

Abstract
Proteolytic processing is a ubiquitous and irreversible post-translational modification involving limited and highly specific hydrolysis of peptide and isopeptide bonds of a protein by a protease. Cleavage generates shorter protein chains displaying neo-N and -C termini, often with new or modified biological activities. Within the past decade, degradomics and terminomics have emerged as significant proteomics subfields dedicated to characterizing proteolysis products as well as natural protein N and C termini. Here we provide an overview of contemporary proteomics-based methods, including specific quantitation, data analysis, and curation considerations, and highlight exciting new and emerging applications within these fields enabling in vivo analysis of proteolytic events.
Proteolysis involves the breakdown of proteins into smaller polypeptides or amino acids through the hydrolysis of peptide bonds by a protease. This represents a remarkably significant, but often underappreciated, post-translational modification (PTM)1 in that is it irreversible yet also ubiquitous. Consequently, the functional sequence of a protein can very rarely be predicted from its transcript, as proteolysis products form new (neo-) N and C termini. These cleavage events, or proteolytic processing events, can result in activation, inactivation, completely altered protein function, and even excision of “neo-proteins” with growth factor activity from an extracellular matrix parent molecule, and they regulate a vast array of biological processes (1). These include DNA replication, cell cycle progression, cell proliferation, and cell death, as well as pathological processes such as inflammation, cancer, arthritis, and cardiovascular disease. For example, in protein synthesis and maturation, precise selective removal of the N-terminal methionine and the signal peptide is essential for correct protein maturation and secretion. In some proteins, scission of the chain forms a molecule with four termini when linked by disulfide bridges. Through the removal of signal, nuclear, and mitochondrial localization sequences and ectodomain shedding, proteases regulate protein localization, and in viral infection, via cleavage of pre- and pro-domains and polyprotein processing, inactive proteins are converted into their active form(s), are inactivated, or change receptor-binding affinity. Thus, proteolysis is involved in much more than the mere degradation and turnover of proteins, important though these processes are in homeostasis.
Proteases exist in all orders of life and constitute one of the largest enzyme families in humans (2), and more than 30 drugs targeting these enzymes are currently approved for clinical use (3). However, in order to fully comprehend the cellular function(s) of a given protease, one must have knowledge of the proteins processed by that protease, as well as the functions of these substrates and specific processing events. This is currently far from the case, as half of all human proteases have no known substrates (4). Degradomics is the application of high-throughput approaches to study proteases, their substrates, and their inhibitors on a system-wide scale (4). More specifically, terminomics is the specific characterization of protein N and C termini and, as such, forms a subfield of degradomics. This review provides an overview of current proteomics-based methods for characterizing protease cleavage events and protein termini. The quantitation, analysis, and curation of proteomics data, as well as exciting new applications within these fields, are also considered.
Methods for Characterizing Active Site Specificity
Several array- and library-based methods have been developed to identify protease active site specificities. These include substrate phage display (5) and bacterial substrate display (6), whereby bacteriophages or bacteria express a chimeric cell surface protein containing a peptide of random sequence and an affinity tag. Proteolysis enables selection based on the affinity tag, and cleavable sequences are determined via DNA sequencing. However, these approaches do not provide the exact cleavage site in the random sequence; for this, a second step is required. Similarly, peptide libraries and microarrays have been used. For microarrays, arrayed peptide libraries are incubated with a test protease and cleavage is detected via methods such as loss of fluorophore binding or the removal of a fluorescent quencher (7–12). Library-based approaches are similar except that peptide mixtures are typically sequenced via Edman degradation or mass spectrometry (MS). One example is mixture-based oriented peptide libraries, which was the first approach used successfully to sequence the prime-side residues of the cleavage site in a library (13). The prime-side cleavage motif (sequence C-terminal to the cleavage site) is determined by proteolysis of a library of N-terminally acetylated dodecamers sequenced via Edman degradation. Subsequently, a second library containing this predetermined prime-side sequence, a random unblocked N terminus, and a C-terminal biotin tag is generated and a second incubation with the protease is performed. Undigested peptides and C-terminal fragments are removed by means of avidin capture, and a second round of Edman degradation determines nonprime-side specificity. In view of the multiple time-consuming steps involved in generating custom second libraries in this otherwise very successful approach, new approaches have been sought to rapidly determine the prime-side and nonprime-side sequences in combination. Proteomic identification of protease cleavage sites (PICS) is one such approach (14). PICS employs a diverse, biologically relevant, and database-searchable peptide library generated from a cellular proteome using trypsin or Glu-C (14, 15). Primary amines (N-terminal α-amines and lysine ε-amines) are blocked, and this forms the library. A test protease is added, and the new terminal α-amines generated by proteolysis are selectively biotinylated and affinity purified. Purified peptides are sequenced via liquid-chromatography tandem mass spectrometry (LC-MS/MS) to determine prime-side cleavage motifs, whereas sequences N-terminal to cleavage sites are extracted bioinformatically. This can be done because the peptide library is accessible to conventional proteomics bioinformatics, whereas randomized synthetic peptide libraries are not. Thus, PICS enables the determination of both prime and nonprime cleavage site residues in the same experiment and so has the advantage of being very rapid.
These peptide library-based techniques have been used to elucidate the cleavage site specificity of many proteases from all catalytic classes. However, a significant limitation is that they depend solely on amino acid sequences of relatively short peptides. Contributions of exosites and protein folding to cleavage site specificity cannot be observed, and as for all techniques that determine only the active site specificity, relevant in vivo protease substrates cannot be reliably identified solely from a cleavage site.
Proteomics Methods for Identifying Substrates
Several proteomics methods have been developed to identify protease substrates. These include both two-dimensional polyacrylamide gel electrophoresis (PAGE)- and LC-MS/MS-based techniques. Following two-dimensional PAGE, stained spots are excised and identified via MS. Substrates are identified by a reduction in spot intensity of the intact protein and the appearance of spots corresponding to cleavage products (16–18). However, two-dimensional PAGE is restricted in terms of reproducibility and sensitivity, and it cannot be applied to small cleavage fragments or those differing by only a few residues.
