

Abstract
The plasma membrane (PM) is a highly dynamic interface that contains detergent-resistant microdomains (DRMs). The aim of this work was to determine the main functions of such microdomains in poplar through a proteomic analysis using gel-based and solution (iTRAQ) approaches. A total of 80 proteins from a limited number of functional classes were found to be significantly enriched in DRM relative to PM. The enriched proteins are markers of signal transduction, molecular transport at the PM, or cell wall biosynthesis. Their intrinsic properties are presented and discussed together with the biological significance of their enrichment in DRM. Of particular importance is the significant and specific enrichment of several callose [(1→3)-β-glucan] synthase isoforms, whose catalytic activity represents a final response to stress, leading to the deposition of callose plugs at the surface of the PM. An integrated functional model that connects all DRM-enriched proteins identified is proposed. This report is the only quantitative analysis available to date of the protein composition of membrane microdomains from a tree species.
The plasma membrane (PM)1 is considered as one of the most interactive and dynamic supramolecular structures of the cell (1, 2). It forms a physical interface between the cytoplasm and the extracellular environment and is involved in many biological processes such as metabolite and ion transport, gaseous exchanges, endocytosis, cell differentiation and proliferation, defense against pathogens, etc. (3). Various combinations of biochemical and analytical approaches have been used to characterize the PM proteome in different organisms such as yeast, plants, and animals (4–8). Typically, PM proteins are either embedded in the phospholipid bilayer through transmembrane helices or less tightly bound to the membrane through reversible or irreversible surface interactions. In eukaryotic cells, some PM proteins are enriched in lateral lipid patches that form microdomains within the membrane (9, 10). These microdomains are considered to act as functional units that support and regulate specific biological processes associated with the PM (9, 10). Often referred to as “membrane (lipid) rafts” in animals and other organisms, they are typically described as being enriched in sphingolipids, sterols, and phospholipids that contain essentially saturated fatty acids (9–11). Early work on PM microdomains has suggested that their specific lipid composition confers resistance to certain concentrations of nonionic detergents, such as Triton X-100 and Nonidet P-40 (10, 11). Although this property has been exploited experimentally to isolate so-called detergent-resistant microdomains (DRMs), the relationship between DRMs and membrane rafts remains controversial (12). Indeed, the relation between the two is much debated, essentially because the use of Triton X-100 at 4 °C to prepare DRMs has been proposed to potentially induce the artificial formation of detergent-resistant structures whose composition may not fully reflect that of physiological membrane rafts (12). Nonetheless, DRM preparations represent an excellent system for the isolation and identification of groups of proteins—eventually associated in complexes—that tend to naturally interact with specific sets of lipids, thereby forming specialized functional units. Their biochemical characterization is therefore most useful in attempts to better understand the mode of interaction of specific proteins with sterols and sphingolipids and to gain insight into the protein composition and biological activity of subdomains from the PM.
Plant DRMs have been understudied relative to their animal counterparts. Indeed, proteomic studies have been undertaken on DRM preparations from only a limited number of plant species. These include tobacco (13–15), Arabidopsis (16), barrel clover (Medicago truncatula) (17), rice (18), oat, and rye (19). These studies, essentially based on qualitative or semi-quantitative proteomics, led to the identification of hundreds of proteins involved in a large range of mechanisms, functions, and biochemical activities (15–19). Depending on the report considered, a variable proportion of the identified proteins can be intuitively linked to DRMs and potentially to PM microdomains. However, many proteins that are clearly not related to the PM and its microdomains co-purify with DRM. These include, for instance, soluble proteins from cytoplasmic metabolic pathways; histones; and ribosomal, chloroplastic, and mitochondrial proteins (15–19). Thus, there is a need to obtain a more restricted list of proteins that are specifically enriched in DRMs and that define specialized functional structures. One way to tackle this problem is through quantitative proteomics, eventually in combination with complementary biochemical approaches. Although quantitative techniques have been increasingly applied to the proteomic analysis of complex mixtures of soluble proteins, their exploitation for the characterization of membrane samples remains challenging. As a result, very few studies of plant DRMs have been based on truly quantitative methods. For instance, stable isotope labeling combined with the selective disruption of sterol-rich membrane domains by methylcyclodextrin was performed in Arabidopsis cell cultures (20). A similar approach was used to study compositional changes of tobacco DRMs upon cell treatment with the signaling elicitor cryptogenin (21). In another study, 64 Arabidopsis proteins were shown to be significantly enriched in DRMs in response to a pathogen-associated molecular pattern protein (22). Together, these few quantitative proteomics analyses suggest a role of plant membrane microdomains in signal transduction, as in mammalian cells.
Although several reports describe the partial characterization of DRMs from higher plants (13–23), there are no data available to date on the protein composition of DRMs from a tree species. We have therefore employed a quantitative proteomic approach for the characterization of DRMs from cell suspension cultures of Populus trichocarpa. In addition, earlier work in our laboratory based on biochemical activity assays revealed the presence of cell wall polysaccharide synthases in DRMs from poplar (23), which suggests the existence of DRM populations specialized in cell wall biosynthesis. This concept was further supported by similar investigations performed on DRMs isolated from the oomycete Saprolegnia monoica (24). The comprehensive quantitative proteomic analysis performed here revealed enrichment in the poplar DRMs of specific carbohydrate synthases involved in callose polymerization. Consistent with the role of callose in plant defense mechanisms, additional proteins related to stress responses and signal transduction were found to be specifically enriched in the poplar DRMs, together with proteins involved in molecular transport. To date, our report is the only analysis available of the DRM proteome of a tree species based on quantitative proteomics. The specific biochemical properties of the 80 proteins significantly enriched in DRMs are described and examined in relation to their localization in membrane microdomains. The relationship between poplar DRMs and molecular transport, signal transduction, stress responses, and callose biosynthesis is discussed, with support from a hypothetical model that integrates the corresponding enriched proteins.
EXPERIMENTAL PROCEDURES
Plant Cell Cultures
Poplar (Populus trichocarpa) cell suspension cultures, established as previously described (25), were a gift from Drs. Ohlsson and Berglund (KTH Biotechnology). Cells were grown at 24 °C in a modified Murashige and Skoog medium containing sucrose (3%), 2,4-dichlorophenoxyacetic acid (1 mg/l), and kinetin (0.02 mg/l) in a 12 h light/12 h dark regime (25). The cells were harvested in logarithmic growth phase seven to nine days after inoculation and washed twice via vacuum filtration with ice-cold MOPS buffer (0.05 m, pH 7.0).
Isolation of PM and DRM
Isolation of the plasma membrane fraction (PMF) and DRMs was performed as described elsewhere (23). All steps were performed at 4 °C. Typically, 100 g of poplar cells recovered via vacuum filtration were disrupted in 100 ml of homogenization buffer (0.05 m MOPS buffer pH 7.0 containing 2 mm EGTA, 2 mm EDTA, 0.33 m sucrose, 1 mm DTT and protease inhibitors from Roche) using a Waring blender. The homogenate was centrifuged at 10,000 × g for 10 min to remove cell debris. The supernatant was filtered through two layers of Miracloth (Calbiochem, Germany) and centrifuged at 50,000 × g for 1 h. The resultant microsomal pellet was resuspended in 6 ml of 5 mm KH2PO4 (pH 7.6), and a two-phase partition system was applied, the final composition of which was 5.8% (w/w) poly(ethylene glycol) 3350 (Sigma), 5.8% (w/w) dextran T-500 (Pharmacosmos A/S, Holbaek, Denmark), 4 mm KCl, and 5 mm potassium phosphate (pH 7.6). After being mixed 10 times, the mixtures were centrifuged at 500 × g for 10 min, and the resulting upper phase was collected and loaded on a newly prepared lower phase. The two-phase separation was repeated and the final upper phase containing the purified PM was diluted 5-fold with MOPS buffer (0.05 m, pH 7.0) and centrifuged at 100,000 × g for 1 h. The PMF pellet was resuspended in 900 μl of MOPS buffer (0.05 m, pH 7.0). The protein concentration was measured using the Bradford dye-binding assay (Bio-Rad) using bovine serum albumin as a standard.
The DRMs were prepared by adding Triton X-100 to the PMF to a final concentration of 1% (detergent-to-protein ratio = 15:1 (w/w)). After incubation for 30 min on ice, a sucrose solution was added to reach a final concentration of 46%. The preparation was overlaid with a continuous sucrose gradient (45%–15%) and centrifuged at 131,000 × g for 20 h at 4 °C using a swinging rotor (SW27; Beckman). The DRMs were recovered as a low-buoyancy white band, and the Triton-solubilized proteins were collected at the bottom of the tube (i.e. in the 46% sucrose layer) (Triton extract (TE)). The DRMs were diluted with MOPS buffer (0.05 m, pH 7.0) and pelleted via centrifugation at 100,000 × g for 2 h at 4 °C. The DRM and TE proteins were resuspended in MOPS buffer (0.05 m, pH 7.0), and the protein content was measured in each sample as described above. A total of four biological replicates were prepared. Out of these, the PMF, DRM, and TE from one biological replicate were used for SDS-PAGE (qualitative analysis), and the PMFs and DRMs from the remaining three replicates were used for the quantitative iTRAQ experiments.
