

Abstract
The cysteine protease caspase-3, best known as an executioner of cell death in apoptosis, also plays a non-apoptotic role in N-methyl-d-aspartate receptor-dependent long-term depression of synaptic transmission (NMDAR-LTD) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor endocytosis in neurons. The mechanism by which caspase-3 regulates LTD and AMPA receptor endocytosis, however, remains unclear. Here, we addressed this question by using an enzymatic N-terminal peptide enrichment method and mass spectrometry to identify caspase-3 substrates in neurons. Of the many candidates revealed by this proteomic study, we have confirmed BASP1, Dbn1, and Gap43 as true caspase-3 substrates. Moreover, in hippocampal neurons, Gap43 mutants deficient in caspase-3 cleavage inhibit AMPA receptor endocytosis and LTD. We further demonstrated that Gap43, a protein well-known for its functions in axons, is also localized at postsynaptic sites. Our study has identified Gap43 as a key caspase-3 substrate involved in LTD and AMPA receptor endocytosis, uncovered a novel postsynaptic function for Gap43 and provided new insights into how long-term synaptic depression is induced.
Synaptic plasticity (the ability of synapses to change in strength) plays an important role in brain development and cognitive function, including learning and memory. N-methyl-d-aspartate receptor (NMDAR)1-dependent long-term depression of synaptic transmission (LTD) is a major form of synaptic plasticity that leads to long-lasting decreases in synaptic strength. In NMDAR-LTD, synaptic depression is mainly mediated by removal of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors from the postsynaptic membrane through endocytosis (1–3).
Caspases are cysteine-dependent proteases that cleave after an aspartate residue (4–6). Primary specificity for aspartate at the cleavage site is so rare in mammalian proteases that only granzyme B, a serine protease derived from lymphocytes, is known to also have such a property (7, 8). Caspases are best known for their pro-apoptotic function in programmed cell death, or apoptosis (9). Caspases activated at the end of a caspase cascade are “effector” caspases, and among them caspase-3 is the dominant one (10). In addition to apoptosis, caspases also play non-apoptotic roles such as in cell differentiation, dendritic development, and memory consolidation (11–14). For instance, our earlier studies show that NMDAR-LTD requires moderate and transient caspase-3 activation, which does not induce cell death (15, 16).
In LTD, caspase-3 is activated by the mitochondrial pathway (15, 16). With the opening of NMDA receptors, calcineurin and protein phosphatase 1 are activated to dephosphorylate the Bcl-2 family protein BAD. Dephosphorylated BAD then translocates to the mitochondria, activating BAX, which is also a member of the Bcl-2 family protein. The subsequent release of cytochrome-c from mitochondria leads to the sequential activation of caspase-9 and caspase-3. Active caspase-3 induces AMPA receptor endocytosis, and therefore depression of synaptic strength. The mechanism by which caspase-3 promotes AMPA receptor endocytosis, however, remains to be determined.
Caspases' cellular functions are primarily mediated by the proteolysis of caspase substrates, resulting in change of their functions. Identification of specific proteins cleaved by caspases is therefore key to understanding the mechanisms mediating their biological functions. Several proteomic approaches, developed specifically for this purpose, have led to the identification of more than 2000 caspase cleavage sites (17–22). In particular, the recently developed subtiligase-based method for isolating proteolytic products has led to the identification of >1,000 putative caspase substrates in human samples (21, 23, 24). Subtiligase is an engineered peptide ligase that ligates esterified peptides onto the N termini of proteins or peptides through free α-amines (25). Because the majority of eukaryotic proteins are N-terminally acetylated—and therefore blocked from subtiligase labeling (26)—subtiligase can couple synthetic tagged peptides selectively to the free N-terminal α-amines of proteins derived from proteolysis. These peptide-conjugated proteolytic products can then be affinity-purified, digested with trypsin and sequenced by mass spectrometry. Identification of peptides ligated to the tagged peptide by subtiligase allows researchers to determine cleavage sites within the substrates.
In this study, the subtiligase-based proteomic method was used to find capase-3 substrates in rat neurons, resulting in the identification of 81 putative aspartate cleavage sites in 56 proteins. Of these, 37 proteins (human and a single rat orthologs) were not previously reported in the CASBAH database (20), and 13 (human orthologs) are not included in the DegraBase data set compiled using the subtiligase methodology in non-neuronal tissue (21). Using complimentary methods, we further confirmed that, both in vivo and in vitro, caspase-3 cleaves three of these candidate substrates: growth associated protein 43 (Gap43), drebrin (Dbn1), and brain acid soluble protein 1 (BASP1). Surprisingly, we also found that AMPA receptor endocytosis and LTD induction both require caspase-3 to cleave Gap43, a protein well known for its presynaptic functions, at the sites identified by our study.
MATERIALS AND METHODS
Antibodies
The following antibodies were obtained commercially: anti-V5 (Invitrogen, Carlsbad, CA; R960), anti-β-tubulin (Developmental Studies Hybridoma Bank, Iowa City, IA; E7), anti-PSD95 (NeuroMab, Davis, CA; clone K28/43), anti-Gap43 (Abcam, Cambridge, MA; ab75810), anti-GluA2 (Millipore, Billerica, MA; MAB397), anti-synaptophysin (Millipore, AB9272), anti-MAP2 (Sigma-Aldrich, St. Louis, MO; m9942), and anti-GluN2A (Covance, Princeton, NJ; PRB-513P), goat anti-mouse IgG (H + L)-HRP conjugate (Bio-Rad, Hercules, CA; 172–1011), IRDye 680 goat anti-mouse IgG (H+L) (Li-Cor, Lincoln, NE; 926–32220), IRDye 800CW goat anti-rabbit IgG (H+L) (Li-Cor, 926–32211), Alexa Fluor 488 goat anti-mouse IgG (H+L) (Invitrogen, Carlsbad, CA; A11001), Alexa Fluor 647 goat anti-mouse IgG (H+L) (Invitrogen, A21235), Alexa Fluor 488 goat anti-rabbit IgG (H+L) (Invitrogen, A11008), and Alexa Fluor 647 goat anti-rabbit IgG (H+L) (Invitrogen, A21244).
Purification of Subtiligase
The subtiligase expression construct, pPW04, was kindly provided by Dr. James A. Wells (UCSF). Recombinant subtiligase was expressed in Bacillus subtilis and purified as described (25). Briefly, B. subtilis cells (ATCC # 47096) transformed with pPW04 were grown in 2xTY broth with vigorous shaking at 37 °C for 24 h. Supernatant containing secreted subtiligase was collected by centrifugation. Proteins in the supernatant were precipitated with 50% (w/v) (NH4)2SO4 on ice, dissolved in 25 mm NaOAc (pH 4.5), 5 mm dithiothreitol (DTT), and precipitated again with 70% ethanol at 4 °C. The precipitated proteins were dissolved in 25 mm NaOAc (pH 4.5), 5 mm dithiothreitol (DTT) and dialyzed against the same buffer for 24 h, with 3 buffer changes. After dialysis, subtiligase was further purified by FPLC (Amersham Biosciences AKTAFPLC) with a HiTrap SP HP column (GE Healthcare). Subtiligase was eluted with a 0–300 mm linear gradient of NaCl. Eluted fractions were examined by SDS-PAGE. Fractions containing subtiligase were pooled and loaded onto a Superdex S200 10/300 GL column (GE Healthcare, Piscataway, NJ) and eluted with 100 mm bicine (pH 8.0), 5 mm DTT for buffer exchange. After measuring protein concentration, aliquots were stored at −80 °C.
N-terminal Labeling and N-terminal Peptide Isolation
N-terminal labeling and N-terminal peptide isolation were performed as described (27), with modifications. For induction of apoptosis, cortical neurons at DIV18 (12 plates, ∼7 million cells/plate) were treated with 1 μm staurosporine for 8 h. Neurons were washed twice with PBS and lysed in lysis buffer (100 mm bicine, pH 8.0, 1% Triton X-100, 5 mm EDTA, 100 μm Z-VAD-FMK, 100 μm E-64, 1 mm PMSF, and 1 mm AEBSF; 0.5 ml/plate). Cell lysates were homogenized using a Dounce tissue grinder (tight pestle, 20 strokes), and incubated at room temperature for 1 h to allow complete inhibition of endogenous protease and esterase activities. The cell lysate was then spun at 20,000g for 15 min at 4 °C. The volume of the supernatant was adjusted to 1.8 ml using the Amicon Ultra-4 3K centrifugal filter unit (Millipore). For N-terminal labeling, DTT, DMSO, subtiligase, and biotinylated peptide ester (New England Peptide, Gardner, MA) were added sequentially to the supernatant to obtain a final concentration of 5 mm, 10%, 1 μm, and 2 mm, respectively. The reaction mix was incubated at 25 °C for 1 h, and stopped by adding iodoacetamide to a final concentration of 1 mm. The unincorporated peptide esters were removed by gel filtration with TBS (pH 7.4), 0.2% Triton X-100. Labeled peptides were denatured with guanidine hydrochloride (6 m), reduced with TCEP (2 mm), and alkylated with iodoacetamide (6 mm). Following alkylation, the sample was diluted to reduce the concentration of guanidine hydrochloride to 2 m, followed by incubation with high capacity NeutrAvidin agarose resins (Thermo Scientific, Rockford, IL) overnight at room temperature. The resins were washed 3 times with wash buffer (4 m guanidine hydrochloride, 100 mm bicine pH 8.0, and 2 mm biotin) and once with digestion buffer (100 mm Tris-HCl, pH 8.0, 10 mm CaCl2). The resins were then resuspended in 1 ml digestion buffer and digested with 2 μg sequencing grade modified trypsin (Promega, Madison, WI) overnight at 37 °C. After digestion, the resins were washed 3 times with wash buffer, twice with 50 mm NH4HCO3 and resuspended in 50 mm NH4HCO3. Bound N-terminal peptides were released from resins by digestion with 20 μg TEV protease (28) for 2 h at room temperature, desalted with C18 tips (Thermo Scientific), and dried using a speedvac before mass spectrometric analysis.