LC-MS/MS now provides vast improvements in throughput and proteome coverage. Shotgun proteomics has been used to identify substrates, including those whose localization has been altered by membrane shedding (19–22). This is done by comparing the secretomes of protease-treated cells to those of control cells. However, this approach cannot be used to determine the actual cleavage site. Nonetheless, these early labeling approaches utilizing isotope-coded affinity tags and isobaric tags for relative and absolute quantitation (iTRAQ) were very successful in easily identifying hundreds of biologically relevant substrates in the cellular context. Whereas these approaches are designed to determine substrates from complex proteomes, amino-terminal-oriented mass spectrometry of substrates is designed to identify multiple cleavage sites in proteins in vitro (23, 24). Amino-terminal-oriented MS involves incubation of a purified substrate with a protease followed by dimethylation of the original and neo-N termini at the whole protein level. Subsequent trypsin digestion generates dimethylated semi-tryptic peptides containing the original N and C termini, as well as neo-N-terminal peptides representing cleavage sites that are identified by the dimethylated termini and their position in the protein sequence. The dimethylated cleavage sites are readily distinguished from the tryptic peptides, which contain a free primary amine at their N terminus. The protein topography and migration analysis platform uses one-dimensional SDS-PAGE in combination with LC-MS/MS to identify cleavage events by peptide mapping (25). Here, proteins from protease-treated and control samples are resolved via one-dimensional SDS-PAGE, and each lane is cut into a number of gel slices, trypsinized, and analyzed via LC-MS/MS. Peptographs representing protein sequence coverage versus SDS migration identify proteolysis products based on shifts from higher to lower molecular weight species. This represents a development of an earlier study using gel slice analysis of isotopically labeled samples separated on one-dimensional SDS-PAGE (26), but with the advantage of employing visually useful software for analysis. Like most gel-based approaches, this is very mass spectrometry intensive, and only occasionally is the exact cleavage site also directly identified. Recently, secretome protein enrichment with click sugars was developed (27). This approach involves the metabolic labeling of N- and O-linked glycans, followed by a click reaction resulting in their biotinylation. Secreted proteins and shed extracellular membrane proteins are purified from contaminating serum proteins by means of avidin capture followed by in-gel digestion and LC-MS/MS. Secretome protein enrichment is particularly useful for cell culture experiments in which the cells have fastidious growth requirements and require serum. The serum glycoproteins are not metabolically labeled, and this enables simplification of the proteomic sample before analysis by separating the cell-derived metabolically labeled proteins from the glycoproteins in serum. However, only glycoprotein substrates can be identified.
The above techniques have been useful in substrate identification. However, as the vast majority of identified peptides are internal, except with the amino-terminal-oriented MS procedure, precise cleavage sites are rarely determined, especially for proteins identified with low sequence coverage.
Methods for Enriching Protein N Termini
N-terminal PTMs, including proteolytic processing, can greatly influence the localization and activity of many proteins (28). For example, N-terminal acetylation (N-Ac) plays important roles in protein function, localization, and stability (29), and N-terminal methylation regulates protein–protein interactions (30). Thus, characterizing protein N termini not only identifies protease cleavage sites, but also is important in determining the functional physiochemical properties of a proteome. Methods employing both positive and negative selection of N-terminal peptides were developed (Table I) following the early recognition that “keeping it simple” approaches aiming to identify rare semi-tryptic terminal peptides within a complex mixture of tryptic peptides without enrichment will not lead to proteome-wide coverage and so will miss most cleavage sites. This is especially relevant for low-abundance but biologically interesting proteins such as cytokines. Thus, a variety of terminal peptide enrichment strategies have been developed to improve both the coverage and the dynamic range of terminal peptide identifications.
Table I. Methods for enriching protein N and C termini.
| Methoda | Advantages/disadvantages | Quantitation | Reference(s) |
|---|---|---|---|
| Selective enzymatic biotinylation of N termini | (+) Positive selection of unmodified N termini | iTRAQ, SILAC, label-free selected reaction monitoring | (32, 33, 61, 86, 87) |
| (+) Does not require chemical modification | |||
| (−) Requires expensive patent protected enzyme | |||
| (−) Requires large amounts of sample | |||
| N-CLAP | (+) Positive selection of unmodified N termini | None to date | (34) |
| (−) Enriched peptides are shortened by one residue | |||
| (−) Not compatible with chemical stable-isotope labeling | |||
| COFRADIC | (+) Negative selection of N and C termini | 12C4 and 13C4 butyric acid, NHS-13C2D3, SILAC, trypsin-catalyzed 18O exchange | (35, 36, 55, 88–90) |
| (−) Extensive fractionation enhances sample loss | |||
| (−) >50 fractions/sample, making it very instrument intensive | |||
| (−) Loss of His- and Arg-containing peptides during strong cation exchange chromatography | |||
| TAILS | (+) Negative selection of modified and unmodified N-termini | Stable-isotope dimethyl labeling, iTRAQ | (37, 79, 80, 91, 92) |
| (+) Very low nonspecific binding to polymer | |||
| (−) Requires commercially available hyperbranched polyglycerol aldehyde polymer | |||
| PTAG | (+) Negative selection of modified and unmodified N termini | None to date | (40) |
| (−) Loss of phosphorylated N-terminal peptides | |||
| (−) Losses due to nonspecific binding to TiO2 material | |||
| Enrichment of modified N termini by selective α-amine biotinylation | (+) Negative selection of modified N termini | None to date | (41, 42) |
| (−) No retention of unmodified N termini | |||
| (−) Loss of His-containing peptides | |||
| C-TAILS | (+) Negative selection of modified and unmodified C termini | Stable-isotope dimethyl labeling | (54) |
| (+) Chemical tag identifies unmodified C termini | |||
| (−) Difficult to achieve complete labeling of carboxyl groups |
a Published name or description of enrichment method.
SILAC, stable isotope labeling by amino acids in cell culture.
Enrichment of N Termini by Positive Selection
Several methods exist for enriching N-terminal peptides by positive selection. Typically ε-amines are blocked and α-amines are tagged (i.e. with biotin), with tagging followed by a secondary proteolysis step and finally enrichment and elution of N-terminal peptides for analysis via LC-MS/MS. Using these methods, unmodified N-terminal and neo-N-terminal peptides can be effectively purified, but modified N termini cannot. This is quite limiting, as ∼85% of soluble proteins are N-terminally acetylated in eukaryotic cells (31). Another challenge is discriminating between α-amines at N termini and ε-amines on lysine residues. This has been termed the lysine problem: when both are blocked, several N termini are excluded from analysis, but failure to block ε-amines results in contamination by the abundant internal peptides in the sample.
Mahrus et al. developed the most useful such approach with an elegant method using an engineered subtiligase to selectively label unblocked α-amines with a biotinylated peptide ester substrate, with labeling followed by trypsinization, avidin capture, and LC-MS/MS (32, 33) (Fig. 1A). The peptide ester substrate contains a virus cleavage site enabling the recovery of enriched peptides. However, up to 50 to 100 mg of protein is required for each sample analysis, which can be very limiting. In a clever use of Edman chemistry, Xu et al. used chemical labeling of the α-amine of proteins (N-CLAP) using phenyl isothiocyanate to block all primary amines (34). Similar to Edman degradation, treatment with trifluoroacetic acid triggers cyclization of phenyl-isothiocyanate-modified α-amines specifically, resulting in peptide bond cleavage after the first amino acid. α-amines are then biotinylated, with biotinylation followed by trypsinization, avidin capture, elution of N termini via reduction, and LC-MS/MS.
Fig. 1.

Representation of select N- and C-terminal peptide enrichment strategies. COFRADIC is shown as a combination of two variations of the method employing either 2,4,6-trinitrobenzenesulfonic acid (TNBS) treatment for enrichment of N-terminal peptides or N-Hydroxysuccinimidyl-butyrate treatment for separation of N- and C-terminal peptides. Refer to the main text for a description of each method.