SDS-PAGE Separation and In-gel Hydrolysis of Proteins
SDS-PAGE analysis was repeated on membrane preparations from five independent experiments. All gels exhibited identical protein profiles. Samples were prepared by mixing the PMF, DRM, and TE proteins (20 μg) with SDS buffer (3% (w/v) SDS, 75 mm Tris-HCl (pH 6.8), 100 mm DTT, 15% (w/v) glycerol, and bromphenol blue). The mixtures were subsequently incubated at 37 °C for 20 min, and proteins were separated on 12% SDS-polyacrylamide gels. Proteins were stained using Coomassie Blue (ThermoScientific, Rockford, IL), and each lane from a single gel was cut from the top to the bottom into 48 bands of similar volume for in-gel digestion (26). Briefly, the gel bands were incubated at 37 °C for 1 h in a solution that consisted of 50% 0.1 m ammonium carbonate, 48.75% acetonitrile, 1% iodoethanol, and 0.25% triethylphosphine (pH 10.0). The liquid was discarded and the gel pieces were successively dehydrated with 100 μl acetonitrile (∼5 min), dried under vacuum, rehydrated in a 30 mm ammonium bicarbonate solution containing 10 ng/μl of sequencing-grade modified trypsin (Promega, Madison, WI), and incubated for 16 h at 37 °C. The resulting peptides were extracted, dried, and redissolved in 0.1% formic acid for mass spectrometric analysis (26).
Protein Hydrolysis in Solution and iTRAQ Labeling
The PMF and DRM proteins (100 μg) were solubilized in 0.05 m triethylammonium bicarbonate containing 1% sodium deoxycholate (Sigma). Their disulfide bonds were reduced for 1 h at 60 °C in the presence of 5 mm tris(2-carboxyethyl)phosphine, and the resulting free thiol groups were alkylated at room temperature for 15 min by methyl methanethiosulfonate (10 mm). The proteins were hydrolyzed for 16 h at 37 °C in the presence of 5% trypsin in 50 mm triethylammonium bicarbonate. The solutions were acidified by the addition of trifluoroacetic acid (TFA) to a final concentration of 0.5% and centrifuged to remove the sodium deoxycholate. The resulting supernatants were transferred to new tubes and dried under vacuum. The dried peptides from the PMF and DRM samples were dissolved in 100 μl of a mixture consisting of 25% 250 mm triethylammonium bicarbonate and 75% (v/v) ethanol and transferred to different vials containing the different iTRAQ reagents (114–117; AB SCIEX, Foster City, CA). After 1 h of incubation at room temperature, the reaction was stopped by the addition of 100 μl of Milli-Q water. The iTRAQ-labeled PMF and DRM samples were pooled, and the mixtures were dried under vacuum. The iTRAQ labeling of the peptides from the other two biological replicates was performed in the same conditions, except that the labels were inverted to reduce bias between samples.
Strong Cation Exchange Fractionation of the iTRAQ-labeled Peptides
The dried iTRAQ-labeled peptides were resuspended in 3 ml of sample loading buffer (10 mm ammonium formate, 20% acetonitrile, pH 3.0) and loaded on a 1-ml NuviaTMS cartridge (Bio-Rad; prepared according to the manufacturer's instructions) at 0.5 ml/min using a syringe pump. After sample loading, the cartridges were washed by 5 ml of sample loading buffer at 0.5 ml/min, with washing followed by elution at the same flow rate with consecutive 1.5-ml ammonium formate salt plugs at pH 3.0 (30, 50, 80, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, and 400 mm in 20% acetonitrile). The eluent from each salt plug was dried using a SpeedVac centrifugal vacuum concentrator, and the peptides were purified on a PepClean C-18 column (Thermo Scientific) prior to mass spectrometry (MS) analysis.
Nano-LC-MS/MS Analysis of the Strong Cation Exchange Fractions
Peptide analysis was performed with reverse-phase LC–electrospray ionization–MS/MS using a nanoACQUITY Ultra Performance Liquid Chromatography system coupled to a Q-TOF mass spectrometer (Xevo Q-TOF, Waters, Milford, MA). The purified strong cation exchange fractions were resuspended in 0.1% TFA and loaded on a C18 trap column (Symmetry 180 μm × 20 mm, 5 μm; Waters, Milford, MA) that was then washed with 1% (v/v) acetonitrile, 0.1% (v/v) formic acid at 15 μl/min for 10 min. The samples eluted from the trap column were separated on a C18 analytical column (75 μm × 100 mm, 1.7 μm; Waters, Milford, MA) at 350 nl/min using 0.1% formic acid as solvent A and 0.1% formic acid in acetonitrile as solvent B in a stepwise gradient: 0.1%–10% B (0–10 min), 10%–30% B (10–110 min), 30%–40% B (110–120 min), 40%–85% B (120–125 min), 85% B (125–130 min), and 85%–0.1% B (130–135 min). The eluting peptides were sprayed in the mass spectrometer (capillary and cone voltages set to 4 kV and 35 V, respectively), and MS/MS spectra were acquired using automated data-directed switching between the MS and MS/MS modes using the instrument software (MassLynx V4.0 SP4). The five most abundant signals of a survey scan (350–1500 m/z range, 0.9-s scan time) were selected by charge state, and collision energy was applied accordingly for sequential MS/MS fragmentation scanning (50–1800 m/z range, 0.9-s scan time).
Data Processing, Protein Identification, and Quantification
An extensive search scheme was used to rigorously profile the MS data following the strategy presented in Fig. 1. The MS raw data files were processed using Mascot Distiller (version 2.4.3.2, Matrix Science, London, UK). The resulting mgf files were converted into the mzXML file format using msconvert (27) and back to the mgf format using mzXML2Other (28) to assign proper titles to each spectrum. The mzXML files were searched by MyriMatch (29) (version 2.1.120) and X!Tandem (30) (version 2011.12.01.1, LabKey, Insilicos, ISB, Seattle, WA) using the Populus protein database (Populus version 3.0; 73,013 entries) and the following settings: trypsin specific digestion with two missed cleavages allowed, peptide tolerance of 100 ppm, fragment tolerance of 0.2 Da, iTRAQ 4-plex for peptide N-t and Lys as fixed modifications, and, in variable mode, iTRAQ 4-plex on Tyr and oxidized Met and methylthio on Cys. For the SDS-gel samples, all search parameters were as above, except that ethanolylated Cys and oxidized Met were used as fixed and variable modifications, respectively. The final mgf outputs were submitted to a local Mascot (Matrix Science, version 2.3.1) server using the same search settings as indicated above. The Mascot search results were exported as Mascot dat files and converted to pep.xml using Mascot2XML (31). The results from the Mascot, MyriMatch, and X!Tandem search engines were validated by PeptideProphet (32) using the parametric (Mascot) and semi-parametric (MyriMatch and X!Tandem) models. For quantitative analysis, all iTRAQ reporter ion intensities were extracted using the TPP tool Libra and the isotopic correction factors from the iTRAQ reagent manufacturer. Normalization of iTRAQ channels was performed by summing all intensities of reporter ions in each iTRAQ channel (for peptides above the Libra probability cutoff) and equalizing each channel contribution by dividing individual reporter ion intensities by the corresponding channel-specific correction factor. All pep.xml files obtained from PeptideProphet were combined using iProphet (33). A protein list was assembled using ProteinProphet (34), and the final protein ratios were calculated using Libra. For the samples from SDS-PAGE, data processing was identical, but without the iTRAQ-related steps. In all searches a concatenated target-decoy database-search strategy was used to check the false positive rate, which was found to be less than 1% in all cases.
Fig. 1.
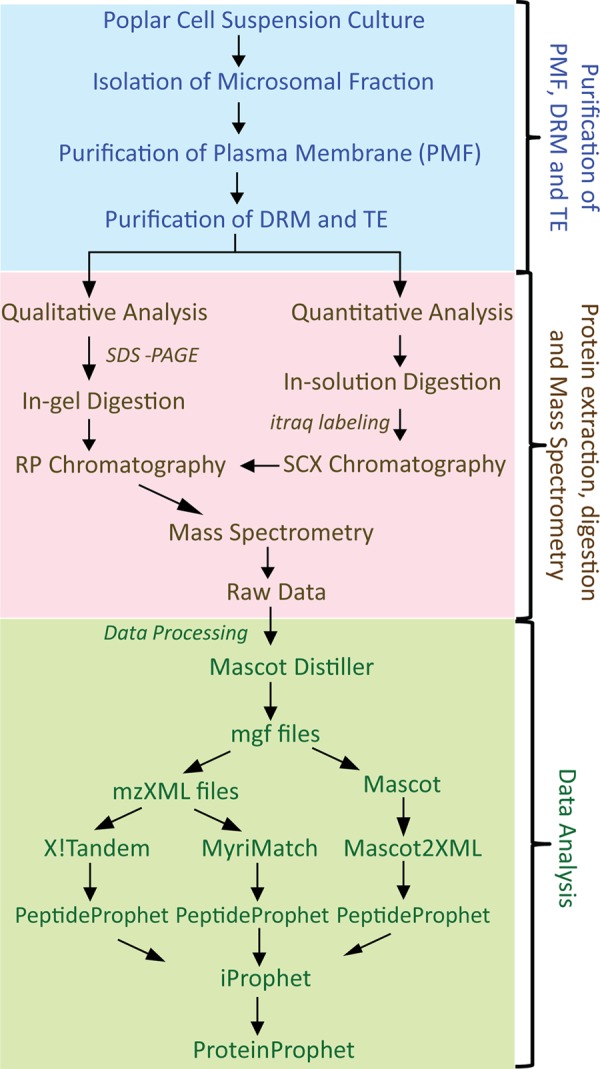
Experimental set-up and strategy used for data analysis. PMF, plasma membrane fraction; DRM, detergent-resistant microdomains; TE, Triton extract; iTRAQ, isobaric tags for relative and absolute quantitation; SCX, strong cation-exchange chromatography.