Plasmid Construction
The ORF cDNAs of Gap43, Dbn1, MARCKSL1, and BASP1 were amplified from rat brain cDNAs (Zyagen) using the following primers: Gap43 (5′-TTG GAT CCG ATA CCA CCA TGC TGT GCT GTA TGA GAA G-3′ and 5′-TTG CGG CCG CCG GCA TGT TCT TGG TCA GCC TCG-3′); Dbn1 (5′-TTG GAT CCC CCC GCA GCA TGG CCG G-3′ and 5′-TTG CGG CCG CCA TCA CCA CCC TCG AAG CCC TCT TCC TC-3′); MARCKSL1 (5′-TTG GAT CCA CCC CCA TCA TGG GCA GC-3′ and 5′-TTG CGG CCG CCC TCA TTC TCA GCA CTG GCA GAT GC-3′); BASP1 (5′-TTG GTA CCA ACT CCA AGA TGG GAA GCA AGC TGA-3′ and 5′-TTG AAT TCC CCT CTT TGA CGG CCA CGC TTT G-3′), and cloned into the modified pEF-ENTR B vector (a SV40 poly(A) terminator was added after the C-terminal tags) (Addgene, Cambridge MA) (29) between the BamHI and NotI sites (for Gap43, MARCKSL1, Dbn1) or the KpnI and EcoRI sites (BASP1). Caspase-resistant mutants were generated by PCR site-directed mutagenesis. The E. coli expression construct for Gap43 was generated by inserting the Gap43 cDNA between the SalI and NotI sites of a modified pET30a vector behind the GB1-NusA-Strep-8xHis-TEV (GNSHT) multidomain tag (a gift from Dr. Zhonghua Hu, NIMH). The GB1 and NusA domains are used to enhance solubility. The Strep and 8xHis tags are used for affinity purification. For the EGFP-GluR2 mammalian expression construct, the pHluorin-GluR2 was firstly amplified by PCR from the pcDNA3.1-pHluorin-T-GluR2 plasmid (a gift from Dr. Richard L. Huganir, Johns Hopkins University) (primers: 5′-TTT GCT AGC CAC CAT GCA AAA GAT TAT GCA TAT TTC TGT CCT CCT TTC TC-3′ and 5′-TTT GCG GCC GCC AAT TTT AAC ACT CTC GAT GCC ATA TAC GTT GTA ACC-3′), and cloned into the pCMV-ENTR B vector (the modified pEF-ENTR B vector, in which the EF1α promoter was replaced by the CMV promoter) between the NheI/SpeI and NotI sites. The pHluorin was then replaced by EGFP using SpeI site. The Gap43-mCherry mammalian expression construct was generated with the FastCloning method (30) using the following primers: Gap43 insert (5′-GAT CGC CGC CAC CAT GCT GTG CTG TAT GAG AAG-3′ and 5′-AAG CTG CAA TAA ACA ATC AGG CAT GTT CTT GGT CAG CC-3′); pCMV-ENTR B vector (5′-TTG TTT ATT GCA GCT TAT AAT GGT TAC AAA TAA AGC-3′ and 5′-CAT GGT GGC GGC GAT CTG ACG GTT CAC TAA ACC AGC TCT G-3′); mCherry insert (5′-AGC GGC GGC GGA AGT GGA ATG GTG AGC AAG GGC GAG GA-3′ and 5′-AAG CTG CAA TAA ACA ATT ACT TGT ACA GCT CGT CCA TGC CG-3′); pCMV-ENTR B-Gap43 vector (5′-TTG TTT ATT GCA GCT TAT AAT GGT TAC AAA TAA AGC-3′ and 5′-ACT TCC GCC GCC GCT GGC ATG TTC TTG GTC AGC CTC G-3′). The pCMV-ENTR B-Gap43 was made firstly from the PCR products of Gap43 insert and pCMV-ENTR B vector. The pCMV-ENTR B-Gap43-mCherry was then generated from the PCR products of mCherry insert and pCMV-ENTR B-Gap43 vector. All constructs were verified by sequencing. The schematic diagrams of these expression constructs are shown in supplemental Fig. S1.
Mass Spectrometry
Purified N-terminal peptides were analyzed on an LTQ Orbitrap Velos (Thermo Fisher Scientific, San Jose, CA) coupled with an Eksigent nanoLC-Ultra 1D plus system (Dublin, CA). Peptides were separated on a PicoFrit analytical column (100 mm long, ID 75 μm, tip ID 10 μm, packed with BetaBasic 5 μm 300Å particles, New Objective, Woburn, MA) using a 120-min linear gradient of 5–35% acetonitrile (ACN) in 0.1% formic acid (FA) at a flow rate of 250 nL/min. Mass analysis was carried out in data-dependent analysis mode, where MS1 scanned full MS mass range from m/z 300 to 2000, at 30,000 mass resolution, and 10 CID MS2 scans were sequentially carried out in the Orbitrap and the ion trap, respectively.
Database Searching
Tandem mass spectra were extracted, charge-state deconvoluted, and deisotoped by Extract_msn from Xcalibur version 2.0. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.4.0) and X! Tandem (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)). Mascot was set up to search the Sp_Trembl_112812 database selected for Mammalia (1026970 entries), assuming the digestion enzymes caspase (any D) and trypsin, and allowing up to 6 missed cleavages. X! Tandem was set up to search same Sp_Trembl database selected for all species (28934417 entries), also assuming trypsin, allowing two missed cleavages. Mascot and X! Tandem were searched with a fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 20 PPM. Carbamidomethyl of cysteine and SerTyr on the N terminus were specified in Mascot and X! Tandem as fixed modifications. Glu->pyro-Glu of the N terminus, ammonia-loss of the N terminus, gln->pyro-Glu of the N terminus, oxidation of methionine and acetyl of the N terminus were specified in X! Tandem as variable modifications. Oxidation of methionine and acetyl of the protein N terminus were specified in Mascot as variable modifications.
Criteria for Protein Identification
Scaffold (version Scaffold_4.0.3, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability. Peptide Probabilities from X! Tandem were assigned by the Peptide Prophet algorithm (31) with Scaffold delta-mass correction. Peptide Probabilities from Mascot were assigned by the Scaffold Local FDR algorithm.
Protein identifications were accepted if they could be established >99.0% probability and contained at least 1 identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm (32). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. The resulting peptide false discovery rate (FDR) and protein FDR were 0.0% using a decoy database.
MALDI Peptide Analysis
MALDI MS analysis was performed using an Applied BioSystems Voyager DE-sSTR (Applied Biosystems, Framingham, MA, USA) MALDI-TOF mass spectrometer. Analysis of Gap43 peptides was performed using standard protocols. Briefly, a 0.5 ml aliquot of matrix, a saturated solution of sinapinic acid in 1:1 acetonitrile/0.1% trifluoroacetic acid (TFA), was applied to the MS sample plate and allowed to dry. Then an equal volume of the protein sample was spotted on top of the dried matrix and was also allowed to dry in air, followed by another equal volume of matrix. Spectra were accumulated in positive linear mode (25,000 V and 750 nsec delayed extraction), over a mass range from 5000 to 50,000 Da and collecting 1000 laser shots/spectra.
Recombinant Gap43 Expression and Purification
E. coli BL21 (DE3) cells transformed with the pET30a-GNSHT-Gap43 construct were grown at 37 °C in LB broth supplemented with 50 μg/ml of kanamycin. When the cells reached the mid-log phase (OD600 ∼0.5), the temperature was reduced to 30 °C and IPTG was added to a final concentration of 1 mm to induce protein expression. After 4 h of induction, cells were collected by centrifugation, resuspended in lysis buffer (10 ml/1.5g wet cells) containing 50 mm sodium phosphate (pH 7.4), 300 mm NaCl, 10 mm imidazole, and protease inhibitors (Roche, Indianapolis, IN), and disrupted using a sonicator. The cell lysate was centrifuged at 20,000g for 20 min at 4 °C. The supernatant was applied onto a column packed with HisPur cobalt resins (1 ml/1.5g wet cells). The flow-through was re-applied onto the same column twice to maximize binding. Resins were washed three times with five-bed volumes of the lysis buffer. Bound proteins were eluted with five-bed volumes of elution buffer containing 50 mm sodium phosphate (pH 7.4), 300 mm NaCl, and 150 mm imidazole. After removing imidazole with the Amicon Ultra-4 -10K centrifugal filter unit (Millipore), purified proteins were digested with TEV protease (1:50, w/w) overnight at 4 °C. The GNSH tag and TEV protease were removed with the same column regenerated by washing with a buffer containing 20 mm MES, 100 mm NaCl (pH 5.0). Purified, tagless proteins were examined by SDS-PAGE and stored at −80 °C.