Enrichment of N Termini by Negative Selection
Several methods exist for enriching protein N-terminal peptides by negative selection. Common to each, N-terminal α-amines and ε-amines are blocked at the protein level, and blocking is followed by trypsinization, which exposes α-amines of internal peptides. These unblocked α-amines are used to deplete internal peptides from the sample, enabling enrichment of both modified and unmodified protein N termini. This facilitates higher proteome coverage than positive selection techniques and is particularly useful for intracellular proteomes, where most N termini are acetylated. The utility of purifying the natural N terminus (whether naturally or chemically blocked) has many advantages; in particular, it enables up to 50% of the identified proteins to be identified from two or more peptides (i.e. the original N terminus and the internal cleaved neo-N-terminal peptide). This greatly increases the confidence scores in protein substrate identification relative to those for proteins identified from just one neo-terminal peptide. However, in contrast to positive selection methods in which tags are used as handles to facilitate purification and concentration, negative selection does not allow for highly selective washing, cleanup, and concentration of terminal peptides during the enrichment step(s), other than by conventional peptide concentration (e.g. precipitation or evaporation), which can result in sample losses and dirtier samples for LC-MS/MS.
The most widely reported negative selection methods are combined fractional diagonal chromatography (COFRADIC) and terminal amine isotope labeling of substrates (TAILS). During COFRADIC, α- and ε-amines are blocked by acetylation, with subsequent proteolysis, pre-enrichment of N-terminal peptides via strong cation exchange chromatography, and fractionation via reverse-phase liquid chromatography (35, 36) (Fig. 1B). α-amines are treated with 2,4,6-trinitrobenzenesulfonic acid to form hydrophobic trinitrophenyl-peptides that are separated from N-terminal peptides by an additional round of reverse-phase liquid chromatography. Enzymatic removal of pyroglutamyl peptides has also been employed (36). One advantage of COFRADIC over several other techniques is that all the required materials are commercially available and relatively inexpensive. However, a disadvantage is that it utilizes extensive fractionation steps, providing several opportunities for samples loss and making it very instrument intensive, with up to 100 LC-MS/MS runs per sample.
During TAILS, α- and ε-amines are blocked by dimethylation or iTRAQ labeling, proteins are subjected to trypsinization, and a commercially available water-soluble hyperbranched polyglycerol aldehyde polymer for proteomics is added to covalently bind the internal tryptic peptide α-amines through reductive amination (37, 38) (Fig. 1C). The polymer provides a large contact area with peptides in solution enabling a very efficient reaction and very low nonspecific binding. Its large size (>10 kDa) allows the depletion of internal peptides via filtration. Roche has adopted TAILS and reported a refinement whereby the polymer mixture is applied directly to the precolumn (39). The unbound N-terminal peptides enter the mass spectrometer directly, minimizing handling and consequent losses. Recently, Mommen et al. developed a technique very similar to TAILS, except that instead of the hyperbranched polyglycerol aldehyde polymer, internal α-amines are phosphorylated via treatment with glyceraldehyde-3-phosphate and depleted via titanium dioxide chromatography (40).
Both TAILS and COFRADIC block ε-amines to enable the retention of lysine-containing N-terminal peptides, which has an added advantage of introducing an isotope label for those N-terminally blocked peptides that otherwise would go unlabeled. Zhang et al. developed a method for the specific enrichment of modified N-terminal peptides that does not require this blocking procedure (41, 42). Following proteolysis, CNBr-activated Sepharose, which is specific to α-amines at pH 6, is added to deplete internal peptides. However, Sepharose beads retain peptides nonspecifically, and isoelectric point discrimination of the α- and ε-amines is rarely quantitative, so this can reduce purity and N-terminal peptide yields. Several additional techniques have been developed for the negative selection of N termini (43–50). However, as they have not yet been applied or proven to work in large-scale proteomics workflows, they are not discussed further here.
Methods for Enriching Protein C Termini
Similar to N termini, C-terminal PTMs can also regulate protein function (51). Examples include chemokine and hormone processing, as well as modifications such as prenylation that localize proteins to lipid membranes (52, 53). Methods for enriching protein C termini have lagged behind those for N termini, largely due to a lack of methods with which to selectively modify carboxyl groups in aqueous solution. Thus, it is likely that many important C-terminal modifications and processing events exist but remain unknown or poorly characterized because of our current inability to enrich and identify them.
Two methods currently exist for proteomic analysis of protein C termini: C-terminal amine-based isotope labeling of substrates (C-TAILS), and COFRADIC combined with strong cation exchange chromatography (54, 55) (Table I). During C-TAILS, proteins are dimethylated at α- and ε-amines (Fig. 1D). Carboxyl groups are protected with ethanolamine and then trypsinized to generate free N and C termini on internal peptides. Newly generated α-amines are blocked by a second dimethylation step, and newly generated carboxyl groups are removed by means of covalent coupling to a high-molecular-weight polyallylamine polymer. Like in TAILS, original blocked C termini are unbound and recovered via filtration. One advantage of C-TAILS is that the C-terminal label allows for validation of original versus neo-C termini during data analysis. In the COFRADIC method, α- and ε-amines are blocked by acetylation prior to proteolysis (Fig. 1B). Tryptic peptides are passed over a strong cation exchange column at pH 3, where N- and C-terminal peptides are collected in the flow-through. Free α-amines of C-terminal peptides are butyrylated to increase their hydrophobicity, enabling their separation from N-terminal peptides via reverse-phase liquid chromatography. Sechi and Chait developed an additional method based on the binding of anhydrotrypsin to α-amines (56), but this has not yet been employed on a large scale.
Quantitation
Several quantitative methods have been applied to the techniques described above to discriminate between background proteolysis events that are present in every sample, whether in vivo or in post-sampling handling, and those induced by a specific condition (Table I). In techniques without quantification, this cannot be achieved, and so the data reflect proteolytic events of interest as well as the background, which makes data interpretation difficult. Typically, differentially labeled proteomes are used representing protease-treated or -related condition(s), with one proteome serving as a control (i.e. protease-null) (Fig. 2A). Equal amounts of the samples are mixed and ultimately analyzed via LC-MS/MS. Neo-N- or C-terminal peptides generated by proteolytic processing events appear with either high or low ratios, whereas peptides unaltered by the treatment condition(s) appear with ratios centered on 1.0.
Fig. 2.
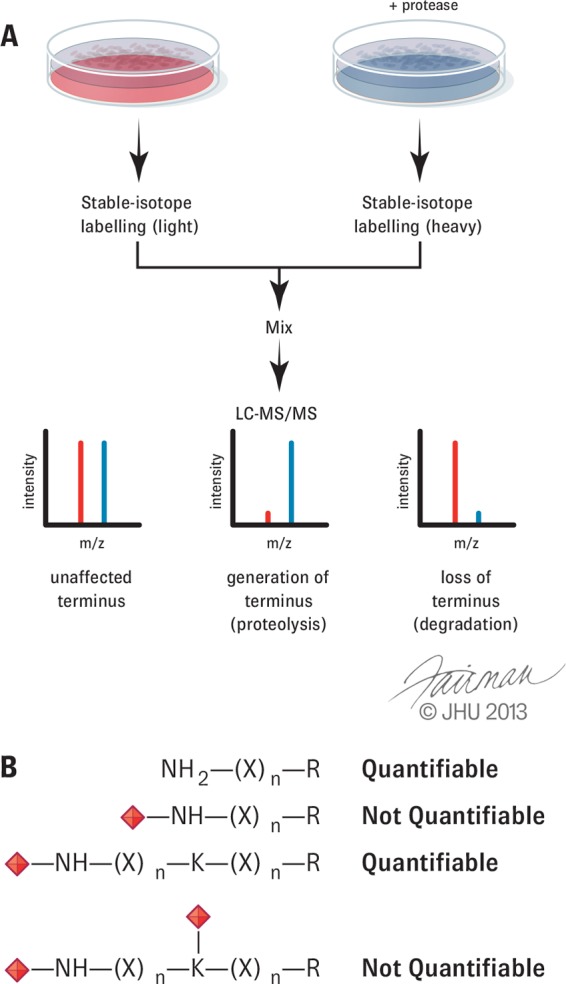
Quantitation and terminomics data. A, flow diagram representing quantitation of terminal peptides between a control and protease-treated condition. B, schematic showing quantifiable N-terminal peptides following stable-isotope labeling methods targeting primary amines. X represents any amino acid except lysine; red diamonds represent an amine modification such as acetylation.
