

Abstract
A two-tiered label-free quantitative (LFQ) proteomics workflow was used to elucidate how salinity affects the molecular phenotype, i.e. proteome, of gills from a cichlid fish, the euryhaline tilapia (Oreochromis mossambicus). The workflow consists of initial global profiling of relative tryptic peptide abundances in treated versus control samples followed by targeted identification (by MS/MS) and quantitation (by chromatographic peak area integration) of validated peptides for each protein of interest. Fresh water acclimated tilapia were independently exposed in separate experiments to acute short-term (34 ppt) and gradual long-term (70 ppt, 90 ppt) salinity stress followed by molecular phenotyping of the gill proteome. The severity of salinity stress can be deduced with high technical reproducibility from the initial global label-free quantitative profiling step alone at both peptide and protein levels. However, an accurate regulation ratio can only be determined by targeted label-free quantitative profiling because not all peptides used for protein identification are also valid for quantitation. Of the three salinity challenges, gradual acclimation to 90 ppt has the most pronounced effect on gill molecular phenotype. Known salinity effects on tilapia gills, including an increase in the size and number of mitochondria-rich ionocytes, activities of specific ion transporters, and induction of specific molecular chaperones are reflected in the regulation of abundances of the corresponding proteins. Moreover, specific protein isoforms that are responsive to environmental salinity change are resolved and it is revealed that salinity effects on the mitochondrial proteome are nonuniform. Furthermore, protein NDRG1 has been identified as a novel key component of molecular phenotype restructuring during salinity-induced gill remodeling. In conclusion, besides confirming known effects of salinity on gills of euryhaline fish, molecular phenotyping reveals novel insight into proteome changes that underlie the remodeling of tilapia gill epithelium in response to environmental salinity change.
Euryhaline fish are capable of living in fresh water (FW),1 brackish water (BW), seawater (SW), and hypersaline water (>SW). They adjust transepithelial ion transport across gill epithelium when challenged by an environmental salinity change (1). Acclimation from hyposmotic (relative to plasma, e.g. FW) to hyperosmotic (relative to plasma, e.g. SW) environments is accompanied by extensive remodeling of gill epithelium, the most prominent feature of which is an increase in the number and size of salt-secretory, mitochondria-rich ionocytes (2). In addition, molecular chaperones and distinct sets of transport proteins are activated when euryhaline fish are challenged by increasing environmental salinity (3–5). A euryhaline fish species in which these physiological responses have been observed is the Mozambique tilapia, Oreochromis mossambicus (6–8). Tilapia have evolved in Africa but have spread to subtropical and tropical freshwater and marine habitats throughout the world as a result of escaping from aquaculture farms and their high environmental adaptability. These cichlids tolerate salinities ranging from fresh water to almost 4× seawater (120 ppt) and they inhabit freshwater and hypersaline desert lakes as well as coastal marine and brackish habitats (9). This high salinity tolerance may have been selected for during tilapia evolution by frequent seasonal droughts and intermittent flooding events in their native African habitat containing salt-rich bedrock and soil (10). Tilapia are highly abundant in the California Salton Sea, which is a large hypersaline desert lake with an average salinity of 50 ppt and seasonal salinity increases up to 100 ppt in some parts (11–13). Thus, studies investigating the mechanisms that enable tilapia to cope with extreme and diverse osmotic stress are of great interest from an ecophysiological perspective and for understanding the basis of their high invasiveness in novel habitats.
Moreover, because of their outstanding osmotolerance tilapia are excellent models for studying the mechanisms of body water and electrolyte homeostasis in vertebrates. O. mossambicus is a very close relative of (and readily hybridizes with) the Nile tilapia, Oreochromis nilotics, for which a complete reference proteome is available in major databases, including UniProtKB (14, 15). Therefore, tilapia are well suited for proteomics studies directed at identifying, quantifying, and explaining molecular phenotypes (alterations in the proteome) induced by environmental stress. Because higher-order phenotypes (physiology, morphology, behavior) associated with salinity acclimation are well documented for tilapia, knowledge of the underlying molecular phenotypes will provide insight into the mechanisms that govern salinity acclimation of euryhaline fish (13, 16, 17). The main purpose of this study is to optimize and use a label-free quantitative proteomics (LFQ) workflow for molecular phenotyping of tilapia gill responses to salinity stress.
The workflow consists of initial protein identification and global label-free quantitative (LFQ) profiling followed by subsequent targeted LFQ of particular proteins based on quantitatively diagnostic, validated peptide ions. High resolution and high retention-time reproducibility in nano-flow liquid chromatography in combination with fast, high mass accuracy and high resolution mass spectrometers have enabled large-scale LFQ of proteins (18). Both relative and absolute LFQ of proteins are possible (19, 20) and protein quantities can be inferred from either spectral counts or ion currents and chromatographic peak intensity (21, 22). Spectral counting procedures have been used to roughly approximate relative protein quantities in different samples (23). In the present study, the other approach for LFQ, quantitation of ion current intensity, is used for relative quantitation of protein abundances in gill tissue from salinity stressed fish compared with FW handling controls. The quantitative precision of carefully optimized ion current intensity-based LFQ approaches is comparable to that of isotopic label-based quantitation (24, 25). Ion current intensity can be measured as peak height (maximum ion current) or peak area (integral of extracted ion chromatogram) (20–22). Because peak area provides a more accurate measure of peptide (and correspondingly protein) quantity this approach is used in the present study (21, 22).