Substrate Cleavage Assay
For in vivo caspase cleavage assay, Hela cells were transfected for 4–5 h with vectors expressing candidate caspase-3 substrates alone or cotransfected with the caspase-3 construct using GenJet in vitro DNA transfection reagent (Ver. II) (SignaGen Laboratories, Rockville, MD). For the control experiment, the caspase inhibitor Z-VAD-FMK (200 μm) was then added to the medium. Approximately 16 h after transfection, cells were treated with 10 ng/ml TNF-α and 20 μg/ml cycloheximide for 0, 3, and 6 h to induce apoptosis. Cells were washed with PBS, harvested, and lysed with lysis buffer (TBS, 0.5% Triton X-100, and protease inhibitors). Lysates were spun at 16,100 × g for 10 min at 4 °C and analyzed by immunoblotting against V5.
For in vitro caspase-3 cleavage assay, proteins synthesized using TNT Quick Coupled Transcription/Translation System (Promega) were incubated with active recombinant human caspase-3 (R&D Systems, Minneapolis, MN) with or without the caspase inhibitor Z-VAD-FMK (200 μm) in the reaction buffer (25 mm HEPES, pH 7.3, 0.1% CHAPS, and 10 mm DTT) at 37 °C for the times indicated. Cleavage was determined by SDS-PAGE and immunoblot.
Subcellular Fractionation
Subcellular fractionation of mouse whole-brain tissues was carried out as described (33), with modifications. Brain tissues (C57BL/6 mouse, 1 month old) were homogenized in buffer A (0.32 m sucrose, 1 mm NaHCO3, 1 mm MgCl2, 0.5 mm CaCl2, 5 mm NaF, and 1 mm Na3VO4) supplemented with protease inhibitors using a Dounce tissue grinder (tight pestle, 20 strokes). Cell debris and nuclei were removed by a low speed centrifugation at 1,400g for 10 min to obtain the post-nuclear protein fraction (T). The T fraction was centrifuged again at 13,800 × g for 10 min. The resulting supernatant was collected as the cytosolic fraction (Cyt). The pellet was re-suspended in buffer B (0.32 m sucrose and 1 mm NaHCO3) and separated on a discontinued sucrose gradient (0.85, 1.0 and 1.2 m) by centrifugation at 82,500 × g with a Beckman SW28 rotor at 4 °C for 2 h. Following centrifugation, the band between 1.0 and 1.2 m sucrose was recovered, diluted 1:10 with buffer B, and centrifuged at 82,500 × g with a Beckman SW28 rotor at 4 °C for 30 min. The resulting pellet was resuspended in buffer B as the synaptosome fraction (Syn). The Syn fraction was centrifuged again and extracted twice with 20 mm Tris-HCl (pH 6.0, 1% Triton X-100), followed by another two extractions with 20 mm Tris-HCl (pH 8.0, Triton X-100). The extracted proteins (the supernatants) were pooled, precipitated with 10 volumes of cold acetone (−20 °C) for overnight at 4 °C, centrifuged, and resuspended in buffer B to obtain the Triton X-100 soluble fraction (Triton). The final pellet after Triton X-100 extractions was the PSD fraction and was resuspended in buffer B. Protein concentration was assessed with the BCA protein assay kit (Thermo Scientific) using BSA as standard. 30 μg of each fraction was applied for immunoblot analysis.
Primary Neuronal Culture, Neuron Transfection, Immunocytochemistry and Imaging
Cultures of hippocampal and cortical neurons were prepared from embryonic (day E18-E19) rat embryos and grown on cover slips in Neurobasal medium supplemented with 2% B27, 2 mm glutamax, and antibiotics (Penicillin Streptomycin). All reagents for neuronal cultures were purchased from Invitrogen. Neurons were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For immunocytochemistry, neurons at DIV17∼18 were fixed with 4% formaldehyde and 4% sucrose in PBS at room temperature for 15 min and permeabilized with cold methanol (−20 °C) for 1 min. For the staining of PSD95, neurons were fixed with cold methanol (−20 °C) for 10 min. After washing with PBS, neurons were incubated with primary antibodies diluted in GDB buffer (0.2% gelatin, 0.5% Triton X-100 and 0.8 m NaCl in 30 mm phosphate buffer, pH 7.4) overnight at 4 °C, followed by rinse with PBS for three times. Neurons were then incubated with secondary antibodies in GDB buffer for 1 h at room temperature, washed with PBS, and mounted with the mounting medium (Vector labs, Burlingame, CA). Fluorescence images were taken using a Ziess LSM 510 confocal microscope with a 63x objective (N.A. 1.40). For live imaging, neurons (DIV14–15) were transfected with the Gap43-mCherry or Gap43 (D23/84/90/167A)-mCherry along with EGFP-GluR2 constructs, and imaged at 3 days after transfection using an Olympus FLUOVIEW FV1000 confocal microscope with a 60× objective (N.A. 0.90).
Hippocampal Slice Culture
Hippocampal slice cultures were prepared from Sprague-Dawley rats (P6–8). After decapitation, the brain was transferred immediately to icy cutting solution (238 mm sucrose, 2.5 mm KCl, 26 mm NaHCO3, 1 mm NaH2PO4, 5 mm MgCl2, 11 mm d-glucose, and 1 mm CaCl2) and cut with a vibratome (Leica, Buffalo Grove, IL). Hippocampal slices (350 μm) were incubated on semipermeable membrane inserts (Millipore) in a six-well plate containing culture medium (78.8% minimum essential medium, 20% heat-inactivated horse serum, 25 mm HEPES, 10 mm d-glucose, 26 mm NaHCO3, 2 mm CaCl2, 2 mm MgSO4, 0.0012% ascorbic acid, 1 μg/ml insulin; pH 7.3; 320–330 mOsm). Neurons were biolistically transfected using the gene gun (Helios Gene-gun system, Bio-Rad) at DIV3–4. Electrophysiological recordings were performed at 2–3 days after transfection.
Electrophysiology
Hippocampal slices were recorded under 30 °C in ACSF (bubbled with 95% O2/5% CO2) at the rate of 2 ml/min. 100 μm bicuculline and 2 μm 2-chloroadenosine were applied to ACSF for blocking GABAA receptors and preventing bursting. EPSCs of CA1 pyramidal cells evoked by a stimulating electrode on the stratum radiatum in CA2 region were recorded in the whole-cell voltage-clamp mode at the holding potential of -70 mV. The patch pipette (4–7 MΩ) solution contained 130 mm cesium methanesulfonate, 8 mm NaCl, 4 mm Mg-ATP, 0.3 mm Na-GTP, 0.5 mm EGTA, 10 mm HEPES, and 5 mm QX-314 at pH 7.3.
LTD was induced by pairing low-frequency stimulations (1 Hz) with a postsynaptic depolarization to -45 mV for 300 s, whereas LTP was induced by pairing 2 Hz stimulations with a postsynaptic depolarization to 0 mV for 100 s. To measure the input-output curve, the averaged amplitude of eight consecutive EPSCs recorded at 0.05 Hz was plotted against the intensity of test stimuli. Series resistance and input resistance were monitored on-line and analyzed with the Clampex program off-line. Only cells with a series resistance of < 25 mΩ and a < 10% drift in both series, and input resistance during the recording period, were included. The student's two-tailed t test was used for statistical analysis (p < 0.05 considered significant).
AMPA Receptor Internalization Assay
Neurons were transfected with the β-galactosidase construct along with the Gap43 constructs at DIV14. At 3 days after transfection, neurons were treated with TTX (1 μm) for 30 min before the internalization assay. After TTX treatment, neurons were incubated with an antibody against the N-terminal region of GluA2 (10 μg/ml in neural medium containing 1 μm TTX) for 15 min at 37 °C to label surface AMPA receptors. After rinse with warm DMEM, neurons were stimulated with NMDA (30 μm) for 5 min at 37 °C to induce AMPA receptor endocytosis. Neurons were rinsed in warm DMEM, and returned to neural medium containing 1 μm TTX to allow AMPA receptor endocytosis for 10 min at 37 °C, followed by fixation with 4% formaldehyde (in PBS containing 4% sucrose) at room temperature for 7 min. After washing with PBS, neurons were incubated with Alexa-555 conjugated secondary antibodies (diluted in the ADB buffer, 3% goat serum and 0.1% BSA in PBS, pH 7.4) overnight at 4 °C to label surface remaining GluA2. Neurons were then permeabilized with methanol at −20 °C for 90 s, and incubated with the β-galactosidase antibody for 2 h at room temperature. After washing with PBS, neurons were incubated with the Alexa-488 conjugated secondary antibody against the GluA2 antibody to label internalized GluA2 and Alexa-647-conjugated secondary against the β-galactosidase antibody for 1 h at room temperature, then mounted in the mounting medium. Confocal images of transfected neurons were acquired using a Zeiss LSM510 laser scanning confocal microscope (63× objective, N.A. 1.40), and analyzed using the Metamorph software. Image acquisition and image analysis were performed blindly to the treatment. The student's two-tailed t test was used for statistical analysis (p < 0.05 considered significant).