Specific quantitation methods include MS1-based stable-isotope dimethyl labeling and stable isotope labeling by amino acids in cell culture, and MS2-based iTRAQ and tandem mass tags (57–60). Specific to N-terminomics, it is important to note that the chemical labeling approaches mentioned above target primary amines. Thus, there are instances when α-amines and ε-amines are otherwise modified (i.e. N-Ac), rendering these peptides unquantifiable (Fig. 2B). Although SILAC offers the advantage of complete quantitation, it is not compatible with all sample types and is impossible to use in human tissues. Label-free approaches have been used to a lesser extent, but they generally suffer from poor accuracy (25, 27, 61). They are also very instrument intensive when done properly to generate the numbers of spectra needed for reliable and statistically significant quantification. Thus, improperly performed spectral counting can be easy, but the resulting data will be misleading. Furthermore, like in analyses of many other PTMs including phosphoproteomics, N- and C-terminomics samples are expected to identify proteins based on a single peptide. MS1-based methods acquire quantitation information from several spectra collected across a chromatographic peak as a measure of technical variance. MS2-based quantitation relies on the generation of higher order scans, and so for single peptide identifications the result is often quantitation based on a single spectrum. Given the possibility of isotope interference if careful MS acquisition methods are not employed (62, 63), the opportunity for unreliable quantitation is somewhat higher when using MS2-based approaches.
Data Acquisition and Analysis
Because each protein is often represented by a single or small number of terminal peptides, terminomics methods reduce sample complexity. However, this also presents challenges to complete proteome coverage, as not all N- and C-terminal peptides are amenable to LC-MS/MS. Many will have suboptimal lengths or poor physiochemical properties for fragmentation and/or ionization. Several groups employ dimethylation or acetylation of α- and ε-amines prior to digestion to increase the lengths of N- and C-terminal peptides (35, 37, 40, 47, 49). This results in trypsin cleaving with ArgC specificity. However, if peptide masses between 600 and 4000 Da are considered suitable for MS/MS, only 63.4% and 62.9% of peptides from a theoretical ArgC digested human proteome are identifiable by N- and C-terminal peptides, respectively (55). If other properties such as hydrophobicity are considered, these numbers will undoubtedly drop. Also, similar to tryptic peptides with missed cleavages, ArgC-generated peptides are typically long and highly charged. Higher energy collision-induced dissociation and electron-transfer dissociation are more effective fragmentation methods for these peptides (64). Thus, acquisition methods employing collision-induced dissociation on smaller low-charge peptides and electron-transfer dissociation or higher energy collision-induced dissociation on longer high-charge peptides may prove beneficial for terminomics. Also, following dimethylation, the positive charge on α- and ε-amines is retained, whereas during acetylation it is lost. This likely offers advantages in maintaining protein solubility and improving the ionization efficiency of these peptides. To increase the percentage of MS-amenable peptides, several groups employ Glu-C and chymotrypsin digests in parallel to trypsinization (37, 41). However, especially in combination with several N-terminal modifications (i.e. N-Ac), these peptides are expected to carry only a single positive charge, rendering them less amenable to conventional acquisition methods that exclude singly charged ions as contaminants. Including singly charged ions for fragmentation has been shown to significantly increase the number of N-Ac peptides identified, particularly in the low mass range (42).
Several software tools can be used to analyze terminomics data. These include search engines such as Mascot and X! Tandem and analysis suites such as MaxQuant, Proteome Discoverer, and the Trans-Proteomic Pipeline (65–68). However, these applications are universally designed to analyze shotgun datasets containing tryptic peptides. N- and C-terminal peptides exhibit semi-enzyme specificities and thus increase the search space considerably. Also, chemical labeling for quantitation is typically achieved at the protein level for terminomics, meaning that α-amines of internal peptides are unlabeled. Current software suites such as MaxQuant and Proteome Discoverer assume labeling at the peptide level, and as a result, peptides with blocked and labeled N termini cannot be quantified within the same analysis. Currently, CLIPPER is the only software application designed specifically for N-terminomics data (69, 70). Specific for MS2-based quantitation, CLIPPER is an add-on to the Trans-Proteomic Pipeline and generates quantitation confidence and isoform assignment scores as well as automated annotation of N-terminal peptides to determine their position within a protein sequence.
Databases
Several efforts are being made to curate emerging high-throughput datasets into online resources (Table II). Created in 1999, the MEROPS database is the gold standard for protease classification and stores information about proteases from all species, as well as their substrates, cleavage sites, and inhibitors (71). However, MEROPS does not easily support meta-analyses such as comparing substrates or protease specificities. Community-based resources such as CutDB rely on users to input data (72). Each entry contains a proteolytic event relating a protease, a substrate, and a cleavage site and a description of the biological context. CutDB is part of the larger PMAP website, which also contains protease and substrate databases and tools for determining protease specificities and mapping signaling pathways (73). The termini-oriented protein function inferred database (TopFIND) is also open to user contributions (74). TopFIND integrates data from UniprotKB, MEROPS, and experimental terminomics studies from humans, mice, bacteria, and yeast, focusing on both translated and neo protein termini, as well as upstream proteases and PTMs. TopFIND provides information on substrate structure, topology, and interaction networks and contains several filtering tools for data manipulation. One limitation of the abovementioned repositories is that they do not provide easy access to original data. The Online Protein Processing Resource (TOPPR) houses a database of proteolysis sites from human and mouse, as well as links to Mascot scores and MS/MS spectra (75). It also houses a meta-analysis platform enabling filtering and analyses of individual substrates and protease specificities. However, TOPPR only accepts N- and C-terminal COFRADIC data from the Gevaert lab. A similar database called DegraBase, from the Wells lab (76), houses data generated via the subtiligase-based enrichment method applied to normal and apoptotic cell lines and also provides links to original MS data. Two cell-death-specific databases also exist: CASBAH (77), which contains all reported mammalian caspase substrates with links to UniprotKB, cleavage sites, and references, and Cell Death Proteomics database (CDP) (78), which houses proteomics data from 73 cell death studies in human, mouse, and rat.