Peptide sequences were exported for each protein, with a protein and peptide probability cutoff of 0.95. Peptides matching two or more proteins (shared peptides) were excluded from the analysis. Proteins with no unique peptides (i.e. only identified by shared peptides) were also excluded. A protein was considered as identified if it contained at least one unique peptide. Only proteins identified by two or more unique peptides were used for quantification.
The method of Ross et al. (35) was used for statistical analysis of the quantitative data. Briefly, the DRM/PMF ratio of each protein was calculated for each of the three biological replicates (supplemental Table S2) and log2 transformed to obtain a normal distribution. The values were then normalized to the median log values, and global means and standard deviations were calculated for each biological replicate. Proteins whose average ratios fell outside a standard deviation of ±1 from the global mean in at least two out of three biological replicates were considered significantly enriched.
Prediction of Topology and Post-translational Modifications
The sequences of all identified proteins were used for the prediction of transmembrane domains (TMDs), sites of glycosylphosphatidylinositol (GPI) modification, myristoylation, and palmitoylation. The number of TMDs was determined by the HMMTOP program. GPI modification, N-terminal myristoylation, and palmitoylation sites were analyzed using the PredGPI predictor, Plant-Specific Myristoylation Predictor, and CSS-Palm 3.0 (with high threshold) algorithms, respectively. Theoretical molecular mass and isoelectric points (pI) were determined by the Protein Molecule Weight and Isoelectric Point Calculator. Gene ontology information was retrieved from TAIR.
RESULTS
Following our previous work on the isolation of DRMs from poplar cell suspension cultures and enzymatic assays (23), we have undertaken a proteomic approach for profiling the composition of DRMs and gaining further insight into the function of these microdomains. In the first instance, a gel-based qualitative analysis was performed on the PMF, DRM, and TE fractions to obtain an extended list of proteins present in each of these fractions. This was followed by an in-depth quantitative analysis on three biological replicates to determine the subset of proteins that are truly enriched in DRMs.
Qualitative Profiling of DRMs
A total of 2396 different proteins were identified from PMF, DRM, and TE (supplemental Table S1). The analysis of the PMF, DRM, and TE in-gel hydrolysates allowed the identification of a total of 917, 267, and 342 proteins in these three samples, respectively (Fig. 2 and supplemental Table S1). Out of these, 30 only were common to the DRM and TE (Fig. 2), which demonstrates the efficiency of the Triton X-100 treatment for separating proteins associated with the DRM from the rest of the PM-bound proteins. As expected, a significantly greater proportion of proteins were common to PMF and DRM (166 (= 141 + 25)) and to PMF and TE (177 (= 152 + 25)) (Fig. 2). In addition, each of the three samples contained proteins that were not identified in the two other samples; 599, 96, and 160 proteins were detected in only the PMF, DRM, and TE, respectively (Fig. 2).
Fig. 2.
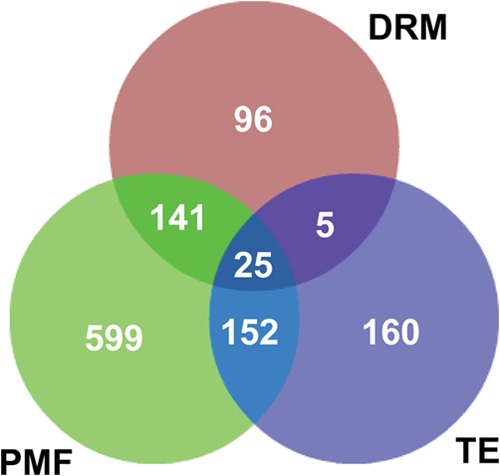
Qualitative proteomic analysis of plasma membrane fraction (PMF), detergent-resistant microdomains (DRM), and Triton extract (TE). The Venn diagram represents the number of proteins identified from SDS-PAGE of PMF, DRM, and TE. In total, 917, 267, and 342 proteins were identified through MS analysis of PMF, DRM, and TE, respectively.
Proteins from the DRM, PMF, and TE were classified based on their predicted cellular localization (Figs. 3A and 3B, supplemental Fig. S1A) and the biological processes in which they are involved (Figs. 3C and 3D, supplemental Fig. S1B). Interestingly, 90% of the 267 DRM proteins identified after in-gel hydrolysis were classified as membrane-bound proteins, and only minor proportions of these (∼5%) were predicted to be nonmembrane proteins located in the cytoplasm or more specifically associated to ribosomes (Fig. 3A). The rest of the identified DRM proteins either were extracellular (2%) or had an unknown cellular localization (3%). Both the PMF and the TE contained a significantly smaller proportion of identified proteins predicted to be associated with cell membranes. Indeed, the percentages of proteins that were not membrane bound were ∼40% and 60% in the PMF and TE, respectively (Fig. 3B and supplemental Fig. S1A). These data show that the PM was not purified to homogeneity in the PMF and that the greatest proportion of nonmembrane proteins that initially co-segregated with the PMs was recovered in TE. Thus, in addition to allowing the enrichment of DRMs, the Triton X-100 treatment is an efficient step for removing contaminating soluble proteins from membranous fractions.
Fig. 3.
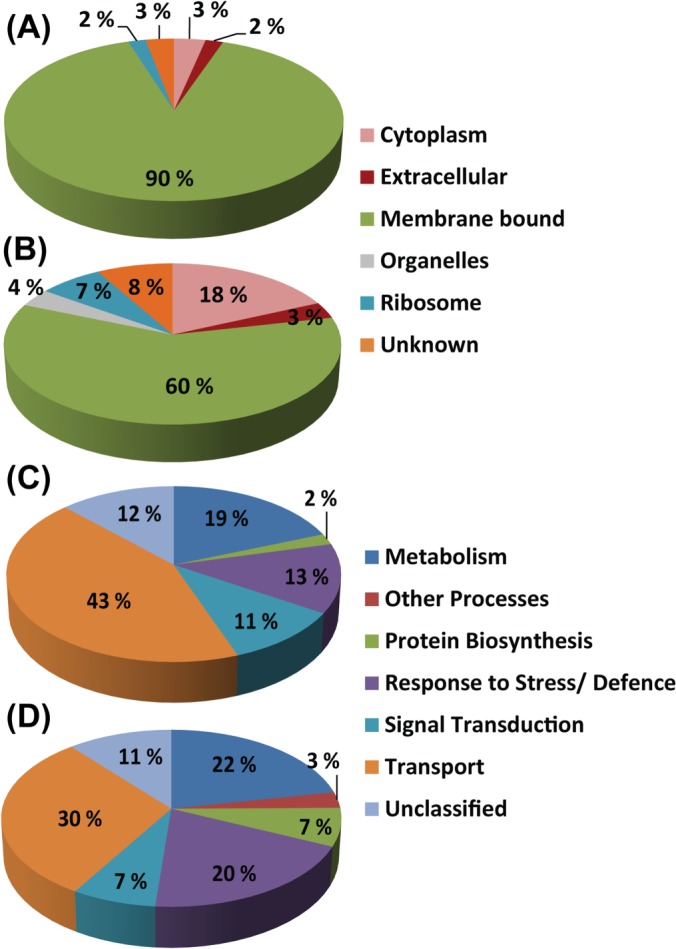
Predicted cellular localization (A, B) and functional classification (C, D) of proteins identified after SDS-PAGE analysis of DRM and PMF. A, C, DRM. B, D, PMF. Gene ontology information was retrieved from TAIR.
The proteins identified in DRM were associated with transport (43%), metabolism (19%), response to stress (13%), and signal transduction (11%) (Fig. 3C). Each fraction also contained a relatively small proportion (7% to 12%) of unclassified proteins of unknown function (Figs. 3C and 3D, supplemental Fig. S1B). The proportions of the different classes of proteins were moderately modified in the PMF, the most important differences being an increase in proteins involved in response to stress (20%) and a decrease in transporters (30%) relative to the DRM (Fig. 3D). The most distinguishable fraction was TE, which, as opposed to DRM and PMF, contained a large proportion (34%) of proteins involved in protein biosynthesis. These are related to ribosomes, which were already identified as more abundant in TE (supplemental Fig. S1B). In addition, TE contained a significantly smaller proportion of proteins involved in transport (19%) and signal transduction (2%) (supplemental Fig. S1B).
Together, these data most likely reflect an initial co-purification of endoplasmic reticulum with PM, whose components, including ribosomes, segregate preferentially in TE after the detergent treatment of PMF. However, we cannot completely rule out the possibility that some of the proteins we have identified may occur in multiple cellular compartments.
SDS-PAGE Profiles and Quantitative Proteomic Analysis
SDS-PAGE analysis revealed that the protein profiles of the DRM, PMF, and TE fractions are clearly distinguishable (Fig. 4). Many PMF proteins were preferentially extracted by the detergent (Triton X-100) and thus were expected to be essentially recovered in the high-density TE fraction. Consistent with this, DRMs exhibited a much simpler profile than the PMF (Fig. 4). In addition, SDS-PAGE analysis revealed that at least 14 protein bands seemed to be significantly enriched in the DRMs, as judged by their intensity after Coomassie Blue staining (arrowheads in Fig. 4). The iTRAQ-based quantitative analysis revealed that the actual total number of proteins enriched in DRM relative to PMF was 80 (Table I). Gene ontology analysis revealed that 77 of them are predicted to be membrane bound, whereas only three have an unidentifiable cell location (Table I). Functional classification of the proteins enriched in DRM shows that they can be grouped in three main families: 50% are transporters, 19% are related to stress responses, and 20% are involved in signal transduction (Fig. 5). The remaining 11% cannot be confidently linked to any function. The latter group corresponds to either uncharacterized proteins or proteins that do have an annotation but whose molecular function is unknown. Although data from cell suspension cultures may not fully reflect the situation in intact plant tissues, the remarkably simple distribution of the enriched proteins into three main functional families most likely reflects specialization of the isolated DRMs in cell-surface-related processes, especially transport, responses to stress, and signal transduction. Furthermore, our data are consistent with other reports describing the qualitative protein composition of DRMs isolated from plant tissues (13, 14, 17, 19).