The present study applies this LFQ workflow to identify the specific isoforms of (a) proteins involved in transepithelial ion transport and (b) molecular chaperones that are regulated by environmental salinity in tilapia gills. Such information is very difficult and often impossible to obtain with antibody-based approaches because isoform-specific antibodies for fish proteins are rare and none are available for the tilapia proteins of interest. Therefore, most quantitative analyses of fish proteins by Western blot use antibodies made against a different species (or even a mammalian or other more distantly evolutionarily related homolog) that are not suitable to distinguish individual isoforms (e.g. 3, 26, 27). The present study also investigates whether salinity-induced changes in ionocyte number and size are reflected in abundances of mitochondrial proteins, whether there is disparity in how different mitochondrial proteins are regulated in response to salinity stress, and which mitochondrial proteins are most affected by salinity stress. In addition, the initial global profiling step of the LFQ proteomics workflow described and the deposition of corresponding identification and quantitation data in the public PRIDE repository (28, 29) provides quantitative information on many proteins for which no prior information about effects of salinity on their abundance is available.
EXPERIMENTAL PROCEDURES
Animals and Salinity Acclimation
All animal procedures were approved by the UC Davis IACUC under protocol numbers 13468 and 15013. Mozambique tilapia (Oreochromis mossambicus) were raised from laboratory brood stock to between 5 and 6 months of age in dechlorinated Davis tap water for all experiments. Davis tap water chemistry has been reported previously (30), having a salinity < 0.5 ppt (fresh water, FW). Three different types of salinity acclimation were conducted in which fish were exposed to either acute salinity increase from FW to 34 ppt (in two 17 ppt/day steps) or gradual salinity increase from FW to 70 ppt and from FW to 90 ppt (in 7 ppt/day steps) (Fig. 1A). All acclimation experiments were performed at 26 ± 1 °C in 30 gallon closed circulation tanks equipped with appropriate aeration, filtration, and heating devices. Daily changes of 10% of the tank volume of water with a stock solution of sea salt (Instant Ocean) were performed to increase salinity by appropriate increments. Handling controls were run in parallel in each experiment. These controls consisted of fish from the same clutch that were kept in identical tanks and exposed to the same daily water changes except that FW was used instead of a concentrated sea salt solution. Fish were fed ad libitum and continued to grow well during salinity stress except at very high salinities (90 ppt exposure) when body mass (but not total length) was significantly less than that of the corresponding FW handling controls (Figs. 1B, 1C).
Fig. 1.
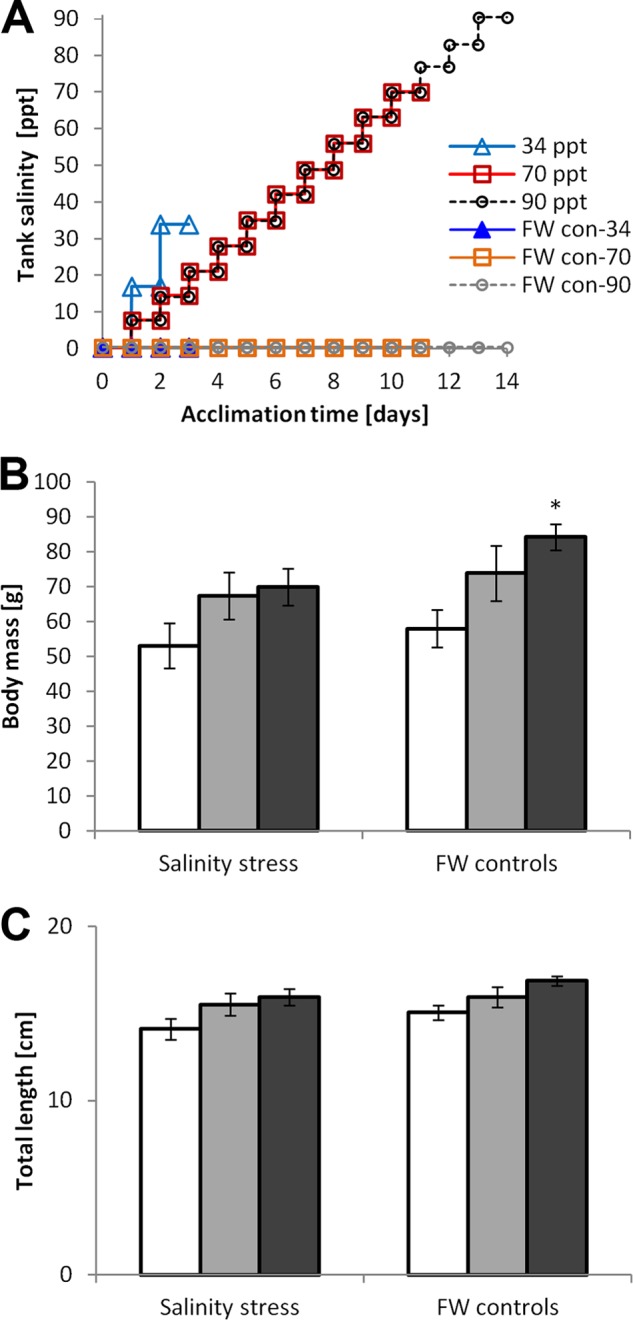
Tilapia salinity acclimation regimens. Tilapia were acclimated to elevated salinity in three independent experiments before gill epithelial samples were collected for proteomics. A, Salinity in treatment and control tanks as a function of acclimation time; B, Body mass and C, Body length of tilapia exposed to different types of salinity stress (white = 34 ppt, light gray = 70 ppt, dark gray = 90 ppt) compared with FW handling controls (mean ± S.E., n = 6, an asterisk indicates a significant effect of salinity stress based on t test p < 0.05).