Table II. Publicly accessible proteolytic databases.
| Name | Descriptiona | Contentsb | Reference |
|---|---|---|---|
| MEROPS | An information resource for peptidases and the proteins that inhibit them | 3000 individual peptidases and inhibitors | (71) |
| CutDB | Annotation of individual proteolytic events, both actual and predicted | 11,081 proteolytic events from 601 proteases and 3387 substrates | (72) |
| TopFIND | Public knowledgebase for protein termini and protease processing | >120,000 N and C termini and 10,000 cleavage sites | (74) |
| TOPPR | High-quality processed events available in an easy and intuitive analysis platform | 2234 substrates, 18 studied treatments or peptidases, and 27,147 cleavage sites | (75) |
| DegraBase | Non-biased description of all possible caspase substrates found in healthy and apoptotic human cells | >8000 α-amines detected via mass spectrometry | (76) |
| CASBAH | Compository of all reported mammalian caspase substrates and known cleavage sites | >400 caspase substrates | (77) |
| CDP | Collection of papers, proteins, and meta-information linked to cell death and proteomics | 6550 records of proteins and cleavage sites reported changed upon cell death | (78) |
a The stated purpose of each database, often taken directly from information on the database website.
b A description of the contents of each database, either current or at the time of publication.
Applications
Over the past decade, degradomics and terminomics have emerged as significant proteomics subfields within the broader arena of PTM analysis. Thus, it is considerably beyond the scope of this review to cover all applications of the methods described above. Instead, we focus here on two exciting new avenues: application of terminomics-based techniques to tissues, and further characterization of protein N termini.
Until very recently, the vast majority of terminomics data were derived from in vitro and cell-culture-based systems. These have provided valuable insights into cleavage events and other PTMs at protein termini, but whether these observations hold true in a complex tissue environment has remained elusive. In recent studies from auf dem Keller et al. and Tholen et al., protein N termini were isolated from murine skin (79, 80). In the former, skin of wild-type (WT) and Mmp2−/− mice was treated with 12-O-tetradecanoylphorbol 13-acetate (TPA) to induce inflammation. 4-plex iTRAQ analysis compared WT and Mmp2−/− mice ± TPA, and peptides extracted from murine skin were analyzed before and after enrichment of N termini. Global analyses showed increased abundance of several inflammatory proteins following TPA treatment and reduced exudation of acute-phase proteins from TPA-treated Mmp2−/− mice. Due to protease–protease and protease–inhibitor interactions that modify proteolytic activity in vivo, it is extremely difficult to directly assign in vivo detected substrates to a protease, even with a knockout. Here, direct MMP2 targets were discerned in vivo by establishing the following criteria: a neo-N terminus increased in TPA-treated versus untreated WT and also TPA-treated WT versus Mmp2−/− mice, while an original N terminus was unchanged between TPA-treated WT and Mmp2−/− mice. Unaltered ratios of the intact mature N-terminal peptide reflect unchanged protein abundance. Otherwise, altered synthesis or import of proteins in the exudate can be revealed as apparent increases in neo-N termini, when in fact these might have been due to steady-state turnover, but with increased (or decreased) synthesis or import. Using this approach, researchers identified an inactivating MMP2 cleavage site within C1 inhibitor and unveiled novel roles for MMP2 in regulating vascular permeability and complement activation. Reduced MMP2 cleavage of C1 inhibitor led to reduced complement activation and a lessening in the normal increase in vascular permeability due to reduced bradykinin excision and release (78).
In another study, 1191 skin proteins from WT versus Ctsb−/− and 1317 proteins from WT versus Ctsl−/− were identified via whole proteome analysis, with 15 and 32 proteins differing significantly in abundance between WT and Ctsb−/− or Ctsl−/− mice, respectively. The authors inferred direct Cstl and Ctsb substrates by comparing the sequences surrounding these cleavage sites to their previously established specificities. This revealed that the majority of cleavages stem from the altered activity of proteases other than Ctsl or Ctsb. Interestingly, periostin, which is implicated in skin physiology and cancer, increased in Ctsl−/− but not Ctsb−/− skin, and a Ctsb-dependent cleavage site was identified in dermokine, a known marker for colorectal cancer.
Terminomics data from humans, mice, and bacteria reveal that >30% of all N termini do not originate from classical protein maturation events involving the removal of signal and pro-peptides and the initiator methionine (74, 79). Several of these likely represent stable cleavage products, while others arise from alternative translation initiation sites (TIS), which exist for >65% of murine proteins and primarily drive translation of upstream open reading frames (uORFs) (81). While the UniProtKB and Ensembl databases do not contain these sequences, a method termed RIBO-seq involves deep sequencing of ribosome-protected mRNA fragments to determine in vivo translation products (82). Menschaert et al. created a novel database combining 16,570 protein sequences from UniProtKB with 7785 from RIBO-seq data (83). Lysates from mouse embryonic stem cells were analyzed via conventional shotgun proteomics and following enrichment of N termini. Matching shotgun data to the combined database identified 3% and 5% more peptides and proteins, respectively, while matching N termini identified 1835 TIS. 84% map to canonical TIS, 14% start beyond position two, indicating alternative or wrongly annotated TIS, 16 correspond to N-terminal extensions, and 4 correspond to uORFs. Interestingly, the majority of TIS identified for N-terminal extensions and uORFs contain near cognate start codons.
According to TopFIND, 12 PTMs occur at N termini (74). By far the most extensive is N-Ac, whereby the α-amine of the initiator methionine (iMet)—or the second residue if iMet is removed—is acetylated. The function of N-Ac is uncertain, but it has been reported to regulate protein function and localization and to be both protein stabilizing and destabilizing (29). Five N-acetyltransferase (NAT) complexes exist in eukaryotes, NatA–NatE, the majority of which have defined sequence specificities. Interestingly, N-Ac is much more prominent in higher eukaryotes, suggesting that it may contribute to their complexity. Van Damme et al. compared N-Ac levels between yeast and humans (84). 648 yeast and 1345 human N-Ac sites were identified within 868 and 1497 N termini. Several dipeptide sequences were preferentially acetylated in humans, including Met-Lys, which is not known to be acetylated by the existing NATs. A novel NAT, NatF, was identified with close homologs in higher eukaryotes but not in yeast. Library-based cleavage assays identified unique sequence specificity for NatF that includes Met-Lys termini. When NatF was expressed in yeast, N-Ac levels increased significantly, particularly at Met-Lys sites, indicating that it accounts for the increased N-Ac observed in higher eukaryotes.
We compared the amino acid specificity of iMet removal with N-acetylation specificity preferences. Interestingly, the residue at position two that is important in defining the preference for iMet removal matches very closely the in vivo acetylation preference found in published datasets (79). Thus Met removal is preferred for the sequences commencing MA, MG, MS, MT, and MP, and N-terminal A, G, S, and T are the preferred residues for N-Ac. That is, when Met is removed, the exposed residue at position two is also preferred for acetylation. The exception is proline. Acetylation of N termini blocks aminopeptidase activity to protect the N terminus from ragging. Pro is a special case and is resistant to most aminopeptidases. Thus, one could view N-Ac as a sequential system involving Met removal and subsequent acetylation to protect chains from aminopeptidase activity.