Fig. 4.
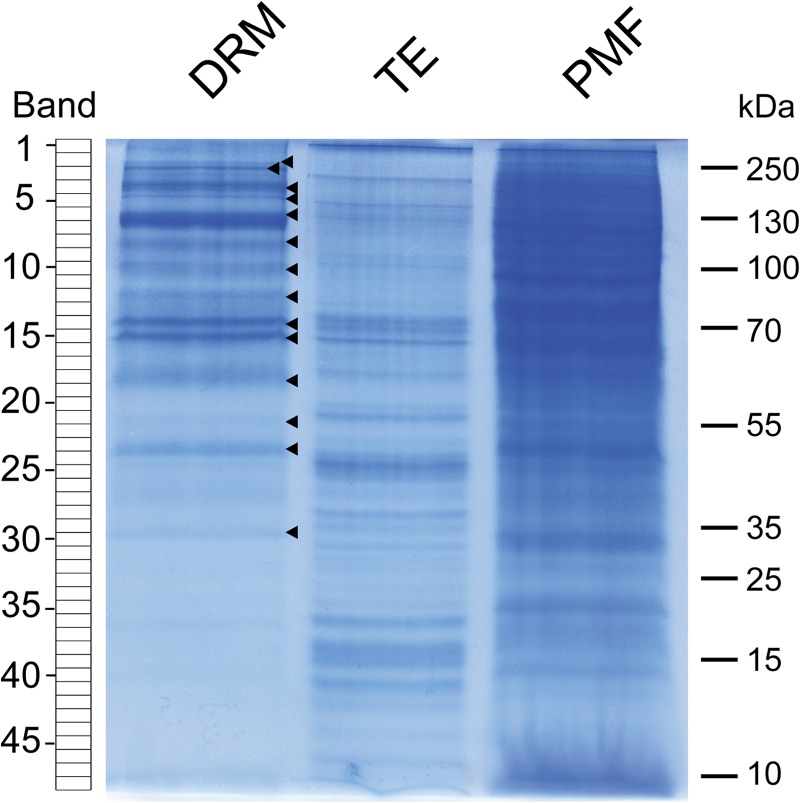
SDS-PAGE analysis of proteins present in detergent-resistant microdomains (DRM), Triton extract (TE), and plasma membrane fraction (PMF). A total of 20 μg of protein was loaded in each lane. Each lane of the Coomassie Blue–stained gel was cut into 48 bands as shown on the left-hand side of the picture. Arrowheads point to protein bands of a higher intensity in DRM relative to TE and PMF.
Table I. List of proteins enriched in DRMs relative to the PMF.
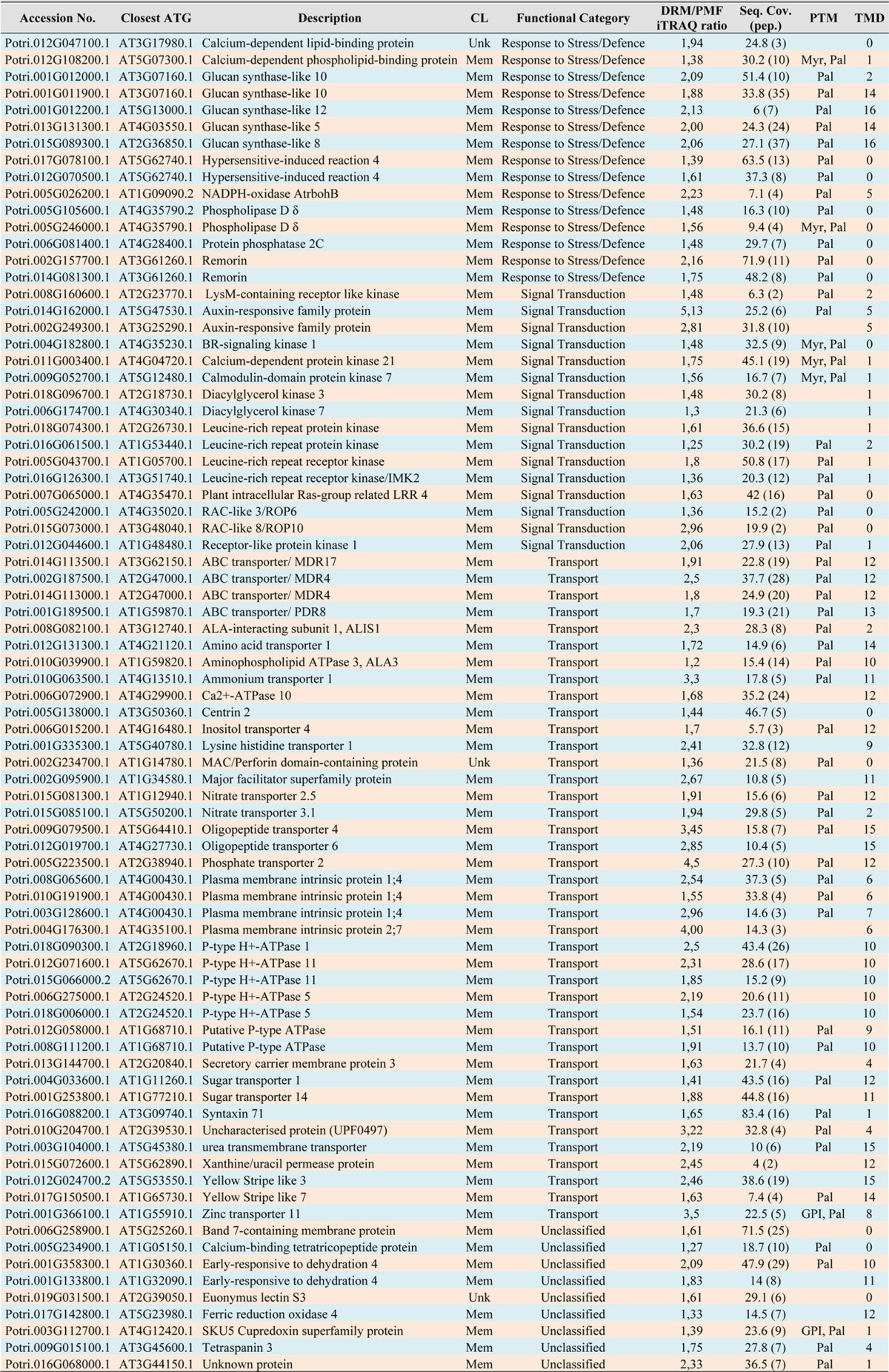
Notes: CL, cellular location; Mem, membrane-bound; Unk, unknown; DRM/PMF iTRAQ ratio, quantitative ratio between DRM and PMF based on iTRAQ; seq. cov., percent sequence coverage; pep., number of unique peptides identified; PTM, predicted post-translational modifications; Pal, palmitoylation; Myr, myristoylation; GPI, glycosylphosphatidylinositol; TMD, number of transmembrane domains.
Fig. 5.
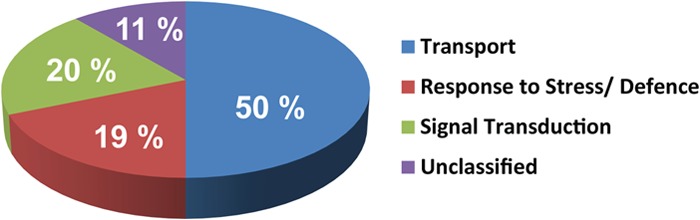
Functional classification of the 80 proteins enriched in the poplar DRM based on gene ontology prediction. Gene ontology information was retrieved from TAIR.
Biochemical Properties of Proteins Enriched in DRM
As expected, a large majority (95%) of the 80 DRM-enriched proteins are predicted to contain up to 16 TMDs and/or membrane-anchoring acylation sites. Only a minority (4 out of 80) contain neither membrane spanning domains nor post-translational modifications responsible for attachment to membranes. Of all enriched proteins, about 64% are predicted to contain at least 2 TMDs, and 15% exhibit one such domain only (Table I). Most interesting, proteins enriched in DRM are characterized on average by a higher number of TMDs compared with all PM proteins (Fig. 6A). In addition, their TMDs have a significantly greater length than those of PM proteins (Fig. 6B). The molecular weights and isoelectric points of total PM and DRM enriched proteins were also analyzed. Proteins below 18 kDa were not present in DRMs (Fig. 6C). Instead, DRMs contained a greater proportion of high-molecular-weight proteins than the PMF (Fig. 6C). Approximately 59% of the DRM-enriched proteins exhibited predicted pIs in the range of 8–10, and the remaining 41% were less alkaline (pI 4–7) (Fig. 6D). Comparison with PMF proteins revealed that acidic proteins (pI < 6) were more abundant in PMF, whereas basic proteins (pI > 8) were present in greater proportions in DRM. These features are comparable to those reported for DRM proteins from tobacco (15).
Fig. 6.

Classification of DRM-enriched (green bars) and total plasma membrane (blue bars) proteins based on their number of transmembrane domains (TMD) (A), average number of amino acids in TMD (B), molecular mass (C), and isoelectric point (D).