RESULTS
Identification of Caspase-3 Substrates in Neurons
As demonstrated in our earlier study, in NMDAR-LTD, caspase-3 activity is required for AMPA receptor endocytosis (16). Because caspase-3 is a protease, we postulated that caspase-3 promotes AMPA receptor endocytosis by cleaving and thereby changing the function of proteins involved in endocytosis. To test this hypothesis, we adapted a method developed by Mahrus et al. to selectively capture emergent peptides derived from proteolysis to identify proteins cleaved by caspases in neurons (23). To increase the chance of detecting caspase-3 cleavage products, we used apoptotic neurons in which caspase-3 was robustly activated. Cultured cortical neurons (DIV18) were treated with staurosporine (STS, 1 μm) for 8 h to induce apoptosis. Using subtiligase, polypeptides with unblocked α-amines at the N terminus including those derived from caspase-3 cleavage were tagged with biotinylated peptides containing a TEV protease cleavage site (Figs. 1A and 1B). Because the majority of mammalian proteins are modified by acetylation at the N terminus, subtiligase-labeled peptides are enriched for proteolytic products. The labeled polypeptides were isolated by affinity purification with avidin beads, followed by on-bead trypsin digestion and then TEV protease elution to obtain their N-terminal portions. The eluted peptides, which had a characteristic N-terminal SerTyr (SY)-dipeptide resulting from TEV protease cleavage, were analyzed by LC-MS/MS.
Fig. 1.

Subtiligase-based identification of peptides derived from caspase cleavage. A, Schematic diagram of subtiligase-based substrate identification method. B, Sequence of synthesized peptide glycolate ester and cleavage sites for subtiligase and TEV protease. C, Nomenclature for residues in the cleavage sites of endopeptidase substrates. For caspase substrates, the P1 position is Asp.
Proteomic analysis was replicated in two different cultures of cortical neurons. A large number (950–2000 for each sample) of unique SY-tagged N-terminal peptides were identified by Mascot. As caspase-3 cleaves after Asp, peptides derived from caspase-3 cleavage contain Asp residues in the P1 position (Fig. 1C). Using the constraints of Asp at the P1 site and more than a single spectral match with scores exceeding 95% confidence, 25 unique peptides (24 unique cleavage sites) in the untreated control, and 85 unique peptides (81 unique cleavage sites) in the STS-treated cells were identified as putative caspase-cleaved products (supplemental Table S1 and S2). All P1-Asp cleavages identified in untreated neurons were also found in STS treated cells. The presence of caspase-3 cleavage products in untreated cells is consistent with our previous observation that cultured neurons contain a low level of active caspase-3 (16).
The identified peptides align to 56 proteins, 19 of which are cleaved in both control and STS treated cells. Of these 56 proteins, 19 (18 human orthologs) are reported on the CAspase Substrate dataBAse Homepage (CASBAH, http://bioinf.gen.tcd.ie/casbah/), and 43 are also in the recently established DegraBase (http://wellslab.ucsf.edu/degrabase/index.htm) derived from human samples analyzed with the same methods used in this study (20, 21). We analyzed the P4 to P1′ motif composition of putative caspase-3 substrates identified from STS treated neurons. As with apoptotic Jurkat cells (23), only ∼30% of the motifs we identified conform to the canonical sequence (loosely defined as DXXD, P4-P1) for caspase-3 cleavage sites. Rather, the most frequently found amino acid is aspartate (30%) at P4, leucine (17%) at P3, threonine (17%) at P2, and glycine (36%) at P1′ position (Fig. 2A). Taken together, using the subtiligase-based method, we identified a large number of putative caspase-3 substrates in neurons, including a number of newly recognized ones.
Fig. 2.

Motif composition, Gene Ontology analysis and Ingenuity Pathway Analysis of putative caspase substrates. A, Sequence-logo plot representing amino acid frequency in P4 to P1′ at 81 unique putative caspase cleavage sites identified in STS-treated neurons (generated by WebLogo, http://weblogo.berkeley.edu/logo.cgi). B, Biological Process Gene Ontology (GO) terms annotating the 56 putative caspase substrates identified in STS-treated neurons (NCBI annotations, analyzed by Scaffold). The numbers of substrates allocated to each GO term are indicated. It should be noted that one substrate could be annotated by multiple GO terms. C, The highest-scored network (score 63, focus molecular 25) identified by Ingenuity Pathway analysis of the 56 putative substrates. Colored nodes indicate putative substrates used in the pathway analysis.
Cellular Processes Regulated by Neuronal Caspase-3 Substrates
The large number of caspase-3 substrates we identified suggests that caspase-3 activation is likely to have pleiotropic effects on cell physiology. To comprehensively interrogate the biological functions and pathways influenced by caspase-3-mediated proteolysis, we analyzed all 56 putative caspase-3 substrates found in this study with the Gene Ontology (GO) and the Ingenuity Pathway Analysis software (IPA, Ingenuity® Systems, www.ingenuity.com). The Biological Process GO terms annotating these putative substrates are shown in Fig. 2B. The network with the highest association score (score 63, focus molecule 25) revealed by the Ingenurity Pathway analysis is that devoted to “Cellular Assembly and Organization, Cellular Function and Maintenance, Nervous System Development and Function.” Key molecules in this network include: calmodulin, F-actin, actin, MAP2, Gap43, tubulin, calpain, and ERK1/2 (Fig. 2C). The other two functional networks we identified are: “Cellular Assembly and Organization, Cellular Development, Embryonic Development” (score 38, focus molecule 17), and “Cellular Assembly and Organization, Cell-To-Cell Signaling and Interaction, Cellular Function and Maintenance” (score 22, focus molecule 11) (supplemental Fig. S2). Interestingly, all three of these networks are related to cellular assembly and organization, suggesting that a major function of neuronal caspase substrates is the maintenance of cellular integrity.
Gap43, Dbn1, and BASP1 are True Caspase-3 Substrates
To validate our mass spectrometry results, we tested whether caspase-3 does indeed cleave the identified proteins. For this test, we selected growth associated protein 43 (Gap43), drebrin (Dbn1), MARCKS-related protein (MARCKSL1), and brain acid soluble protein 1 (BASP1), because they interact with the cellular machinery and signaling molecules potentially related to endocytosis, such as cytoskeleton and PIP2 (34–37), and as suggested by our IPA analysis, they act in common signaling networks (Fig. 2C).
To test whether these proteins are cleavable by caspase-3, we first conducted an in vivo caspase cleavage assay in apoptotic Hela cells. Cells transfected with constructs expressing V5 tagged Gap43, Dbn1, BASP1, or MARCKSL1 along with a construct expressing caspase-3 were treated with TNF-α and cycloheximide (CHX) to induce apoptosis. At 3 and 6 h after apoptosis induction, cell lysates were collected for immunoblotting against V5. Although the levels of all candidate substrates were only slightly decreased by TNF-α/CHX stimulation when transfected alone, overexpression of caspase-3 dramatically increased this effect (Fig. 3A). Addition of the caspase inhibitor Z-VAD-FMK, moreover, obliterated the cleavage of transfected candidates, indicating that it resulted from cleavage by caspases (Fig. 3A). Not surprisingly, apoptosis induction had no effect on β-tubulin, a protein neither identified by our mass spectrometric analysis nor previously reported as a caspase-3 substrate (Fig. 3A). There were no fragments of the C-terminal V5-tagged proteins detected on gel. It is likely that abundant active proteases in apoptotic Hela cells further cleave the caspase-3 fragments, removing the V5 tag.
Fig. 3.

Caspase-3 cleavage assay for Gap43, Dbn1, BASP1, and MARCKSL1. A, In vivo caspase-3 cleavage assay. Hela cells were transfected for 4–5 h with constructs expressing V5-tagged candidate caspase-3 substrates alone (Gap43, Dbn1, BASP1, or MARCKSL1) or cotransfected with a construct expressing caspase-3. To confirm the reduction of the tested protein is because of caspase activation, the caspase inhibitor Z-VAD-FMK (200 μm) was applied to the medium throughout the same test right after transfection. About 16 h after transfection, the cells were treated with TNF-α and cycloheximide (CHX) to induce apoptosis. Cell lysates were collected at 0, 3, and 6 h after apoptosis induction for immunoblotting against V5. B, In vitro caspase-3 cleavage assay. The V5 tagged candidate genes were expressed in vitro. Synthesized proteins were incubated with recombinant active caspase-3 (50 ng) alone or along with the caspase inhibitor Z-VAD-FMK (200 μm) for 16 h at 37 °C, then analyzed by immunoblotting against V5.
