular integration, which might explain the limited off-target mutagenesis that we observed in vivo. Moving forward, there is clearly a need for other unbiased in vivo techniques to measure off-target editing events and for greater control in maintaining Cas9 expression, perhaps through the use of inducible systems. In sum, our results demonstrate both the efficiency and simplicity of altering unique genomic loci in a variety of settings using the retroviral-based “all-in-one” Cas9–sgRNA plasmid, with seemingly no apparent innate negative selection or off-target effects following Cas9 expression in vivo.
Materials and methods
Vector design
The wild-type Cas9 gene sequence from S. pyogenes was mammalian codon-optimized using an online tool (http://www.encorbio.com/protocols/Codon.htm) and synthesized by GenScript into pUC57 (pUC57/Cas9) (Supplemental Fig. 1). The Cas9 D10A mutation was engineered into pUC57/Cas9 by replacing the NTD NcoI/KpnI fragment with a corresponding fragment harboring a GAC-to-GCC change. The Cas9 H840A mutation was engineered into pUC57/Cas9 by replacing the BglII/HindIII fragment by a corresponding gene fragment harboring the appropriate CAC-to-GCC alteration. The C-terminal Flag-tagged Cas9 variant was generated by removing the 3xFlag/NLS signals from pUC57/Cas9 by replacing the NcoI/KpnI fragment with an oligonucleotide and rebuilding the native Cas9 NTD. A synthetic gene construct harboring the Cas9 CTD fused in-frame to 3xFlag and SV40 NLS signals was obtained from GenScript, digested with PmlI/ClaI, and inserted into the corresponding sites in the untagged Cas9 variant. Full-length Cas9 variants were transferred from pUC57 (NcoI [Klenow-repaired]–EcoRV fragment]) into LeGo-iG (StuI–BsrGI [Klenow-repaired]) (http://www.lentigo-vectors.de/vectors.htm) to generate pLC (Fig. 1A).
The pQ vector series was developed by gutting pQCXIX (Clontech) using BglII/PuvII and inserting the following polylinker: 5′-AGATCTCTAGACAACAATGGCACTCGAGCCCAACCAGCACAGTGGATCGAGGTGTGGAAAGGATATCCAATTGCATGACAATTTAGTAAAATCGATTTGAATTCCGTAGCAAAAGAACTGTTTAAACCAGCTG-3′ (BglII and PvuII sites are underlined). A BstXI/StuI fragment harboring the SV40 promoter was isolated from MLS and inserted into the BstXI/EcoRV sites of the above-described polylinker to generate pQ/SV40. The XbaI/XhoI U6–sgRNA fragment was excised from pLC and inserted into the same sites upstream of the SV40 promoter in pQ/SV40, to generate pQ/U6/SV40. An EcoRI/ClaI (Klenow-repaired) fragment harboring the EMCV IRES/GFP fragment was isolated from MSCV-IRES-GFP and cloned into the EcoRI/PvuII site of pQ/U6/SV40 to generate pQ/U6/SV40-iG. Finally, the AflII (Klenow-repaired)/ClaI Cas9 gene from pUC57/Cas9 was inserted into the BamHI (Klenow-repaired)/ClaI sites of pQ/U6/SV40-iG to generate pQCiG.
sgRNA were ordered as gBlocks from IDT and cloned into the HpaI–XhoI sites downstream from the human U6 promoter in pLC.
Cell culture and lentiviral/retroviral infections
All retroviral packaging was performed using ecotropic Phoenix cells according to established protocols (http://www.stanford.edu/group/nolan/retroviral_systems/retsys.html). All murine lymphomas used in this study were maintained in B-cell medium (45% DMEM, 45% IMDM, 55 μM β-mercaptoethanol, 10% fetal bovine serum, 100 U/mL penicillin/streptomycin, 100 U/mL GLN) on γ-irradiated Arf−/− MEF feeder layers. Feeder layers were comprised of ∼25% confluent γ-irradiated Arf−/− MEFs preincubated with B-cell medium for 3 d prior to addition of lymphoma cells. Lymphomas were routinely split 1:3 every 2–3 d. MLP-p53.1224, an MSCV-based retrovirus that expresses a potent p53 shRNA, has been described previously (Dickins et al. 2005). U2OS cells were maintained in McCoy's 5A medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin/streptomycin, and 100 U/mL Gln.