Bioinformatics tools were used to predict the number of TMDs and the type of membrane-anchoring post-translational modifications that the DRM-enriched proteins may contain (see “Experimental Procedures”). The data suggest that most of the 21% of DRM-enriched proteins that are devoid of membrane spanning domains contain acylation sites (Table I). The in silico analyses also indicated that about 54% of DRM-enriched proteins predicted to be associated to the membrane contained at least one TMD and an acylation site. Among the 56 proteins predicted to be acylated, 5 contained both myristoylation and palmitoylation sites, and all others contained palmitoylation as the single predicted modification. Together, these bioinformatics predictions tend to confirm that the isolated DRMs exhibited the expected enrichment in transmembrane and palmitoylated proteins.
DISCUSSION
Suspension cultures of tree cells represent a convenient alternative system to resilient woody tissues for the isolation of membrane structures and their proteomic characterization. Populus is a well-established model system for woody plants, and as such it provides an opportunity to study processes for which herbaceous models are less adapted (36). In order to shed light on the identity and function of proteins specifically enriched in plasma membrane DRMs from a tree species, we have undertaken the proteomic characterization of such microdomains from Populus trichocarpa cell suspension cultures using both qualitative and quantitative approaches (Fig. 1). First, we qualitatively analyzed the composition of the PMF and DRMs after protein separation by SDS-PAGE (Fig. 4). This allowed the identification of hundreds of proteins in each fraction by mass spectrometry (Fig. 2). Sodium deoxycholate was used for the solubilization of the PMF and DRM proteins prior to their partial enzymatic hydrolysis in solution. This detergent has the advantage of increasing the solubility of membrane proteins, which is accompanied by an enhanced efficiency of trypsin and increased recovery of hydrophobic peptides, while also being easy to remove via acidification after the proteolysis step (37). Peptides/proteins were quantified using the iTRAQ approach, which allows the concomitant analysis of proteins in up to eight samples, limits sample complexity, and reduces experimental variability between samples (35, 38, 39).
Our strategy has allowed the identification of a total of 2396 unique proteins in the poplar samples, out of which nearly 48% (1151 proteins) are predicted to contain at least one TMD (supplemental Table S1). These numbers are significantly higher than those reported in the only proteomics study available on poplar PM (8). Indeed, in the latter case only 22% of the total 956 identified proteins were predicted to contain at least one TMD. Similar analyses on Arabidopsis PM revealed that ∼38% (4) and 51% (40) of the total identified proteins contain one or more putative TMDs. Interestingly, nearly 80% of the 80 proteins enriched in poplar DRM have one or more predicted TMDs (Table I). In tobacco cells, 59% of the DRM identified proteins showed one or more putative TMDs (15), similar to the percentage (∼60%) reported in DRMs from oat and rye (19). Further, in our study, ∼46% of all the DRM-enriched proteins exhibited more than seven TMDs. This is significantly more than in Arabidopsis (∼29%) (40) and tobacco (40%) (15). As reported earlier in tobacco (15), we observed a greater average number of amino acids per TMD in the DRM-enriched proteins than in the total proteins from the PM (Fig. 6B). Together, our data further support the earlier suggestion that DRMs are thicker than the detergent-soluble parts of the PM and, consequently, tend to contain a greater proportion of proteins with longer TMDs (41). In addition to TMDs, acylation and glypiation are involved in anchoring proteins in membrane structures. Palmitoylation is a reversible type of post-translational modification responsible for the membrane association of proteins involved in multiple cell-surface processes (42). The observed enrichment of the poplar DRMs in palmitoylated proteins is consistent with the functional specialization of such microdomains in signal perception and transduction, as well as in transport at the PM. GPI-anchored proteins represent another type of protein expected to be enriched in DRMs (43). It is noteworthy that our analyses of the 80 proteins enriched in DRMs revealed that only 2 proteins are predicted to contain a GPI post-translational modification (Table I). This might be due to the naturally low abundance of such proteins in biological membranes and the consequent need to use an enrichment protocol specific for GPI proteins to facilitate their detection. Indeed, mass spectrometric identification of GPI-anchored proteins from plant cells is greatly facilitated by the use of a specific Triton X-114-based two-phase partitioning step, combined with the action of phosphatidylinositol phospholipase C, to specifically enrich and release the protein moieties from their GPI anchors (44, 45). Even when such specific approaches are used, the total number of GPI-anchored proteins identified typically corresponds to only a fraction of the actual GPI-modified proteins, as illustrated by the relatively low number (<50) of GPI-anchored proteins experimentally identified from total plasma membranes of Arabidopsis cell suspension cultures (44).
Quantitative proteomic analyses revealed that the poplar DRM-enriched proteins correspondd to three main functional categories, namely, transport (50%), responses to stress (19%), and signal transduction (20%) (Fig. 5). The relevance of their presence in DRM is discussed below.
Remorins
Remorins have been previously identified in DRMs from tobacco (15), barrel clover (17), oat, and rye (19), as well as in the proteome of Arabidopsis plasmodesmata (46). In tobacco, remorins accumulate preferentially within the cytosolic leaflet of thick (∼70 nm) plasmodesmatal membrane domains associated with plant defense mechanisms (47–49). Furthermore, quantitative proteomics in Arabidopsis involving metabolic labeling and the specific disruption of sterol-rich membrane domains by methyl-β-cyclodextrin strongly suggests that remorins are sterol-dependent proteins (20). Together, these data and our finding that remorins are also enriched in the poplar DRMs (Table I) further support the concept that remorins are functional markers of this type of microdomain.
Intracellular Trafficking
Six proteins involved in membrane trafficking were enriched in the poplar DRMs (Table I). Out of these, one is a secretory carrier membrane protein (SCAMP) containing four putative TMDs (Table I). In animal cells, SCAMPs are post-Golgi integral membrane proteins mediating endocytosis (50). Interestingly, a rice homolog of an animal SCAMP was found to be targeted to the PM, trans-Golgi network, and early endosomes when expressed in tobacco (51). Although plant SCAMPs have never been linked to DRMs in any proteomics studies, it is tempting to speculate that SCAMP-mediated endocytic pathways involve specific membrane microdomains. It remains, however, to be determined via imaging and other complementary techniques whether SCAMPs are located in such thick domains of the PM. A Q-SNARE (syntaxin 71), a phospholipid translocase (ALA3), and ALA-interacting protein (ALIS1) were also enriched in the poplar DRMs (Table I). These types of proteins participate in intracellular membrane fusion, vesicle formation, and trafficking (52, 53). The association of ALIS1 to ALA3 is required for phospholipid translocase activity, and the resulting complex is a key component of the Golgi secretory machinery (53). It is noteworthy that DRM proteomic studies in tobacco (15), rice (18), oat, and rye (19) have also reported the presence of similar proteins involved in trafficking.
Transport Proteins
Several types of transporters, including ABC transporters, aquaporins, sugar, metal, inorganic and organic solute transporters, and ATPases, were significantly enriched in the poplar DRMs (Table I). The occurrence of the same classes of transport proteins has also been reported in DRMs from other plants such as tobacco (15) and Arabidopsis (16), and in monocots such as rice (18), oat, and rye (19). Out of the eight ATPases enriched in the poplar DRMs, one was a Ca2+-ATPase, and others were P3-type ATPases (Table I). P-ATPases transport different compounds across the PM upon ATP hydrolysis, including ions and phospholipids (54). In contrast to other studies in tobacco (15), Arabidopsis (16), and the monocots oat and rye (19), no V-type ATPase was identified in the poplar DRMs. Interestingly, the relative abundance of P-type H+-ATPases was increased in Arabidopsis DRMs during cold acclimation, accompanied with a decrease in V-type H+-ATPase subunits (55), consistent with the observation that the expression of H+-ATPase genes changes as a result of abiotic and biotic stresses (56). Similarly, the expression of plant Ca2+-ATPases has been associated with salt stress (57) and the maintenance of calcium homeostasis within the cell (58).
Aquaporins are members of the PM intrinsic protein family and facilitate the movement of water through membranes. The level of some of them is increased during cold acclimation in Arabidopsis (55). Interestingly, four such proteins were enriched in the poplar DRMs (Table I). Plant ABC transporters have been associated with, for instance, auxin transport, lipid catabolism, disease resistance, and the function of the stomata (59). Four proteins from ABC transporter subfamilies (i.e. multidrug resistance (three proteins) and pleiotropic drug resistance (one protein) were enriched in the poplar DRMs (Table I). The Arabidopsis AtPDR protein is localized in the PM and involved in the removal of toxic compounds (Pb) from the cytoplasm (60), and AtMDR4 is an auxin transporter (61). Several of the poplar DRM-enriched transporters were also enriched in DRMs from Arabidopsis in response to flagellin, a pathogen-associated molecular pattern protein (22). In addition, the expression of some sugar transporters is affected in response to biotic and abiotic stresses (62). Together, these observations and our data show that even for proteins that are typically classified in functional groups other than “response to stress,” a link to the stress response can be established.
Signal Transduction and Response to Stress
Rop/Rac GTPases are important regulators of signal transduction in plants (63). It has been suggested that acylation regulates ROP signaling in PM microdomains (64). Two of this type of protein, ROP6 and ROP10, were enriched in the poplar DRMs (Table I). In Arabidopsis, ROP10 is a PM-localized protein that negatively regulates the sensitivity of stress-responsive genes to abscisic acid (65). Unlike its inactive prenylated form, which was recovered in detergent-soluble fractions, the active form of ROP6 was specifically acylated and localized in the Arabidopsis DRMs (64). Interestingly, it was also shown that the rice GTPase Rac1 and its effector RACK1A segregate in DRM upon elicitation with chitin and are involved in the plant's innate immunity (18). In addition to Rop/Rac GTPases, the poplar DRMs were also enriched in other types of proteins that are directly involved in the signaling cascade, such as leucine-rich repeat protein kinases and receptor kinases (Table I).