Because the cleavage of transfected proteins in apoptotic cells can be caused by caspases other than caspase-3, we next performed an in vitro assay to assess proteolysis by caspase-3 directly. V5-tagged candidate genes expressed in vitro (using the TNT Quick Coupled Transcription/Translation System) were incubated with recombinant active caspase-3. Following incubation with active caspase-3, the amount of Gap43, Dbn1, and BASP1, but not of MARCKSL1, was reduced, and z-VAD-FMK abolished this reduction (Fig. 3B). These results confirm that Gap43, Dbn1, and BASP1 are indeed caspase-3 substrates, whereas MARCKSL1 is likely to be a substrate of other caspases.
Gap43 Mutants Deficient in Caspase-3 Cleavage Inhibit NMDAR-LTD
We have previously shown that casapse-3 activity is required for the induction of NMDAR-LTD (16). To determine whether the caspase-3 substrates identified by our proteomics study are involved in LTD, we replaced P1 Asp at their caspase-3 cleavage sites with Ala to generate caspase-resistant mutants. Our proteomics analysis identified two P1 Asp in Gap43 (D84, D167), three in BASP1 (D135, D159, D166), and one in Dbn1 (D529) (supplemental Table S1). We mutated these Asp residues, and tested the mutants with in vitro caspase-3 cleavage assay. The mutant Gap43 (D84/167A), Dbn1 (D529A), and BASP1 (D135/159/166A) were proteolytically cleaved to a lesser extent than their wild-type proteins (supplemental Fig. S3), confirming that the caspase cleavage sites we identified are indeed valid. The remaining cleavability observed in the mutant proteins suggests the existence of unidentified caspase-3 cleavage sites.
Next, we biolistically transfected cultured hippocampal slices with constructs expressing either wild-type or mutant caspase-3 substrates along with a plasmid expressing Venus (a YFP mutant) (38). NMDAR-LTD was induced by stimulating the Schaffer collateral pathway with the pairing low-frequency stimulation protocol (see Materials and Methods). Excitatory postsynaptic currents (EPSCs) were examined by whole-cell recordings in the CA1 region. As shown in Figs. 4A, 4D, supplemental Fig. S4, and supplemental Table S3, LTD was comparable in control plasmid, Gap43 (WT), Dbn1 (WT and D529A), and BASP1 (WT and D135/159/166A) transfected cells as revealed by a reduction of EPSCs measured at 30 min after stimulation. Transfection of wild-type MARCKSL1, which is not a casapse-3 substrate, or its mutant, which is mutated in the cleavage site identified by proteomics, also left LTD intact (supplemental Fig. S4 and supplemental Table S3). By contrast, LTD was reduced in cells transfected with Gap43 single and double mutants (Figs. 4B–4D and supplemental Table S3). To further investigate the effects of Gap43 overexpression on electrophysiological properties, we analyzed basal synaptic transmission in hippocampal slices transfected with a construct expressing wild-type Gap43 or the control plasmid. As shown in supplemental Fig. S5A, EPSCs elicited by stimulation at various intensities are comparable in Gap43 overexpressing and control neurons, suggesting that basal synaptic strength is not affected by Gap43 overexpression. Taken together, these results indicate that LTD induction requires caspase-3 cleavage of Gap43, but not of Dbn1or BASP1.
Fig. 4.

Cleavage of Gap43 by caspase-3 is required for LTD induction. Cultured hippocampal slices (Div 6–7) were biolistically transfected with a construct expressing the visual marker Venus along with an empty vector, or constructs expressing wild-type or mutant caspase-3 substrates. At 2–3 days after transfection, LTD was induced by low-frequency stimulation of the Schaffer collateral pathway. For clear visualization of EPSC data collected from different conditions, EPSCs recorded from CA1 neurons transfected with the empty vector or wild-type Gap43 were shown in (A), those from cells transfected with the empty vector or single Gap43 mutants in (B), and those from cells transfected with the empty vector, double or quadruple Gap43 mutants in (C). D, EPSCs at 30 min after LTD induction were normalized to pre-induction baselines. Sample traces of EPSCs collected at 10 min before (1) and at 30 min after LTD induction (2) are displayed and superimposed (1 + 2) on the top of histograms in (A–C). For each condition, the EPSC amplitude, which was normalized to baseline before LTD induction, was plotted as mean ± S.E. Two-tailed Student's t test was used for statistical analysis. n = 9–10 transfected neurons for each group; *p < 0.05, **p < 0.01, compared with cells transfected with the empty vector.
Having established that Gap43 is a caspase-3 substrate and that it is required for LTD, we next sought to identify additional caspase-3 cleavage sites possibly missed using the subtiligase-based method. Toward that end, we analyzed peptides derived from in vitro caspase-3 cleavage of recombinant Gap43. Two methods were used to identify Gap43 peptides: matrix-assisted-laser-desorption-ionization mass spectrometry (MALDI/MS) and LC-MS/MS. For MALDI/MS, the theoretical peptide mass fingerprint of Gap43 was generated in silico (choosing “microwave-assisted hydrolysis with formic acid” option), which cleaves proteins specifically after Asp residues and therefore mimics caspase-3 cleavage. Peptides were identified using PeptideMass (http://web.expasy.org/peptide_mass/) allowing 5 missed cleavages, because caspase-3 does not cleave at every Asp. MALDI analysis confirmed the cleavage at D167 and revealed four additional cleavage sites at: D23, D76, D90, and D140. For LC-MS/MS, recombinant Gap43 was sequentially digested with active caspase-3 and trypsin, and then subjected to LC-MS/MS analysis. This analysis identified three cleavage sites at: D23, D167, and D202.
Using all three methods (subtiligase-based method, MALDI, and double digestion followed by LC-MS/MS), we identified seven caspase-3 cleavage sites in Gap43 (supplemental Fig. S6 and supplemental Table S4). We also found that in the in vitro cleavage assay, mutant Gap43 with mutations at four of these sites (23/84/90/167) was more resistant to proteolysis by caspase-3 than the Gap43 (D84A), Gap43 (D167A), or Gap43 (D84/167A) mutants (supplemental Fig. S3), suggesting that caspase-3 does indeed cleave Gap43 at these sites. In addition, LTD was reduced in neurons transfected with Gap43 (D23/84/90/167A) to a larger extent than in those transfected with singly- or doubly mutated Gap43 (Figs. 4C, 4D and supplemental Table S3).
To test whether Gap43 mutants also affect other forms of synaptic plasticity, we analyzed long-term potentiation of synaptic transmission (LTP, a form of synaptic plasticity characterized by increases in synaptic strength) in cultured hippocampal slices transfected with the control vector or Gap43 (D23/84/90/167A). LTP in the CA1 region was induced by stimulating the Schaffer collateral pathway with two trains of high frequency stimulations (100 pulses at 100 Hz, 20 s intervals). EPSCs were recorded in CA1 neurons by whole-cell patch. After high frequency stimulations, there were comparable increases in EPSCs in neurons transfected with Gap43 (D23/84/90/167A) and controls (supplemental Fig. S5B). This suggests that the cleavage of Gap43 by caspase-3 is specifically involved in LTD, but not in LTP.
Taken together, these results indicate that the cleavage of Gap43 by caspase-3 is required for LTD induction.
Gap43 Mutants Deficient in Caspase-3 Cleavage Inhibit AMPA Receptor Endocytosis
AMPA receptor endocytosis is a critical step in NMDA receptor-dependent LTD and requires caspase-3 activity (1–3). Hence, we examined whether cleavage of Gap43 by caspase-3 is involved in AMPA receptor endocytosis. An antibody feeding assay was used to analyze the endocytosis of AMPA receptor subunit GluA2 (16). Dissociated hippocampal neurons (DIV14) were transfected with the control plasmid, wild-type Gap43 or Gap43 mutants (D84A, D167A, D84/167A, or D23/84/90/167A). For visualization of transfected cells, these were all cotransfected with a plasmid expressing β-galactosidase. At 2–3 days after transfection, neurons were stimulated with NMDA (30 μm, 5 min), a protocol inducing “chemical LTD” which shares mechanisms with electrically induced LTD (39). As shown in Fig. 5, all Gap43 mutants inhibit NMDA-induced GluA2 internalization. Basal GluA2 internalization without NMDA stimulation, by contrast, was not affected by transfection with Gap43 constructs (supplemental Fig. S7). These results indicate that cleavage of Gap43 by caspase-3 is required for NMDA-induced but not basal AMPA receptor endocytosis.
Fig. 5.