Lentiviral transductions were performed using second-generation vectors. Essentially, 10 μg of pLC, 7.5 μg of packaging plasmid psPAX2, and 3 μg of envelope vector (pMDG2) were combined and transfected by calcium phosphate into 293T/17 (American Type Culture Collection) cells. The medium was changed the following day, and, 48 h post-transfection, lentiviral supernatant was collected every 8–12 h up until 72 h post-transfection. The lentiviral supernatant (0.5 mL) was mixed with an equal volume of medium and added to a well of a six-well dish containing U2OS target cells that had been prepared at 60% confluency the previous day. Following transduction, ∼5–7 d were allowed to pass to allow genome-editing events to occur, at which point assays were started.
TLR
The TLR assay was performed essentially as described by Certo et al. (2011) and is described in the Supplemental Material.
Western blots
Total extracts were prepared by lysis in NuPAGE LDS sample buffer. Subcellular fractionation was performed according to a differential detergent fractionation procedure, as previously described (Ramsby and Makowski 2011). Protein concentrations were determined using a DC protein assay (Bio-Rad), and extracts were resolved by SDS-PAGE, transferred to PVDF membranes, probed with the indicated antibodies, and visualized using enhanced chemiluminescence (Perkin Elmer). The antibodies used were directed against murine p53 (NCL-p53-505, Leica Microsystems), human p53 (DO-1, Santa Cruz Biotechnology), Flag tag (M2, Sigma), eEF2 (#2332, Cell Signaling), GFP (JL-8, Clontech), GAPDH (14C10, Cell Signaling), VDAC (Ab-5, Calbiochem), and hnRNPA1 (K350, Cell Signaling).
Flow cytometry
Tumors were harvested, and single-cell suspensions were prepared by grinding tumors gently between the frosted portion of two microscope slides followed by passing through a 40-μm cell strainer (BD Falcon). These cells were treated with ACK buffer (0.15 M NH4Cl, 0.1 mM EDTA, 0.01 M KHCO3 at pH 7.3) to remove red blood cells, harvested by centrifugation, and washed several times with ice-cold PBS. Cells were stained with propidium iodide (PI), and the forward, side scatter, PI measurements, and GFP content were assessed using a Guava EasyCyte HT. The percent GFP+ cells was determined from the total viable cell population using Guava ExpressPro software (Millipore).
Tumor transplantation studies
A total of 2 × 106 lymphoma cells was injected into the tail vein of 6- to 8-wk-old female C57BL/6 mice. Upon development of well-palpable tumors (auxiliary and inguinal lymph nodes; ∼2 wk post-transplantation), mice were injected once intraperitoneally with DXR. Tumor-free survival was defined as the time between the disappearance and reappearance of a palpable lymphoma following treatment. All animal studies were approved by the McGill University Faculty of Medicine Animal Care Committee.