In plants, calcium is an important secondary messenger involved in the regulation of stress-signaling pathways (66). Consistent with the function of DRMs in these processes, many proteins dependent on calcium and calmodulin for their activity were significantly enriched in the DRMs (Table I). Examples are a calcium-dependent protein kinase that plays an important role in plant defense responses in tobacco (67) and calcium-dependent lipid binding proteins that act as repressors of abiotic stress response in Arabidopsis (68).
Phospholipase D and diacylglycerol kinase, both involved in the biosynthesis of phosphatidic acid (69), were enriched in the poplar DRMs (Table I). Phosphatidic acid acts as a second messenger and is involved in many biotic and abiotic stress responses (70). Protein phosphatase 2C, which is one of the main targets of phosphatidic acid (70), was also enriched in the poplar DRMs (Table I). Likewise, phospholipase D clusters at the site of infection of the PM of rice cells attacked by the pathogenic bacterium Xanthomonas oryzae (71). In addition to being involved in responses to pathogens, phospholipases are associated with abiotic stress, as exemplified by the increased freezing tolerance of Arabidopsis upon overexpression of the PM-associated phospholipase Dδ (72).
Callose Synthases
The poplar DRMs contained several isoforms of glucan synthase-like protein (GSL), namely, GSL 5, 8, 10, and 12 (Table I), which are involved in different biological processes associated with callose deposition at the surface of the PM. These polytopic proteins were among the most enriched in the DRMs, which suggests specialization of the microdomains in callose biosynthesis. Callose deposition and GSL are typically involved in specialized physiological processes, as demonstrated for GSL 5 (pollen development and fertility) (73), 8, 10 (male gametophyte development, cytokenesis, and plant growth) (74, 75), and 12 (plasmodesmata aperture and associated regulation of molecular trafficking) (76). In addition, GSL 5 has been reported to be involved in wound and papillary callose deposition (77), thereby having a dual function in plant development (73) and stress response (77). Aniline blue staining of callose in the poplar cells used for our proteomics analyses revealed a punctate type of pattern for callose deposition (supplemental Fig. S2G). A similar specific pattern has been observed in Arabidopsis cell suspension cultures stressed by plasmolysis or cellulase treatments (78). Our data suggest that the observed callose deposition in the poplar cell suspension cultures (supplemental Fig. S2) is a result of stress, although no specific treatment was used in our experiments to trigger a stress response. The observed punctate pattern reflects localized deposition sites at the surface of the plasma membrane rather than a homogeneous deposition of callose throughout the cell walls. This is consistent with the association and enrichment of callose synthases in defined lateral patches in the plasma membrane (i.e. membrane microdomains (DRMs)). Further to these observations, callose synthase assays performed on the isolated DRMs confirmed the occurrence of active enzymes in the microdomains. Indeed, the poplar DRMs prepared here exhibited a specific callose synthase activity similar to that reported in our earlier work (data not shown) (23). Thus, it can be concluded that some, if not all, of the callose synthases identified here are functional in the isolated microdomains. Together our data indicate that callose synthase activity is confined and enriched in poplar DRMs and that callose deposition in the cell cultures most likely arises from stress. It can be inferred that the isolated DRMs reflect a natural interaction of the identified callose synthases (and the other proteins identified) with the specific types of structural lipids that are typically expected to be enriched in DRMs, namely, sterols, sphingolipids, and phospholipids that preferentially contain saturated fatty acids (9, 10, 23). Interestingly, plasmodesmata have been shown to occur in cell suspension cultures (78, 79). There is also substantiated evidence that callose synthases are plasmodesmal proteins associated with several physiological and developmental processes, cell-to-cell communication, and biotic and abiotic stress responses (80, 81). Thus, consistent with the involvement of callose synthase activity in these processes, we hypothesize that some of the DRMs isolated here may arise from plasmodesmatal structures. The relationship between callose synthase and the other DRM-enriched proteins identified here is presented in an integrated hypothetical model (Fig. 7). Although additional work is needed to confirm the actual function of some of the identified proteins, the model reflects the general proposed function of the isolated microdomains in stress response and connects the different categories of proteins identified in the poplar DRMs.
Fig. 7.
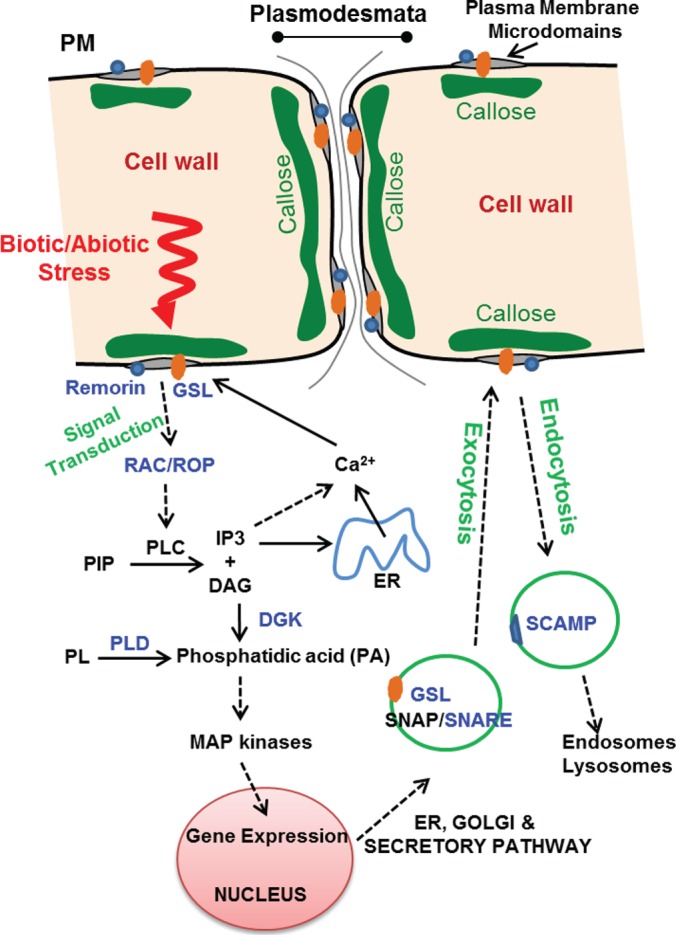
Integrated hypothetical model representing the different proteins enriched in the poplar DRM and the biological processes in which they are involved. Proteins whose names are highlighted in blue were shown to be enriched in plasma membrane DRMs (gray patches) together with other proteins listed in Table I. All proteins enriched in DRMs are markers of signal transduction, molecular transport, and callose biosynthesis and are known to be involved in biotic and/or abiotic stress. Vesicles carrying proteins involved in endocytosis and exocytosis and cell wall biosynthetic enzymes (callose synthases or GSL) are represented by green circles. The represented cascade of events leads to the release of calcium on the cytoplasmic side of the plasma membrane, which is required for the activation of callose synthase as a final response to stress (80). The enrichment of callose synthase in DRMs suggests a preferential deposition of callose plugs at the surface of plasma membrane microdomains, possibly in specialized plasmodesmatal structures and/or other sites of the plasma membrane. DAG, diacylglycerol; DGK, diacylglycerol kinase; ER, endoplasmic reticulum; GSL, glucan synthase-like/callose synthase; IP3, inositol-3-phosphate; PIP, phosphatidyl-inositol-phosphate; PL, phospholipid; PLC, phosphatidyl-inositol phospholipase C; PLD, phospholipase D; PM, plasma membrane; SCAMP, secretory carrier membrane protein; SNAP, soluble NSF attachment protein; SNARE, SNAP receptor.
Supplementary Material
Acknowledgments
The authors thank Drs. Ohlsson and Berglund (KTH Biotechnology) for the generous gift of the poplar cell suspension cultures, which were established in 2007 (25). Confocal microscopy was performed at the Imaging Facility of Stockholm University (IFSU).
Footnotes
* Financial support was received from the Swedish Centre for Biomimetic Fibre Engineering (grant from the Swedish Foundation for Strategic Research to V.B.) and the KTH Advanced Carbohydrate Materials Consortium (grant from the Swedish Research Council FORMAS to V.B.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- DRM
- detergent-resistant microdomain
- GPI
- glycosylphosphatidylinositol
- GSL
- glucan synthase-like protein
- iTRAQ
- isobaric tags for relative and absolute quantitation
- PM
- plasma membrane
- PMF
- plasma membrane fraction
- SCAMP
- secretory carrier membrane protein
- TE
- Triton extract
- TMD
- transmembrane domain.