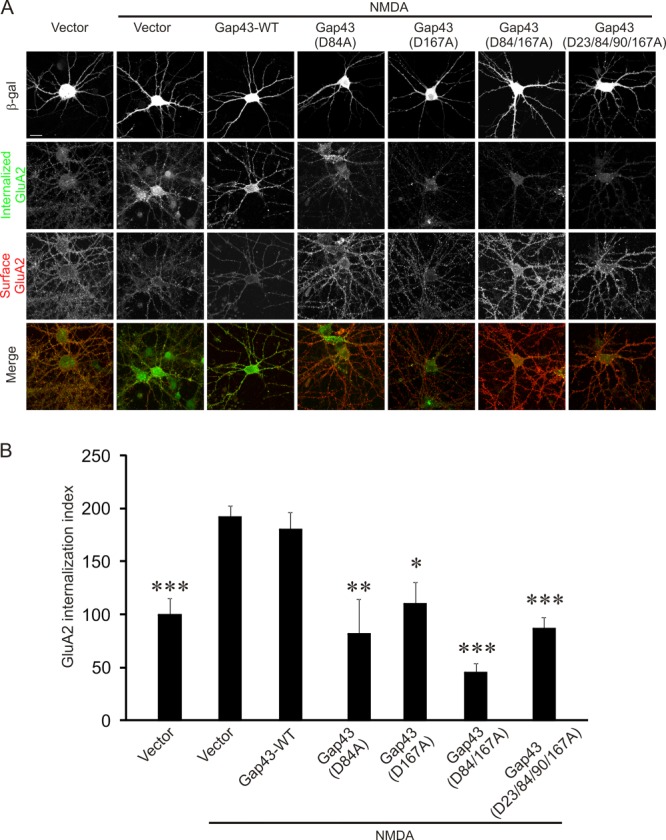
Mutant Gap43 deficient in caspase-3 cleavage blocks NMDA-induced internalization of AMPA receptors. Cultured hippocampal neurons were transfected with constructs expressing wildtype or mutant Gap43 together with a plasmid expressing β-galactosidase for visualizing transfected cells. At 3 days after transfection, NMDA (30 μm, 5 min) was added to induce AMPA receptor endocytosis. A, Representative images of transfected neurons. The last row shows merged images of internalized (green) and surface-remaining (red) GluA2. B, Quantification for transfected neurons in (A). The internalization index (integrated fluorescence intensity of internalized GluA2/integrated fluorescence intensity of internalized GluA2 plus surface GluA2) was normalized to unstimulated control cells transfected with the empty vector. n = 15 neurons for each group. Two-tailed Student's t test was used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001, compared with NMDA treated cells transfected with the control plasmid. The graph shows mean ± S.E. Scale bar: 20 μm.
Gap43 is Localized at the Postsynaptic Site
Gap43 is highly expressed in axonal growth cones during development and axon regeneration (35, 40). Because our electrophysiology and endocytosis analyses show that Gap43 mutants inhibit LTD and AMPA receptor internalization when transfected into postsynaptic neurons, we decided to investigate whether Gap43 is, in fact, also localized postsynaptically. To test this hypothesis, we used subcellular fractionation to examine Gap43 distribution in mouse brains. The brain homogenate was separated into the cytosolic fraction, the synaptosome (enriched for heavy membranes, including both pre- and post-synaptic proteins), the Triton-soluble fraction (enriched for presynaptic proteins), and the postsynaptic density fraction (PSD, enriched for postsynaptic proteins). The specificity of these fractions was confirmed by enrichment of presynaptic (synaptophysin) and postsynaptic (PSD-95, GluA2, and GluN2A) proteins (Fig. 6A). Notably, Gap43 was detected in all fractions, including the PSD fraction and Triton-soluble fraction (Fig. 6A), suggesting that it is localized at both postsynaptic and presynaptic sites.
Fig. 6.
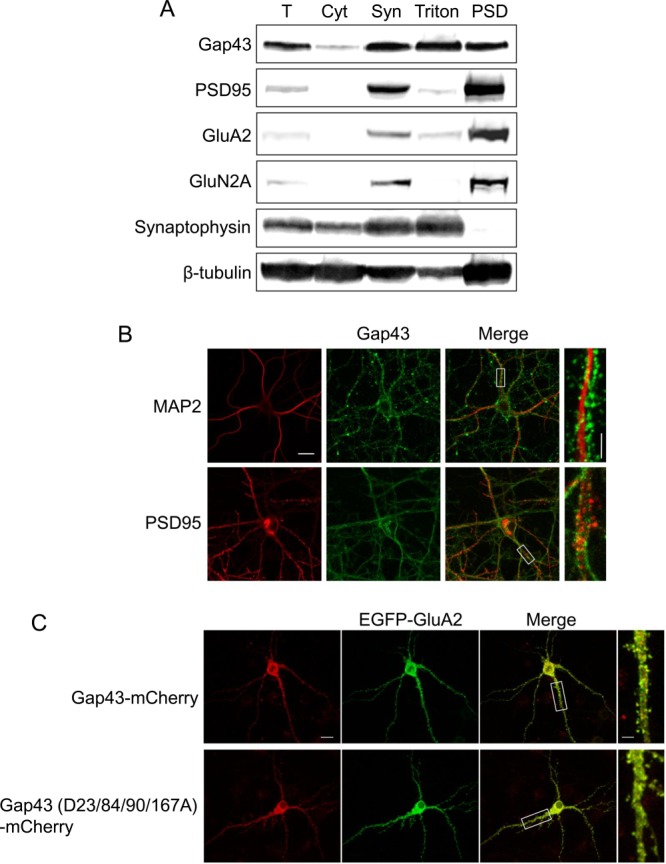
Subcellular distribution of Gap43. A, Immunoblotting of Gap43, the postsynaptic markers GluA2, GluN2A and PSD-95, and the presynaptic marker synaptophysin in subcellular fractions prepared from mouse brains. Fractions: T, total post-nuclear proteins; Cyt, cytosol; Syn, synaptosome; Triton, Triton-soluble presynaptic proteins; and PSD, postsynaptic density. B, Immunostaining of Gap43, MAP2, and PSD95 in hippocampal neurons (DIV17∼18). C, Images of live hippocampal neurons (DIV17∼18) transfected with Gap43-mCherry or Gap43 (D23/84/90/167A)-mCherry along with EGFP-GluA2 constructs. Scale bar: 20 μm in the left three images, and 5 μm in the enlarged image on the right in B and C.
To confirm the postsynaptic localization of Gap43, we stained cultured hippocampal neurons (DIV17∼18) with an antibody against Gap43, as well as with antibodies against the dendritic marker MAP2 and the postsynaptic protein PSD-95 (Fig. 6B). Gap43 was found in somas, axons, and dendrites and displayed a punctate pattern. In dendrites, we found that Gap43 outlines MAP2, suggesting that Gap43 is predominantly localized on the surface of dendrites. This finding is consistent with earlier reports that Gap43 is a membrane-bound protein (35). We also observed some puncta that were doubly-stained by both the Gap43 and PSD-95 antibodies, indicating that Gap43 is partially colocalized with PSD-95.
It should be noted that the punctuate pattern of Gap43 staining we observed could possibly derive from fixation during immunocytochemistry (34). To circumvent this potential problem, we examined the subcellular distribution of Gap43 in live neurons. To this end, we transfected cultured hippocampal neurons (DIV17∼18) with a construct expressing Gap43 fused with mCherry along with a construct expressing EGFP-tagged GluA2, which allowed us to acquire confocal images from live cells. In these transfected cells, we observed both diffuse and clustered Gap43 throughout axons and dendrites. GluA2, by contrast, did not appear in axons and was exclusively localized in dendrites (Fig. 6C and supplemental Fig. S8). Consistent with the immunocytochemistry results, moreover, some puncta contained both Gap43 and GluA2, indicating that a fraction of Gap43 is, indeed, localized postsynaptically. Caspase-resistant mutant Gap43 (D23/84/90/167A), moreover, had a similar subcellular distribution as wild-type Gap43 (Fig. 6C and supplemental Fig. S8), suggesting that the inhibition of LTD and AMPA receptor endocytosis by mutant Gap43 is not caused by ectopic expression of Gap43.
Taken together, these results indicate that Gap43 is present at the postsynaptic site.
DISCUSSION
In this study, we asked how caspase-3 activation promotes AMPA receptor endocytosis and LTD. We addressed this question by identifying caspase-3 substrates with proteomics and testing the identified substrates with electrophysiology and the endocytosis assay. Although earlier studies have identified >1000 putative caspase substrates in nonneuronal cells (20, 21), the identification of neuronal caspase-3 substrates, and their possible involvement in LTD, had yet to be established. To our knowledge, ours is the first systematic study of caspase-3 substrates in neurons.
We applied a subtiligase-based method to capture the newly formed N-terminal peptides generated by proteolysis in neurons (23). These were then subjected to mass spectrometric analysis for identification. The subtiligase method has two advantages: (1) its affinity enrichment step greatly increases sensitivity for the detection of rare substrates, a feature that is especially important for primary neurons because they are far more costly to culture in large quantities than cell lines; (2) the tag added by subtiligase enables us to identify the exact cleavage site, which can then be mutated for the functional validation of the substrates.
We used apoptotic neurons rather than neurons undergoing LTD for our proteomics analysis, because cultured neurons have a low level of basal caspase-3 activity and caspase-3 activity is elevated only moderately (∼two to threefold) and transiently during LTD (16). After STS treatment, by contrast, caspase-3 activity in neurons is increased up to 10–20-fold (16). Hence, more peptides derived from caspase cleavage can be recovered for proteomics from apoptotic than from NMDA treated neurons.