SURVEYOR assay
Genomic DNA was prepared from the indicated samples using TNE buffer (10 mM Tris 8.0, 100 mM NaCl, 25 mM EDTA) containing 0.15 mg/mL proteinase K, 0.15 mg/mL RNase A (DNase-free), and 0.2% SDS. DNA was extracted from nuclei by incubating samples for 12 h at 45°C followed by phenol, phenol/chloroform, and chloroform extraction. Samples were precipitated in 2 vol of EtOH and one-tenth volume of 3 M NaOAc and resuspended in ddH2O. The p53 target locus and predicted off-target loci were amplified from genomic DNA using the primers shown in Supplemental Table 1. PCR products were gel-purified using QiaQuick Spin Columns (Qiagen). Mutations in the amplicons were assessed using the SURVEYOR mutation detection kit following the manufacturer's guidelines (Transgenomic) and resolved on 10% nondenaturing polyacrylamide gels. Gels were stained with ethidium bromide for 20 min followed by washing with ddH2O for 20 min. Relative band intensities were quantified using ImageJ (National Institutes of Health). One predicted off-target locus (5′-…aggtctaggtctAGCTCCTGCATGGAGGttcag…-3′; located 571,069 base pairs (bp) 5′ to Agbl4 and 496,777 bp 3′ from Ntrk3 [Mus musculus strain C57BL/6J, chromosome 7, genomic contig, GRCm38.p1 C57BL/6J MMCHR7_CTG5_2]) was not analyzed in the present study as it consistently failed to yield an amplicon after multiple PCR attempts.
Deep sequencing to assess mutation spectrum and targeting specificity
Genomic regions flanking p53 and predicted CRISPR target sites were amplified using barcoded primers with engineered adaptor regions. Off-target loci (with the exception of #4 and #7) (Fig. 4D) were amplified by PCR using Phusion HiFi polymerase (New England Biolabs) with five cycles of annealing temperature at 56°C followed by 20 cycles with an annealing temperature of 68°C. Conditions used to PCR off-target locus #4 were the same; however, Q5 HiFi polymerase (New England Biolabs) was used. To amplify off-target locus #7, a touchdown PCR protocol with Q5 HiFi polymerase (New England Biolabs) was used. The annealing temperature was stepwise-reduced from 68°C to 56°C with 1°C decrease per cycle over 13 cycles followed by 15 cycles with an annealing temperature at 56°C. PCR products were gel-purified using QiaQuick Spin Columns (Qiagen). DNA samples were quantified using picogreen and pooled in equimolar ratios. Sequencing libraries were then sequenced on an Ion Torrent personal genome machine as recommended by the manufacturer (Life Technologies).
Sequence reads were initially filtered requiring a perfect sequence match to barcodes and to PCR primers used to generate the amplicons. Reads were further filtered by excluding reads containing ≥4 bases with Phred quality scores <25 using NextGENe software (Softgenetics). For all CRISPR target sites, a wild-type sequence at the target site identified a nonedited read. The inclusion of amplicons from cells that had been targeted using the Rosa26 trigger acted as a negative control to gauge the frequency of indels arising from non-CRIPSR/Cas9 events. For amplicons to the p53 pseudogene, a distinguishing nucleotide difference (G in wild-type Trp53 vs. A in the 53 pseudogene) 5 bp downstream from the PAM site was used to identify and bin the reads (Fig. 4C).
Acknowledgments
We thank Patrick Sénéchal and Denis Paquette for excellent technical assistance. We thank Julianna Blagih and other members of the Russell Jones laboratory for their assistance and generosity in allowing us to use their FACS equipment. We are grateful to Dr. Sidong Huang for critical reading of the manuscript. J.D. is a Fonds de la Recherche en Santé du Quebec (FRSQ) Research Scholar. This work is supported by a grant from the Canadian Institutes of Health Research (CIHR: MOP-86716) to J.D. and grants from CIHR (MOP-106530) and the Richard and Edith Strauss Canada Foundation to J.P.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.227132.113.