REFERENCES
- 1. Engelman D. M. (2005) Membranes are more mosaic than fluid. Nature 438, 578–580 [DOI] [PubMed] [Google Scholar]
- 2. Edidin M. (2003) Lipids on the frontier: a century of cell-membrane bilayers. Nat. Rev. Mol. Cell Biol. 4, 414–418 [DOI] [PubMed] [Google Scholar]
- 3. Sprenger R. R., Jensen O. N. (2010) Proteomics and the dynamic plasma membrane: quo vadis? Proteomics 10, 3997–4011 [DOI] [PubMed] [Google Scholar]
- 4. Alexandersson E., Saalbach G., Larsson C., Kjellbom P. (2004) Arabidopsis plasma membrane proteomics identifies components of transport, signal transduction and membrane trafficking. Plant Cell Physiol. 45, 1543–1556 [DOI] [PubMed] [Google Scholar]
- 5. Josic D., Clifton J. G. (2007) Mammalian plasma membrane proteomics. Proteomics 7, 3010–3029 [DOI] [PubMed] [Google Scholar]
- 6. Macher B. A., Yen T. Y. (2007) Proteins at membrane surfaces—a review of approaches. Mol. Biosyst. 3, 705–713 [DOI] [PubMed] [Google Scholar]
- 7. Komatsu S. (2008) Plasma membrane proteome in Arabidopsis and rice. Proteomics 8, 4137–4145 [DOI] [PubMed] [Google Scholar]
- 8. Nilsson R., Bernfur K., Gustavsson N., Bygdell J., Wingsle G., Larsson C. (2010) Proteomics of plasma membranes from poplar trees reveals tissue distribution of transporters, receptors, and proteins in cell wall formation. Mol. Cell. Proteomics 9, 368–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simons K., Ikonen E. (1997) Functional rafts in cell membranes. Nature 387, 569–572 [DOI] [PubMed] [Google Scholar]
- 10. Bhat R. A., Panstruga R. (2005) Lipid rafts in plants. Planta 223, 5–19 [DOI] [PubMed] [Google Scholar]
- 11. Rietveld A., Simons K. (1998) The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochim. Biophys. Acta 1376, 467–479 [DOI] [PubMed] [Google Scholar]
- 12. Tanner W., Malinsky J., Opekarová M. (2011) In plant and animal cells, detergent-resistant membranes do not define functional membrane rafts. Plant Cell 23, 1191–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peskan T., Westermann M., Oelmuller R. (2000) Identification of low-density Triton X-100-insoluble plasma membrane microdomains in higher plants. Eur. J. Biochem. 267, 6989–6995 [DOI] [PubMed] [Google Scholar]
- 14. Mongrand S., Morel J., Laroche J., Claverol S., Carde J. P., Hartmann M. A., Bonneu M., Simon-Plas F., Lessire R., Bessoule J.-J. (2004) Lipid rafts in higher plant cells: purification and characterization of Triton X-100-insoluble microdomains from tobacco plasma membrane. J. Biol. Chem. 279, 36277–36286 [DOI] [PubMed] [Google Scholar]
- 15. Morel J., Claverol S., Mongrand S., Furt F., Fromentin J., Bessoule J.-J., Blein J.-P., Simon-Plas F. (2006) Proteomics of plant detergent-resistant membranes. Mol. Cell. Proteomics 5, 1396–1411 [DOI] [PubMed] [Google Scholar]
- 16. Borner G. H. H., Sherrier D. J., Weimar T., Michaelson L. V., Hawkins N. D., Macaskill A., Napier J. A., Beale M. H., Lilley K. S., Dupree P. (2005) Analysis of detergent-resistant membranes in Arabidopsis. Evidence for plasma membrane lipid rafts. Plant Physiol. 137, 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lefebvre B., Furt F., Hartmann M.-A., Michaelson L. V., Carde J. P., Sargueil-Boiron F., Rossignol M., Napier J. A., Cullimore J., Bessoule J.-J., Mongrand S. (2007) Characterization of lipid rafts from Medicago truncatula root plasma membranes: a proteomic study reveals the presence of a raft-associated redox system. Plant Physiol. 144, 402–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujiwara M., Hamada S., Hiratsuka M., Fukao Y., Kawasaki T., Shimamoto K. (2009) Proteome analysis of detergent-resistant membranes (DRMs) associated with OsRac1-mediated innate immunity in rice. Plant Cell Physiol. 50, 1191–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takahashi D., Kawamura Y., Yamashita T., Uemura M. (2012) Detergent-resistant plasma membrane proteome in oat and rye: similarities and dissimilarities between two monocotyledonous plants. J. Proteome Res. 11, 1654–1665 [DOI] [PubMed] [Google Scholar]
- 20. Kierszniowska S., Seiwert B., Schulze W. X. (2009) Definition of Arabidopsis sterol-rich membrane microdomains by differential treatment with methyl-β-cyclodextrin and quantitative proteomics. Mol. Cell. Proteomics 8, 612–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stanislas T., Bouyssie D., Rossignol M., Vesa S., Fromentin J., Morel J., Pichereaux C., Monsarrat B., Simon-Plas F. (2009) Quantitative proteomics reveals a dynamic association of proteins to detergent-resistant membranes upon elicitor signaling in tobacco. Mol. Cell. Proteomics 8, 2186–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keinath N. F., Kierszniowska S., Lorek J., Bourdais G., Kessler S. A., Shimosato-Asano H., Grossniklaus U., Schulze W. X., Robatzek S., Panstruga R. (2010) PAMP (pathogen-associated molecular pattern)-induced changes in plasma membrane compartmentalization reveal novel components of plant immunity. J. Biol. Chem. 285, 39140–39149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bessueille L., Sindt N., Guichardant M., Djerbi S., Teeri T. T., Bulone V. (2009) Plasma membrane microdomains from hybrid aspen cells are involved in cell wall polysaccharide biosynthesis. Biochem. J. 420, 93–103 [DOI] [PubMed] [Google Scholar]
- 24. Briolay A., Bouzenzana J., Guichardant M., Deshayes C., Sindt N., Bessueille L., Bulone V. (2009) Cell wall polysaccharide synthases are located in detergent-resistant membrane microdomains in oomycetes. Appl. Environ. Microbiol. 75, 1938–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohlsson A. B., Djerbi S., Winzell A., Bessueille L., Ståldal L., Li X. G., Blomqvist K., Bulone V., Teeri T. T., Berglund T. (2006) Cell suspension cultures of Populus tremula x tremuloides exhibit a high level of cellulose synthase gene expression that coincides with increased in vitro cellulose synthase activity. Protoplasma 228, 221–229 [DOI] [PubMed] [Google Scholar]
- 26. Hale J. E., Butler J. P., Gelfanova V., You J.-S., Knierman M. D. (2004) A simplified procedure for the reduction and alkylation of cysteine residues in proteins prior to proteolytic digestion and mass spectral analysis. Anal. Biochem. 333, 174–181 [DOI] [PubMed] [Google Scholar]
- 27. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deutsch E. W., Shteynberg D., Lam H., Sun Z., Eng J. K., Carapito C., Von Haller P. D., Tasman N., Mendoza L., Farrah T., Aebersold R. (2010) Trans-Proteomic Pipeline supports and improves analysis of electron transfer dissociation data sets. Proteomics 10, 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tabb D. L., Fernando C. G., Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 8, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 31. Pedrioli P. G. A. The SASHIMI Project. [Google Scholar]
- 32. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 33. Shteynberg D., Deutsch E. W., Lam H., Eng J. K., Sun Z., Tasman N., Mendoza L., Moritz R. L., Aebersold R., Nesvizhskii A. I. (2011) iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10, M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) ProteinProphet: a statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 35. Ross P. L., Huang Y. N., Marchese J., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 36. Jansson S., Douglas C. J. (2007) Populus: a model system for plant biology. Annu. Rev. Plant Biol. 58, 435–458 [DOI] [PubMed] [Google Scholar]
- 37. Masuda T., Tomita M., Ishihama Y. (2008) Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 7, 731–740 [DOI] [PubMed] [Google Scholar]
- 38. Aggarwal K., Choe L. H., Lee K. H. (2006) Shotgun proteomics using the iTRAQ isobaric tags. Brief. Funct. Genomic. Proteomic. 5, 112–120 [DOI] [PubMed] [Google Scholar]
- 39. Ow S. Y., Salim M., Noirel J., Evans C., Wright P. C. (2011) Minimising iTRAQ ratio compression through understanding LC-MS elution dependence and high-resolution HILIC fractionation. Proteomics 11, 2341–2346 [DOI] [PubMed] [Google Scholar]
- 40. Marmagne A., Rouet M.-A., Ferro M., Rolland N., Alcon C., Joyard J., Garin J., Barbier-Brygoo H., Ephritikhine G. (2004) Identification of new intrinsic proteins in Arabidopsis plasma membrane proteome. Mol. Cell. Proteomics 3, 675–691 [DOI] [PubMed] [Google Scholar]
- 41. McIntosh T. J., Vidal A., Simon S. A. (2003) Sorting of lipids and transmembrane peptides between detergent-soluble bilayers and detergent-resistant rafts. Biophys. J. 85, 1656–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hemsley P. A., Grierson C. S. (2008) Multiple roles for protein palmitoylation in plants. Trends Plant Sci. 13, 295–302 [DOI] [PubMed] [Google Scholar]
- 43. Simon-Plas F., Perraki A., Bayer E., Gerbeau-Pissot P., Mongrand S. (2011) An update on plant membrane rafts. Curr. Opin. Plant Biol. 14, 642–649 [DOI] [PubMed] [Google Scholar]
- 44. Elortza F., Nühse T. S., Foster L. J., Stensballe A., Peck S. C., Jensen O. N. (2003) Proteomic analysis of glycosylphosphatidylinositol-anchored membrane proteins. Mol. Cell. Proteomics 2, 1261–1270 [DOI] [PubMed] [Google Scholar]