Our study identified 56 putative caspase substrates. Because it was impossible to conduct functional studies for all of them in a single study, we conducted a literature-based IPA analysis to determine cellular processes and signaling pathways associated with them. This analysis revealed their associations with three networks whose major functions include cellular assembly, organization, development, function, signaling, and maintenance. Interestingly, the highest-scored network identified by IPA mainly comprises neuronal cytoskeletal components and their regulators (such as actin, MAP2, Gap43, Dbn1, and calmodulin), some of which are known to be important for AMPA receptor endocytosis. From this network, four putative caspase substrates—Gap43, Dbn1, BASP1, and MARCKSL1—were selected for further study.
Of the four substrates selected, Gap43 and BASP1 are localized in lipid rafts (34, 41); Dbn1 is a substrate of caspase-6 (42) whose relationship to caspase-3 is unknown; and MARCKSL1 was previously identified as a putative caspase substrate in apoptotic Jurkat cells (23). Although we found that all four substrates were cleaved by caspases in apoptotic Hela cells, in the in vitro caspase-3 cleavage assay, only Gap43, Dbn1, and BASP1 were cleaved by caspase-3. These results suggest that, because multiple caspases are activated in apoptotic cells, a direct cleavage test is necessary to validate the substrates.
Although we mutated P1 Asp in the cleavage sites identified by the subtiligase method, the mutants were still proteolytically cleaved by caspase-3, but to a lesser degree than the wild-type proteins. This may be due to cleavage at unidentified sites, because LC-MS/MS tends to miss peptides that are unusually long or short (23). To make up for this limitation of the subtiligase-based method, we subjected the peptides derived from direct cleavage of recombinant Gap43 by caspase-3 to MALDI-MS and LC-MS/MS analysis, which revealed five additional cleavage sites. The predicted N-terminal peptides derived from cleavage at these sites in the subtiligase assay are 8, 7, 6, 5, and 4 residues in length (without SY), likely too short to have been identified in the subtiligase-based data analysis.
Confirming our hypothesis that if proteolysis of caspase-3 substrates is required for LTD, obliteration of this process should impair LTD, we observed precisely this result in neurons transfected with mutant Gap43 resistant to casapse-3-mediated cleavage. Perhaps most surprising is our finding that Gap43 mediates caspase-3's function in AMPA receptor endocytosis and LTD induction, both of which occur postsynaptically. Gap43 has been known for its high levels in neuronal growth cones during development and axon regeneration, and therefore thought to primarily reside in presynaptic terminals. But results from our subcellular fractionation, immunocytochemical analyses, and live-imaging experiments all indicate that Gap43 is also present at the postsynaptic site. These findings are also consistent with reports from several other groups that Gap43 is a component of the PSD proteome (43–48).
After Gap43 is cleaved by caspase-3, Gap43 may become dysfunctional (loss-of-function), or gain new function (gain-of-function). Both mechanisms could result in AMPA receptor endocytosis and LTD. Gap43, which binds to the plasma membrane via palmitoylation at Cys3 and Cys4, is a component of lipid rafts, where it binds to the lipid second messenger PIP2 (34, 35, 49). The first 10 amino acids of Gap43 are responsible for membrane targeting, activating heterotrimeric GTP-binding proteins, and promoting filopodial formation and cell expansion (50, 51). Gap43 also binds to calmodulin, and the binding of Gap43 to PIP2 and calmodulin affects actin cytoskeleton dynamics (34, 52). Both interaction with PIP2 and calmodulin is inhibited by phosphorylation of Gap43 by protein kinase C (PKC). Although none of the caspase-3 cleavage sites in Gap43 identified in this study are within the first 10 amino acids or the basic effector domain (a.a. 32–52) that binds to PIP2, calmodulin, and PKC (34, 53), cleavage of Gap43 by casapse-3, even outside these regions, may still perturb the interaction of Gap43 with these proteins.
In conclusion, our data show that Gap43 is an important substrate mediating the function of caspase-3 in synaptic plasticity, and identified a new, postsynaptic function of Gap43 in LTD and AMPA receptor endocytosis. This is particularly interesting because Gap43 has been implicated in mental disorders. Brains of patients with schizophrenia, for example, show altered Gap43 expression (54–57), and social interaction in heterozygous Gap43 knockout mice is impaired (58). Our identification of Gap43 as a caspase-3 substrate with a novel function in LTD suggests a new potential mechanism for its connection to mental health.
Supplementary Material
Acknowledgments
We thank Dr. James A. Wells for providing the expression construct of subtiligase; Dr. Pete Wildes (in Dr. Wells' laboratory) for technical supports for subtiligase purification and N-terminal labeling, and to NIMH editor Dr. Elizabeth J. Sherman for her close reading of the manuscript for language, expression, and content. The advice and assistance of Jim Makusky, Ronald Finnegan (scientific applications and virtual machine services), Dr. Brian Martin, Dr. Michael Strader, Dr. Adele Blackler, and Dr. Jeff Kowalak in NIMH contributed to this work, as did Dr. Peter Backlund in NICHD.
Footnotes
* This work was supported by the Intramural Research Program of the National Institute of Mental Health, project 1ZIAMH000274-35, 1ZIAMH00279-27 and 1ZIAMH002881.
 This article contains supplemental Figs. S1 to S8 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S8 and Tables S1 to S4.
1 The abbreviations used are:
- NMDAR
- N-methyl-D-aspartate receptor
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- LTD
- long-term depression
- Bcl-2
- B-cell lymphoma 2
- BAD
- Bcl2-associated agonist of cell death
- BAX
- BCL2-associated X protein
- Gap43
- growth associated protein 43
- Dbn1
- drebrin
- BASP1
- brain acid soluble protein 1
- DIV
- days in vitro
- STS
- staurosporine
- TEV
- tobacco etch virus
- ORF
- open reading frame
- GO
- Gene Ontology
- IPA
- Ingenuity Pathway Analysis software
- MARCKSL1
- MARCKS-related protein
- MAP2
- microtubule-associated protein 2
- ERK
- extracellular signal-regulated kinase
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- TNF-α
- tumor necrosis factor-alpha
- CHX
- cycloheximide
- EPSC
- excitatory postsynaptic current
- WT
- wild-type
- GluA2
- glutamate receptor 2
- PSD
- postsynaptic density
- PSD-95
- postsynaptic density protein 95
- GluN2A
- glutamate receptor, ionotropic, N-methyl D-aspartate 2A
- dark
- plants grown in darkness for 1 day
- darksuc
- plants grown in darkness for 1 day and treated with 30 mm sucrose for 4 h
- darkman
- plants grown in darkness for 1 day and treated with 30 mm mannitol for 4 h
- H2O2t1/2
- treatment with 500 μm H2O2 for 8 min
- H2O2tmax
- treatment with 500 μm H2O2 for 18 min
- mant1/2
- treatment with 200 mm mannitol for 1 h
- NaClt1/2
- treatment with 100 mm NaCl for 45 min
- NaCltmax
- treatment with 100 mm NaCl for 2 h
- NOt1/2
- treatment with 125 μm DEANO for 8 min
- Pstarv
- starved in phosphate for 6 days.