References
- Bassett AR, Tibbit C, Ponting CP, Liu JL 2013. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 4: 220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, Scharenberg AM 2011. Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods 8: 671–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Elledge SJ, Hannon GJ 2006. Lessons from nature: MicroRNA-based shRNA libraries. Nat Methods 3: 707–714 [DOI] [PubMed] [Google Scholar]
- Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ 2013. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res 23: 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS 2013. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232 [DOI] [PubMed] [Google Scholar]
- Chylinski K, Le Rhun A, Charpentier E 2013. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol 10: 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Fine EJ, Antico CJ, Bao G 2013. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 41: 9584–9592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR–Cas systems. Nucleic Acids Res 41: 4336–4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, Lowe SW 2005. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 37: 1289–1295 [DOI] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B 2013. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods 10: 1028–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K 2013. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12: 393–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE, Lowe SW 2012. Life in the fast lane: Mammalian disease models in the genomics era. Cell 148: 1099–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund K, Larsson O, Ameur A, Bunikis I, Gyllensten U, Leroy B, Sundstrom M, Micke P, Botling J, Soussi T 2012. Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc Natl Acad Sci 109: 9551–9556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Ortega-Molina A, Velasco-Miguel S, Herranz D, Vassilev LT, Serrano M 2007. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res 67: 7350–7357 [DOI] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL 1999. Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 13: 2658–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhang B, Ding W, Liu X, Yang DL, Wei P, Cao F, Zhu S, Zhang F, Mao Y, et al. 2013. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23: 1229–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. 2011. COSMIC: Mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 39: D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, Calarco JA 2013. Heritable genome editing in C. elegans via a CRISPR–Cas9 system. Nat Methods 10: 741–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF III 2013. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154: 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O'Connor-Giles KM 2013. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194: 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Kaini P, Sander JD, Joung JK, Peterson RT, Yeh JR 2013a. Heritable and precise zebrafish genome editing using a CRISPR–Cas system. PLoS ONE 8: e68708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK 2013b. Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat Biotechnol 31: 227–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao LE, Wente SR, Chen W 2013. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci 110: 13904–13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA 2013a. RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat Biotechnol 31: 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP 2013b. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res doi: 10.1093/nar/gkt780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J 2013. RNA-programmed genome editing in human cells. Elife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katic I, Grosshans H 2013. Targeted heritable mutation and gene conversion by Cas9–CRISPR in Caenorhabditis elegans. Genetics doi: 10.1534/genetics.113.155754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A 2007. Tumor growth need not be driven by rare cancer stem cells. Science 317: 337. [DOI] [PubMed] [Google Scholar]
- Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J 2013a. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31: 688–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Teng F, Li T, Zhou Q 2013b. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR–Cas systems. Nat Biotechnol 31: 684–686 [DOI] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. 2008. Rapid ‘open-source’ engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31: 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Guo D, Zhang J, Huang Q, Qin G, Zhang X, Wan J, Gu H, Qu LJ 2013. Targeted mutagenesis in rice using CRISPR–Cas system. Cell Res 23: 1233–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PA, Vousden KH 2013. p53 mutations in cancer. Nat Cell Biol 15: 2–8 [DOI] [PubMed] [Google Scholar]
- Nekrasov V, Staskawicz B, Weigel D, Jones JD, Kamoun S 2013. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 691–693 [DOI] [PubMed] [Google Scholar]
- Oren M, Rotter V 2010. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2: a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR 2013. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31: 839–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M 2007. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum Mutat 28: 622–629 [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152: 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsby M, Makowski G 2011. Differential detergent fractionation of eukaryotic cells. Cold Spring Harb Protoc 2011: prot5592. [DOI] [PubMed] [Google Scholar]
- Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V 2011. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39: 9275–9282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci 108: 10098–10103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, et al. 2013. Targeted genome modification of crop plants using a CRISPR–Cas system. Nat Biotechnol 31: 686–688 [DOI] [PubMed] [Google Scholar]
- Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, Zhang X, Zhang P, Huang X 2013. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23: 720–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Lawrence CM, Wiedenheft B 2013. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 82: 237–266 [DOI] [PubMed] [Google Scholar]
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. 2006. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc Natl Acad Sci 103: 1888–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K, Bartsch U, Stocking C, Fehse B 2008. A multicolor panel of novel lentiviral ‘gene ontology’ (LeGO) vectors for functional gene analysis. Mol Ther 16: 698–706 [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154: 1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]