- 45. Borner G. H. H., Lilley K. S., Stevens T. J., Dupree P. (2003) Identification of glycosylphosphatidylinositol-anchored proteins in Arabidopsis: a proteomic and genomic analysis. Plant Physiol. 132, 568–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fernandez-Calvino L., Faulkner C., Walshaw J., Saalbach G., Bayer E., Benitez-Alfonso Y., Maule A. (2011) Arabidopsis plasmodesmal proteome. PLoS One 6, e18880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Raffaele S., Bayer E., Lafarge D., Cluzet S., German Retana S., Boubekeur T., Leborgne-Castel N., Carde J.-P., Lherminier J., Noirot E., Satiat-Jeunemaître B., Laroche-Traineau J., Moreau P., Ott T., Maule A. J., Reymond P., Simon-Plas F., Farmer E. E., Bessoule J.-J., Mongrand S. (2009) Remorin, a solanaceae protein resident in membrane rafts and plasmodesmata, impairs potato virus X movement. Plant Cell 21, 1541–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raffaele S., Bayer E., Mongrand S. (2009) Upregulation of the plant protein remorin correlates with dehiscence and cell maturation: a link with the maturation of plasmodesmata? Plant Signal. Behav. 4, 915–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jarsch I. K., Ott T. (2011) Perspectives on remorin proteins, membrane rafts, and their role during plant-microbe interactions. Mol. Plant Microbe Interact. 24, 7–12 [DOI] [PubMed] [Google Scholar]
- 50. Fernandez-Chacon R., Südhof T. C. (2000) Novel SCAMPs lacking NPF repeats: ubiquitous and synaptic vesicle-specific forms implicate SCAMPs in multiple membrane-trafficking functions. J. Neurosci. 20, 7941–7950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lam S. K., Siu C. L., Hillmer S., Jang S., An G., Robinson D. G., Jiang L. (2007) Rice SCAMP1 defines clathrin-coated, trans-Golgi-located tubular-vesicular structures as an early endosome in tobacco BY-2 cells. Plant Cell 19, 296–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lipka V., Kwon C., Panstruga R. (2007) SNARE-ware: the role of SNARE-domain proteins in plant biology. Annu. Rev. Cell Dev. Biol. 23, 147–174 [DOI] [PubMed] [Google Scholar]
- 53. Poulsen L. R., López-Marqués R. L., McDowell S. C., Okkeri J., Licht D., Schulz A., Pomorski T., Harper J. F., Palmgrena M. G. (2008) The Arabidopsis P4-ATPase ALA3 localizes to the Golgi and requires a β-subunit to function in lipid translocation and secretory vesicle formation. Plant Cell 20, 658–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arango M., Gevaudant F., Oufattole M., Boutry M. (2003) The plasma membrane proton pump ATPase: the significance of gene subfamilies. Planta 216, 355–365 [DOI] [PubMed] [Google Scholar]
- 55. Minami A., Fujiwara M., Furuto A., Fukao Y., Yamashita T., Kamo M., Kawamura Y., Uemura M. (2009) Alterations in detergent-resistant plasma membrane microdomains in Arabidopsis thaliana during cold acclimation. Plant Cell Physiol. 50, 341–359 [DOI] [PubMed] [Google Scholar]
- 56. Janicka-Russak M. (2011) Plant plasma membrane H+-ATPase in adaptation of plants to abiotic stresses. In Abiotic Stress Response in Plants—Physiological, Biochemical and Genetic Perspectives (Shanker Arun, ed.), pp. 197–218, InTech, Rijeka, Croatia [Google Scholar]
- 57. Wimmers L. E., Ewing N. N., Bennett A. B. (1992) Higher plant Ca(2+)-ATPase: primary structure and regulation of mRNA abundance by salt. Proc. Natl. Acad. Sci. U.S.A. 89, 9205–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Geisler M., Axelsen K. B., Harper J. F., Palmgren M. G. (2000) Molecular aspects of higher plant P-type Ca(2+)-ATPases. Biochim. Biophys. Acta 1465, 52–78 [DOI] [PubMed] [Google Scholar]
- 59. Rea P. A. (2007) Plant ATP-binding cassette transporters. Annu. Rev. Plant Biol. 58, 347–375 [DOI] [PubMed] [Google Scholar]
- 60. Lee M., Lee K., Lee J., Noh E. W., Lee Y. (2005) AtPDR12 contributes to lead resistance in Arabidopsis. Plant Physiol. 138, 827–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Terasaka K., Blakeslee J. J., Titapiwatanakun B., Peer W. A., Bandyopadhyay A., Makam S. N., Lee R., Richards E. L., Murphy A. S., Sato F., Yazaki K. (2005) PGP4, an ATP-binding cassette P-glycoprotein, catalyzes auxin transport in Arabidopsis thaliana roots. Plant Cell 17, 2922–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Williams L. E., Lemoine R., Sauer N. (2000) Sugar transporters in higher plants—a diversity of roles and complex regulation. Trends Plant Sci. 5, 283–290 [DOI] [PubMed] [Google Scholar]
- 63. Temple B. R. S., Jones A. M. (2007) The plant heterotrimeric G-protein complex. Annu. Rev. Plant Biol. 58, 249–266 [DOI] [PubMed] [Google Scholar]
- 64. Sorek N., Poraty L., Sternberg H., Bar E., Lewinsohn E., Yalovsky S. (2007) Activation status-coupled transient S acylation determines membrane partitioning of a plant Rho-related GTPase. Mol. Cell. Biol. 27, 2144–2154 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65. Xin Z., Zhao Y., Zheng Z. (2005) Transcriptome analysis reveals specific modulation of abscisic acid signaling by ROP10 small GTPase. Plant Physiol. 139, 1350–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang T., Poovaiah B. W. (2003) Calcium/calmodulin-mediated signal network in plants. Trends Plant Sci. 8, 505–512 [DOI] [PubMed] [Google Scholar]
- 67. Romeis T., Ludwig A. A., Martin R., Jones J. D. (2001) Calcium-dependent protein kinases play an essential role in a plant defence response. EMBO J. 20, 5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. De Silva K., Laska B., Brown C., Sederoff H. W., Khodakovskaya M. (2011) Arabidopsis thaliana calcium-dependent lipid-binding protein (AtCLB): a novel repressor of abiotic stress response. J. Exp. Bot. 62, 2679–2689 [DOI] [PubMed] [Google Scholar]
- 69. Arisz S. A., Testerink C., Munnik T. (2009) Plant PA signaling via diacylglycerol kinase. Biochim. Biophys. Acta 1791, 869–875 [DOI] [PubMed] [Google Scholar]
- 70. Testerink C., Munnik T. (2005) Phosphatidic acid: a multifunctional stress signaling lipid in plants. Trends Plant Sci. 10, 368–375 [DOI] [PubMed] [Google Scholar]
- 71. Young S. A., Wang X., Leach J. E. (1996) Changes in the plasma membrane distribution of rice phospholipase D during resistant interactions with Xanthomonas oryzae pv oryzae. Plant Cell 8, 1079–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li W., Li M., Zhang W., Welti R., Wang X. (2004) The plasma membrane-bound phospholipase Dδ enhances freezing tolerance in Arabidopsis thaliana. Nat. Biotechnol. 22, 427–433 [DOI] [PubMed] [Google Scholar]
- 73. Enns L. C., Kanaoka M. M., Torii K. U., Comai L., Okada K., Cleland R. E. (2005) Two callose synthases, GSL1 and GSL5, play an essential and redundant role in plant and pollen development and in fertility. Plant Mol. Biol. 58, 333–349 [DOI] [PubMed] [Google Scholar]
- 74. Chen X. Y., Liu L., Lee E., Han X., Rim Y., Chu H., Kim S.-W., Sack F., Kim J. Y. (2009) The Arabidopsis callose synthase gene GSL8 is required for cytokinesis and cell patterning. Plant Physiol. 150, 105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Töller A., Brownfield L., Neu C., Twell D., Schulze-Lefert P. (2008) Dual function of Arabidopsis glucan synthase-like genes GSL8 and GSL10 in male gametophyte development and plant growth. Plant J. 54, 911–923 [DOI] [PubMed] [Google Scholar]
- 76. Vatén A., Dettmer J., Wu S., Stierhof Y. D., Miyashima S., Yadav S. R., Roberts C. J., Campilho A., Bulone V., Lichtenberger R., Lehesranta S., Mähönen A. P., Kim J. Y., Jokitalo E., Sauer N., Scheres B., Nakajima K., Carlsbecker A., Gallagher K. L., Helariutta Y. (2011) Callose biosynthesis regulates symplastic trafficking during root development. Dev. Cell 21, 1144–1155 [DOI] [PubMed] [Google Scholar]
- 77. Jacobs A. K., Lipka V., Burton R. A., Panstruga R., Strizhov N., Schulze-Lefert P., Fincher G. B. (2003) An Arabidopsis callose synthase, GSL5, is required for wound and papillary callose formation. Plant Cell 15, 2503–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bayer E., Thomas C. L., Maule A. J. (2004) Plasmodesmata in Arabidopsis thaliana suspension cells. Protoplasma 223, 93–102 [DOI] [PubMed] [Google Scholar]
- 79. Lee J.-Y., Taoka K.-I., Yoo B.-C., Ben-Nissan G., Kim D.-J., Lucas W. J. (2005) Plasmodesmal-associated protein kinase in tobacco and Arabidopsis recognizes a subset of non-cell-autonomous proteins. Plant Cell 17, 2817–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Brownfield L., Doblin M., Fincher G. B., Bacic A. (2009) Biochemical and molecular properties of biosynthetic enzymes for (1,3)-β-glucans in embryophytes, chlorophytes and rhodophytes. In Chemistry, Biochemistry and Biology of (1,3)-β-Glucans and Related Polysaccharides (Bacic A., Fincher G. B., Stone B. A., eds.), pp. 283–326, Academic Press, San Diego, CA [Google Scholar]
- 81. Zavaliev R., Ueki S., Epel B. L., Citovsky V. (2011) Biology of callose (β-1,3-glucan) turnover at plasmodesmata. Protoplasma 248, 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.