REFERENCES
- 1. Shepherd J. D., Huganir R. L. (2007) The cell biology of synaptic plasticity: AMPA receptor trafficking. Ann. Rev. Cell Develop. Biol. 23, 613–643 [DOI] [PubMed] [Google Scholar]
- 2. Collingridge G. L., Isaac J. T., Wang Y. T. (2004) Receptor trafficking and synaptic plasticity. Nat. Rev. 5, 952–962 [DOI] [PubMed] [Google Scholar]
- 3. Malenka R. C., Bear M. F. (2004) LTP and LTD: an embarrassment of riches. Neuron 44, 5–21 [DOI] [PubMed] [Google Scholar]
- 4. Alnemri E. S., Livingston D. J., Nicholson D. W., Salvesen G., Thornberry N. A., Wong W. W., Yuan J. (1996) Human ICE/CED-3 protease nomenclature. Cell 87, 171. [DOI] [PubMed] [Google Scholar]
- 5. Thornberry N. A., Lazebnik Y. (1998) Caspases: enemies within. Science 281, 1312–1316 [DOI] [PubMed] [Google Scholar]
- 6. Nicholson D. W. (1999) Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differentiation 6, 1028–1042 [DOI] [PubMed] [Google Scholar]
- 7. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 8. Kaiserman D., Bird C. H., Sun J., Matthews A., Ung K., Whisstock J. C., Thompson P. E., Trapani J. A., Bird P. I. (2006) The major human and mouse granzymes are structurally and functionally divergent. J. Cell Biol. 175, 619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McStay G. P., Salvesen G. S., Green D. R. (2008) Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differentiation 15, 322–331 [DOI] [PubMed] [Google Scholar]
- 11. Kuo C. T., Zhu S., Younger S., Jan L. Y., Jan Y. N. (2006) Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 51, 283–290 [DOI] [PubMed] [Google Scholar]
- 12. Williams D. W., Kondo S., Krzyzanowska A., Hiromi Y., Truman J. W. (2006) Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234–1236 [DOI] [PubMed] [Google Scholar]
- 13. Li J., Yuan J. (2008) Caspases in apoptosis and beyond. Oncogene 27, 6194–6206 [DOI] [PubMed] [Google Scholar]
- 14. Huesmann G. R., Clayton D. F. (2006) Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron 52, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiao S., Li Z. (2011) Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 70, 758–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Z., Jo J., Jia J. M., Lo S. C., Whitcomb D. J., Jiao S., Cho K., Sheng M. (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agard N. J., Wells J. A. (2009) Methods for the proteomic identification of protease substrates. Curr. Opin. Chem. Biol. 13, 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van den Berg B. H., Tholey A. (2012) Mass spectrometry-based proteomics strategies for protease cleavage site identification. Proteomics 12, 516–529 [DOI] [PubMed] [Google Scholar]
- 19. Demon D., Van Damme P., Vanden Berghe T., Vandekerckhove J., Declercq W., Gevaert K., Vandenabeele P. (2009) Caspase substrates: easily caught in deep waters? Trends Biotechnol. 27, 680–688 [DOI] [PubMed] [Google Scholar]
- 20. Luthi A. U., Martin S. J. (2007) The CASBAH: a searchable database of caspase substrates. Cell Death Differentiation 14, 641–650 [DOI] [PubMed] [Google Scholar]
- 21. Crawford E. D., Seaman J. E., Agard N., Hsu G. W., Julien O., Mahrus S., Nguyen H., Shimbo K., Yoshihara H. A., Zhuang M., Chalkley R. J., Wells J. A. (2013) The DegraBase: A Database of Proteolysis in Healthy and Apoptotic Human Cells. Mol. Cell. Proteomics 12, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rawlings N. D., Barrett A. J., Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Agard N. J., Maltby D., Wells J. A. (2010) Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell. Proteomics 9, 880–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abrahmsen L., Tom J., Burnier J., Butcher K. A., Kossiakoff A., Wells J. A. (1991) Engineering subtilisin and its substrates for efficient ligation of peptide bonds in aqueous solution. Biochemistry 30, 4151–4159 [DOI] [PubMed] [Google Scholar]
- 26. Brown J. L., Roberts W. K. (1976) Evidence that approximately eighty per cent of the soluble proteins from Ehrlich ascites cells are Nalpha-acetylated. J. Biol. Chem. 251, 1009–1014 [PubMed] [Google Scholar]
- 27. Wildes D., Wells J. A. Sampling the N-terminal proteome of human blood. Proc. Natl. Acad. Sci. U.S.A. 107, 4561–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kapust R. B., Tozser J., Fox J. D., Anderson D. E., Cherry S., Copeland T. D., Waugh D. S. (2001) Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Engineering 14, 993–1000 [DOI] [PubMed] [Google Scholar]
- 29. Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., Kaufman P. D. (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PloS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li C., Wen A., Shen B., Lu J., Huang Y., Chang Y. (2011) FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 11, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 32. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 33. Talbot K., Cho D. S., Ong W. Y., Benson M. A., Han L. Y., Kazi H. A., Kamins J., Hahn C. G., Blake D. J., Arnold S. E. (2006) Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Human Mol. Genetics 15, 3041–3054 [DOI] [PubMed] [Google Scholar]
- 34. Laux T., Fukami K., Thelen M., Golub T., Frey D., Caroni P. (2000) GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell Biol. 149, 1455–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Denny J. B. (2006) Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Current Neuropharmacol. 4, 293–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mosevitsky M. I. (2005) Nerve ending “signal” proteins GAP-43, MARCKS, and BASP1. Int. Rev. Cytol. 245, 245–325 [DOI] [PubMed] [Google Scholar]
- 37. Shirao T. (1995) The roles of microfilament-associated proteins, drebrins, in brain morphogenesis: a review. J. Biochem. 117, 231–236 [DOI] [PubMed] [Google Scholar]
- 38. Nagai T., Ibata K., Park E. S., Kubota M., Mikoshiba K., Miyawaki A. (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20, 87–90 [DOI] [PubMed] [Google Scholar]
- 39. Beattie E. C., Carroll R. C., Yu X., Morishita W., Yasuda H., von Zastrow M., Malenka R. C. (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 3, 1291–1300 [DOI] [PubMed] [Google Scholar]
- 40. Benowitz L. I., Routtenberg A. (1997) GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 20, 84–91 [DOI] [PubMed] [Google Scholar]
- 41. Maekawa S., Sato C., Kitajima K., Funatsu N., Kumanogoh H., Sokawa Y. (1999) Cholesterol-dependent localization of NAP-22 on a neuronal membrane microdomain (raft). J. Biol. Chem. 274, 21369–21374 [DOI] [PubMed] [Google Scholar]
- 42. Klaiman G., Petzke T. L., Hammond J., Leblanc A. C. (2008) Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol. Cell. Proteomics 7, 1541–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bayes A., Collins M. O., Croning M. D., van de Lagemaat L. N., Choudhary J. S., Grant S. G. (2012) Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PloS One 7, e46683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nanavati D., Austin D. R., Catapano L. A., Luckenbaugh D. A., Dosemeci A., Manji H. K., Chen G., Markey S. P. (2011) The effects of chronic treatment with mood stabilizers on the rat hippocampal post-synaptic density proteome. J. Neurochem. 119, 617–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trinidad J. C., Thalhammer A., Specht C. G., Lynn A. J., Baker P. R., Schoepfer R., Burlingame A. L. (2008) Quantitative analysis of synaptic phosphorylation and protein expression. Mol. Cell. Proteomics 7, 684–696 [DOI] [PubMed] [Google Scholar]
- 46. Collins M. O., Husi H., Yu L., Brandon J. M., Anderson C. N., Blackstock W. P., Choudhary J. S., Grant S. G. (2006) Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 97, 16–23 [DOI] [PubMed] [Google Scholar]
- 47. Yoshimura Y., Yamauchi Y., Shinkawa T., Taoka M., Donai H., Takahashi N., Isobe T., Yamauchi T. (2004) Molecular constituents of the postsynaptic density fraction revealed by proteomic analysis using multidimensional liquid chromatography-tandem mass spectrometry. J. Neurochem. 88, 759–768 [DOI] [PubMed] [Google Scholar]
- 48. Peng J., Kim M. J., Cheng D., Duong D. M., Gygi S. P., Sheng M. (2004) Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 279, 21003–21011 [DOI] [PubMed] [Google Scholar]
- 49. Arni S., Keilbaugh S. A., Ostermeyer A. G., Brown D. A. (1998) Association of GAP-43 with detergent-resistant membranes requires two palmitoylated cysteine residues. J. Biol. Chem. 273, 28478–28485 [DOI] [PubMed] [Google Scholar]
- 50. Strittmatter S. M., Valenzuela D., Fishman M. C. (1994) An amino-terminal domain of the growth-associated protein GAP-43 mediates its effects on filopodial formation and cell spreading. J. Cell Sci. 107(Pt 1), 195–204 [DOI] [PubMed] [Google Scholar]
- 51. Zuber M. X., Strittmatter S. M., Fishman M. C. (1989) A membrane-targeting signal in the amino terminus of the neuronal protein GAP-43. Nature 341, 345–348 [DOI] [PubMed] [Google Scholar]
- 52. He Q., Dent E. W., Meiri K. F. (1997) Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J. Neurosci. 17, 3515–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chapman E. R., Au D., Alexander K. A., Nicolson T. A., Storm D. R. (1991) Characterization of the calmodulin binding domain of neuromodulin. Functional significance of serine 41 and phenylalanine 42. J. Biol. Chem. 266, 207–213 [PubMed] [Google Scholar]
- 54. Weickert C. S., Webster M. J., Hyde T. M., Herman M. M., Bachus S. E., Bali G., Weinberger D. R., Kleinman J. E. (2001) Reduced GAP-43 mRNA in dorsolateral prefrontal cortex of patients with schizophrenia. Cereb. Cortex 11, 136–147 [DOI] [PubMed] [Google Scholar]
- 55. Perrone-Bizzozero N. I., Sower A. C., Bird E. D., Benowitz L. I., Ivins K. J., Neve R. L. (1996) Levels of the growth-associated protein GAP-43 are selectively increased in association cortices in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 93, 14182–14187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hakak Y., Walker J. R., Li C., Wong W. H., Davis K. L., Buxbaum J. D., Haroutunian V., Fienberg A. A. (2001) Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 98, 4746–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eastwood S. L., Harrison P. J. (1998) Hippocampal and cortical growth-associated protein-43 messenger RNA in schizophrenia. Neuroscience 86, 437–448 [DOI] [PubMed] [Google Scholar]
- 58. Zaccaria K. J., Lagace D. C., Eisch A. J., McCasland J. S. Resistance to change and vulnerability to stress: autistic-like features of GAP43-deficient mice. Genes, Brain, Behavior 9, 985–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.