CONCLUSIONS
Within the past decade, terminomics methods designed to enrich for N- and C-terminal peptides have emerged as the gold standard for identifying protease substrates and cleavage sites and for characterizing protein N and C termini. These methods enable thousands of termini to be identified both in vitro and in vivo, and more recently in complex tissues. However, as these approaches are inherently restricted to single peptide identifications, complete proteome coverage is an extremely challenging, if not impossible, task. Furthermore, several tantalizing questions currently wait to be answered by degradomics methods. While the general focus appears to be the large-scale identification of cleavage sites and PTMs, Agard et al. recently employed selected reaction monitoring to assay cleavage kinetics of hundreds of caspase substrates in cell culture (61, 85). Absolute quantitation by selected reaction monitoring offers the potential to assay kinetics as well as the stoichiometry of cleavage events between hundreds of tissues and disease states. This includes tracking specific substrates as biomarkers in various disease models and, similarly, utilizing propeptide removal to measure protease activity. Such targeted analyses and challenging in vivo studies herald a bright future for terminomics in elucidating novel insights into new proteolytic pathways in vivo and hence new drug targets for disease.
Acknowledgments
C.M.O. holds a Canada Research Chair in Metalloproteinase Proteomics and Systems Biology. We thank Dr. Georgina Butler for editing assistance.
Footnotes
* This work was supported by a Canadian Institutes of Health Research Fellowship and a Michael Smith Foundation for Health Research Trainee Award to L.D.R. This work was also supported by a grant from the CIHR and an Infrastructure Grant from MSFHR.
1 The abbreviations used are:
- COFRADIC
- combined fractional diagonal chromatography
- C-TAILS
- C-terminal amine-based isotope labeling of substrates
- iMet
- initiator methionine
- iTRAQ
- isobaric tags for relative and absolute quantitation
- MS
- mass spectrometry
- N-Ac
- N-terminal acetylation
- NAT
- N-acetyltransferase
- PAGE
- polyacrylamide gel electrophoresis
- PTM
- post-translational modification
- TAILS
- terminal amine isotope labeling of substrates
- TIS
- translation initiation sites
- TPA
- 12-O-tetradecanoylphorbol 13-acetate
- uORF
- upstream open reading frame
- WT
- wild-type.
REFERENCES
- 1. Barret A. J., Rawlings N. D., Woessner J. F. (1998) Handbook of Proteolytic Enzymes, Academic Press, London [Google Scholar]
- 2. Puente X. S., Sanchez L. M., Overall C. M., Lopez-Otin C. (2003) Human and mouse proteases: a comparative genomic approach. Nature Rev. Genet. 4, 544–558 [DOI] [PubMed] [Google Scholar]
- 3. Turk B. (2006) Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 5, 785–799 [DOI] [PubMed] [Google Scholar]
- 4. Lopez-Otin C., Overall C. M. (2002) Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell. Biol. 3, 509–519 [DOI] [PubMed] [Google Scholar]
- 5. Matthews D. J., Wells J. A. (1993) Substrate phage: selection of protease substrates by monovalent phage display. Science 260, 1113–1117 [DOI] [PubMed] [Google Scholar]
- 6. Boulware K. T., Daugherty P. S. (2006) Protease specificity determination by using cellular libraries of peptide substrates (CLiPS). Proc. Natl. Acad. Sci. U.S.A. 103, 7583–7588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salisbury C. M., Maly D. J., Ellman J. A. (2002) Peptide microarrays for the determination of protease substrate specificity. J. Am. Chem. Soc. 124, 14868–14870 [DOI] [PubMed] [Google Scholar]
- 8. Rosse G., Kueng E., Page M. G., Schauer-Vukasinovic V., Giller T., Lahm H. W., Hunziker P., Schlatter D. (2000) Rapid identification of substrates for novel proteases using a combinatorial peptide library. J. Comb. Chem. 2, 461–466 [DOI] [PubMed] [Google Scholar]
- 9. Gosalia D. N., Salisbury C. M., Maly D. J., Ellman J. A., Diamond S. L. (2005) Profiling serine protease substrate specificity with solution phase fluorogenic peptide microarrays. Proteomics 5, 1292–1298 [DOI] [PubMed] [Google Scholar]
- 10. Winssinger N., Damoiseaux R., Tully D. C., Geierstanger B. H., Burdick K., Harris J. L. (2004) PNA-encoded protease substrate microarrays. Chem. Biol. 11, 1351–1360 [DOI] [PubMed] [Google Scholar]
- 11. Petrassi H. M., Williams J. A., Li J., Tumanut C., Ek J., Nakai T., Masick B., Backes B. J., Harris J. L. (2005) A strategy to profile prime and non-prime proteolytic substrate specificity. Bioorg. Med. Chem. Lett. 15, 3162–3166 [DOI] [PubMed] [Google Scholar]
- 12. Loch C. M., Cuccherini C. L., Leach C. A., Strickler J. E. (2011) Deubiquitylase, deSUMOylase, and deISGylase activity microarrays for assay of substrate preference and functional modifiers. Mol. Cell. Proteomics 10, M110.002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Turk B. E., Huang L. L., Piro E. T., Cantley L. C. (2001) Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol. 19, 661–667 [DOI] [PubMed] [Google Scholar]
- 14. Schilling O., Overall C. M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 26, 685–694 [DOI] [PubMed] [Google Scholar]
- 15. Schilling O., Huesgen P. F., Barre O., Auf dem Keller U., Overall C. M. (2011) Characterization of the prime and non-prime active site specificities of proteases by proteome-derived peptide libraries and tandem mass spectrometry. Nat. Protoc. 6, 111–120 [DOI] [PubMed] [Google Scholar]
- 16. Hwang I. K., Park S. M., Kim S. Y., Lee S. T. (2004) A proteomic approach to identify substrates of matrix metalloproteinase-14 in human plasma. Biochim. Biophys. Acta 1702, 79–87 [DOI] [PubMed] [Google Scholar]
- 17. Lee A. Y., Park B. C., Jang M., Cho S., Lee D. H., Lee S. C., Myung P. K., Park S. G. (2004) Identification of caspase-3 degradome by two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization-time of flight analysis. Proteomics 4, 3429–3436 [DOI] [PubMed] [Google Scholar]
- 18. Bredemeyer A. J., Lewis R. M., Malone J. P., Davis A. E., Gross J., Townsend R. R., Ley T. J. (2004) A proteomic approach for the discovery of protease substrates. Proc. Natl. Acad. Sci. U.S.A. 101, 11785–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tam E. M., Morrison C. J., Wu Y. I., Stack M. S., Overall C. M. (2004) Membrane protease proteomics: isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc. Natl. Acad. Sci. U.S.A. 101, 6917–6922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dean R. A., Butler G. S., Hamma-Kourbali Y., Delbe J., Brigstock D. R., Courty J., Overall C. M. (2007) Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol. Cell. Biol. 27, 8454–8465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dean R. A., Overall C. M. (2007) Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell. Proteomics 6, 611–623 [DOI] [PubMed] [Google Scholar]
- 22. Butler G. S., Dean R. A., Tam E. M., Overall C. M. (2008) Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol. Cell. Biol. 28, 4896–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doucet A., Overall C. M. (2011) Broad coverage identification of multiple proteolytic cleavage site sequences in complex high molecular weight proteins using quantitative proteomics as a complement to Edman sequencing. Mol. Cell. Proteomics 10, M110.003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doucet A., Overall C. M. (2011) Amino-terminal oriented mass spectrometry of substrates (ATOMS) N-terminal sequencing of proteins and proteolytic cleavage sites by quantitative mass spectrometry. Methods Enzymol. 501, 275–293 [DOI] [PubMed] [Google Scholar]
- 25. Dix M. M., Simon G. M., Cravatt B. F. (2008) Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 134, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo L., Eisenman J. R., Mahimkar R. M., Peschon J. J., Paxton R. J., Black R. A., Johnson R. S. (2002) A proteomic approach for the identification of cell-surface proteins shed by metalloproteases. Mol. Cell. Proteomics 1, 30–36 [DOI] [PubMed] [Google Scholar]
- 27. Kuhn P. H., Koroniak K., Hogl S., Colombo A., Zeitschel U., Willem M., Volbracht C., Schepers U., Imhof A., Hoffmeister A., Haass C., Rossner S., Brase S., Lichtenthaler S. F. (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 31, 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lange P. F., Overall C. M. (2013) Protein TAILS: when termini tell tales of proteolysis and function. Curr. Opin. Chem. Biol. 17, 73–82 [DOI] [PubMed] [Google Scholar]
- 29. Hollebeke J., Van Damme P., Gevaert K. (2012) N-terminal acetylation and other functions of Nalpha-acetyltransferases. Biol. Chem. 393, 291–298 [DOI] [PubMed] [Google Scholar]
- 30. Chen T., Muratore T. L., Schaner-Tooley C. E., Shabanowitz J., Hunt D. F., Macara I. G. (2007) N-terminal alpha-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat. Cell Biol. 9, 596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polevoda B., Sherman F. (2000) Nalpha-terminal acetylation of eukaryotic proteins. J. Biol. Chem. 275, 36479–36482 [DOI] [PubMed] [Google Scholar]
- 32. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshihara H. A., Mahrus S., Wells J. A. (2008) Tags for labeling protein N-termini with subtiligase for proteomics. Bioorg. Med. Chem. Lett. 18, 6000–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu G., Shin S. B., Jaffrey S. R. (2009) Global profiling of protease cleavage sites by chemoselective labeling of protein N-termini. Proc. Natl. Acad. Sci. U.S.A. 106, 19310–19315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gevaert K., Goethals M., Martens L., Van Damme J., Staes A., Thomas G. R., Vandekerckhove J. (2003) Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat. Biotechnol. 21, 566–569 [DOI] [PubMed] [Google Scholar]
- 36. Staes A., Van Damme P., Helsens K., Demol H., Vandekerckhove J., Gevaert K. (2008) Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC). Proteomics 8, 1362–1370 [DOI] [PubMed] [Google Scholar]
- 37. Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R. K., Starr A. E., Foster L. J., Kizhakkedathu J. N., Overall C. M. (2010) Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 28, 281–288 [DOI] [PubMed] [Google Scholar]
- 38. Kleifeld O., Doucet A., Prudova A., auf dem Keller U., Gioia M., Kizhakkedathu J. N., Overall C. M. (2011) Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 6, 1578–1611 [DOI] [PubMed] [Google Scholar]
- 39. Guryca V., Lamerz J., Ducret A., Cutler P. (2012) Qualitative improvement and quantitative assessment of N-terminomics. Proteomics 12, 1207–1216 [DOI] [PubMed] [Google Scholar]
- 40. Mommen G. P., van de Waterbeemd B., Meiring H. D., Kersten G., Heck A. J., de Jong A. P. (2012) Unbiased selective isolation of protein N-terminal peptides from complex proteome samples using phospho tagging (PTAG) and TiO(2)-based depletion. Mol. Cell. Proteomics 11, 832–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang X., Ye J., Hojrup P. (2009) A proteomics approach to study in vivo protein N(alpha)-modifications. J. Proteomics 73, 240–251 [DOI] [PubMed] [Google Scholar]
- 42. Zhang X., Ye J., Engholm-Keller K., Hojrup P. (2011) A proteome-scale study on in vivo protein Nalpha-acetylation using an optimized method. Proteomics 11, 81–93 [DOI] [PubMed] [Google Scholar]
- 43. Akiyama T. H., Sasagawa T., Suzuki M., Titani K. (1994) A method for selective isolation of the amino-terminal peptide from alpha-amino-blocked proteins. Anal. Biochem. 222, 210–216 [DOI] [PubMed] [Google Scholar]
- 44. Coussot G., Hawke D. H., Mularz A., Koomen J. M., Kobayashi R. (2007) A method for the isolation of blocked N-terminal peptides. Anal. Biochem. 361, 302–304 [DOI] [PubMed] [Google Scholar]
- 45. Mikami T., Takao T. (2007) Selective isolation of N-blocked peptides by isocyanate-coupled resin. Anal. Chem. 79, 7910–7915 [DOI] [PubMed] [Google Scholar]
- 46. Zhao L., Zhang Y., Wei J., Cao D., Liu K., Qian X. (2009) A rapid isolation and identification method for blocked N-terminal peptides by isothiocyanate-coupled magnetic nanoparticles and MS. Proteomics 9, 4416–4420 [DOI] [PubMed] [Google Scholar]
- 47. McDonald L., Robertson D. H., Hurst J. L., Beynon R. J. (2005) Positional proteomics: selective recovery and analysis of N-terminal proteolytic peptides. Nat. Methods 2, 955–957 [DOI] [PubMed] [Google Scholar]
- 48. McDonald L., Beynon R. J. (2006) Positional proteomics: preparation of amino-terminal peptides as a strategy for proteome simplification and characterization. Nat. Protoc. 1, 1790–1798 [DOI] [PubMed] [Google Scholar]
- 49. Shen P. T., Hsu J. L., Chen S. H. (2007) Dimethyl isotope-coded affinity selection for the analysis of free and blocked N-termini of proteins using LC-MS/MS. Anal. Chem. 79, 9520–9530 [DOI] [PubMed] [Google Scholar]
- 50. Sonomura K., Kuyama H., Matsuo E., Tsunasawa S., Futaki S., Nishimura O. (2011) Selective isolation of N-blocked peptide by combining AspN digestion, transamination, and tosylhydrazine glass treatment. Anal. Biochem. 410, 214–223 [DOI] [PubMed] [Google Scholar]
- 51. Gomis-Ruth F. X. (2008) Structure and mechanism of metallocarboxypeptidases. Crit. Rev. Biochem. Mol. Biol. 43, 319–345 [DOI] [PubMed] [Google Scholar]
- 52. Cox J. H., Dean R. A., Roberts C. R., Overall C. M. (2008) Matrix metalloproteinase processing of CXCL11/I-TAC results in loss of chemoattractant activity and altered glycosaminoglycan binding. J. Biol. Chem. 283, 19389–19399 [DOI] [PubMed] [Google Scholar]
- 53. Reznik S. E., Fricker L. D. (2001) Carboxypeptidases from A to Z: implications in embryonic development and Wnt binding. Cell. Mol. Life Sci. 58, 1790–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schilling O., Barre O., Huesgen P. F., Overall C. M. (2010) Proteome-wide analysis of protein carboxy termini: C terminomics. Nat. Methods 7, 508–511 [DOI] [PubMed] [Google Scholar]
- 55. Van Damme P., Staes A., Bronsoms S., Helsens K., Colaert N., Timmerman E., Aviles F. X., Vandekerckhove J., Gevaert K. (2010) Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat. Methods 7, 512–515 [DOI] [PubMed] [Google Scholar]
- 56. Sechi S., Chait B. T. (2000) A method to define the carboxyl terminal of proteins. Anal. Chem. 72, 3374–3378 [DOI] [PubMed] [Google Scholar]
- 57. Hsu J. L., Huang S. Y., Chow N. H., Chen S. H. (2003) Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75, 6843–6852 [DOI] [PubMed] [Google Scholar]
- 58. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 59. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 60. Thompson A., Schafer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A. K., Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 61. Agard N. J., Mahrus S., Trinidad J. C., Lynn A., Burlingame A. L., Wells J. A. (2012) Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc. Natl. Acad. Sci. U.S.A. 109, 1913–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ting L., Rad R., Gygi S. P., Haas W. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wenger C. D., Lee M. V., Hebert A. S., McAlister G. C., Phanstiel D. H., Westphall M. S., Coon J. J. (2011) Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 8, 933–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Frese C. K., Altelaar A. F., Hennrich M. L., Nolting D., Zeller M., Griep-Raming J., Heck A. J., Mohammed S. (2011) Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 10, 2377–2388 [DOI] [PubMed] [Google Scholar]
- 65. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 66. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 67. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 68. Keller A., Eng J., Zhang N., Li X. J., Aebersold R. (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. auf dem Keller U., Prudova A., Gioia M., Butler G. S., Overall C. M. (2010) A statistics-based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol. Cell. Proteomics 9, 912–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Keller U. A., Overall C. M. (2012) CLIPPER: an add-on to the Trans-Proteomic Pipeline for the automated analysis of TAILS N-terminomics data. Biol. Chem. 393, 1477–1483 [DOI] [PubMed] [Google Scholar]
- 71. Rawlings N. D., Barrett A. J., Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Igarashi Y., Eroshkin A., Gramatikova S., Gramatikoff K., Zhang Y., Smith J. W., Osterman A. L., Godzik A. (2007) CutDB: a proteolytic event database. Nucleic Acids Res. 35, D546–D549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Igarashi Y., Heureux E., Doctor K. S., Talwar P., Gramatikova S., Gramatikoff K., Zhang Y., Blinov M., Ibragimova S. S., Boyd S., Ratnikov B., Cieplak P., Godzik A., Smith J. W., Osterman A. L., Eroshkin A. M. (2009) PMAP: databases for analyzing proteolytic events and pathways. Nucleic Acids Res. 37, D611–D618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lange P. F., Overall C. M. (2011) TopFIND, a knowledgebase linking protein termini with function. Nat. Methods 8, 703–704 [DOI] [PubMed] [Google Scholar]
- 75. Colaert N., Maddelein D., Impens F., Van Damme P., Plasman K., Helsens K., Hulstaert N., Vandekerckhove J., Gevaert K., Martens L. (2013) The Online Protein Processing Resource (TOPPR): a database and analysis platform for protein processing events. Nucleic Acids Res. 41, D333–D337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Crawford E. D., Seaman J. E., Agard N., Hsu G. W., Julien O., Mahrus S., Nguyen H., Shimbo K., Yoshihara H. A., Zhuang M., Chalkley R. J., Wells J. A. (2013) The DegraBase: a database of proteolysis in healthy and apoptotic human cells. Mol. Cell. Proteomics 12, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Luthi A. U., Martin S. J. (2007) The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 14, 641–650 [DOI] [PubMed] [Google Scholar]
- 78. Arntzen M. O., Bull V. H., Thiede B. (2013) Cell Death Proteomics database: consolidating proteomics data on cell death. J. Proteome Res. 12, 2206–2213 [DOI] [PubMed] [Google Scholar]
- 79. auf dem Keller U., Prudova A., Eckhard U., Fingleton B., Overall C. M. (2013) Systems-level analysis of proteolytic events in increased vascular permeability and complement activation in skin inflammation. Sci. Signal. 6, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tholen S., Biniossek M. L., Gansz M., Gomez-Auli A., Bengsch F., Noel A., Kizhakkedathu J. N., Boerries M., Busch H., Reinheckel T., Schilling O. (2013) Deletion of cysteine cathepsins B or L yields differential impacts on murine skin proteome and degradome. Mol. Cell. Proteomics 12, 611–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ingolia N. T., Lareau L. F., Weissman J. S. (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ingolia N. T. (2010) Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 470, 119–142 [DOI] [PubMed] [Google Scholar]
- 83. Menschaert G., Van Criekinge W., Notelaers T., Koch A., Crappe J., Gevaert K., Van Damme P. (2013) Deep proteome coverage based on ribosome profiling aids MS-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol. Cell. Proteomics 12, 1780–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Van Damme P., Hole K., Pimenta-Marques A., Helsens K., Vandekerckhove J., Martinho R. G., Gevaert K., Arnesen T. (2011) NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 7, e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shimbo K., Hsu G. W., Nguyen H., Mahrus S., Trinidad J. C., Burlingame A. L., Wells J. A. (2012) Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc. Natl. Acad. Sci. U.S.A. 109, 12432–12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wildes D., Wells J. A. (2010) Sampling the N-terminal proteome of human blood. Proc. Natl. Acad. Sci. U.S.A. 107, 4561–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Agard N. J., Maltby D., Wells J. A. (2010) Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell. Proteomics 9, 880–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Van Damme P., Van Damme J., Demol H., Staes A., Vandekerckhove J., Gevaert K. (2009) A review of COFRADIC techniques targeting protein N-terminal acetylation. BMC Proc. 3 Suppl 6, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Van Damme P., Martens L., Van Damme J., Hugelier K., Staes A., Vandekerckhove J., Gevaert K. (2005) Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat. Methods 2, 771–777 [DOI] [PubMed] [Google Scholar]
- 90. Gevaert K., Van Damme P., Ghesquiere B., Vandekerckhove J. (2006) Protein processing and other modifications analyzed by diagonal peptide chromatography. Biochim. Biophys. Acta 1764, 1801–1810 [DOI] [PubMed] [Google Scholar]
- 91. Prudova A., auf dem Keller U., Butler G. S., Overall C. M. (2010) Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteomics 9, 894–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wilson C. H., Indarto D., Doucet A., Pogson L. D., Pitman M. R., Menz R. I., McNicholas K., Overall C. M., Abbott C. A. (2013) Identifying natural substrates for dipeptidyl peptidase 8 (DP8) and DP9 using terminal amine isotopic labelling of substrates, TAILS, reveals in vivo roles in cellular homeostasis and energy metabolism. J. Biol. Chem. 288, 13936–13949 [DOI] [PMC free article] [PubMed] [Google Scholar]