Protein extraction and in Solution Digestion
A protocol that minimizes sample preparation steps while efficiently and reproducibly extracting membrane-, cytosolic-, and organelle-specific proteins was developed and optimized to maximize quantitative accuracy for comparing protein amounts in multiple samples. Immediately after dissection tissue samples were snap-frozen in liquid nitrogen and transferred to −80 °C. They were crushed to a fine powder by grinding the tissue using a mortar and pestle under liquid nitrogen. The powder was transferred to a low retention Eppendorf tube (LR-MCF) and an ice-cold solution of 10% trichloroacetic acid (TCA)/90% Acetone/0.2% dithiothreitol (DTT) was added (6× the volume of tissue weight). Samples were incubated in this solution while rotating overnight. Samples were then centrifuged at 20,000 × g for 5 min (4 °C) and the precipitated protein pellet washed twice in acetone/0.2% DTT. The protein pellet was dissolved in UT buffer (7 m urea/2 M thiourea/0.2% DTT, 6x the volume of the original tissue weight). After centrifugation at 18,000 × g (5 min) the supernatant was transferred to a clean LR-MCF tube and stored frozen at −80 °C. Before freezing the sample, triplicate 2 μl aliquots were used to determine protein concentration using a 660 nm protein assay that is compatible with 5× diluted UT buffer (Thermo-Pierce, cat. 22660). Protein extracts were thawed and appropriate amounts of LCMS grade water and 10× triethanolamine (TEA) buffer (pH 8.0, final concentration 100 mm) added to dilute samples of 150 μg/100 μl total protein concentration in 0.6 ml LR-MCF tubes. Samples were reduced by adding 16 mm DTT and incubation at 55 °C for 30 min, and alkylated by adding 16 mm iodoacetic acid (IAA) followed by 30 min incubation at room temperature in the dark. Immobilized trypsin (Princeton Separations cat. Nr. EN-251) was added at a 1:25 ratio relative to total protein and the samples incubated in a rotator at 35 °C for exactly 16h. Trypsin beads were removed by centrifugation for 2 min at 14,000 × g and 50% of the total initial volume of supernatant was transferred to a clean 0.6 ml LR-MCF tube. Samples were then dried by speedvac (Thermo-Savant, ISS-110). This step was stopped immediately when urea precipitate started to form to ensure optimal resuspension of peptides, which was done in 1 ml of LCMS grade water containing 0.1% formic acid (FA). Pipetting the solution slowly 50× up and down using a pipetman set at 0.8 ml over a period of 5 min ensured that peptides and urea redissolved. Peptide solutions were then transferred to maximum recovery glass vials (Waters 600000669CV) and stored frozen at −80 °C.
Peptide Separation and Mass Spectrometry
Peptides were injected in 2 μl volume corresponding to 330 ng total peptide amount using a nanoAcquity sample manager (Waters, Milford, MA) and separated after 3 min sample trapping (Symmetry, Waters 186003514) on a 1.7 μm particle size BEH C18 column (250 mm × 75 μm, Waters 186003545) by reversed phase chromatography using a nanoAcquity binary solvent manager (Waters). A 50 min linear gradient ranging from 3% to 35% acetonitrile (ACN) was used. The aqueous solvent contained 0.1% FA, which was omitted from the organic phase to prevent formation of brown ACN aggregates that would otherwise precipitate at low pH at the pico-emitter tip. A dual pico-emitter tip (New Objective FS360–20-10-d-20) nano-spray setup was custom-fitted at the nano-ESI source of a micrOTOF-QII mass spectrometer (Bruker Daltonics, for details see PRIDE AC 28628) to deliver analyte via two independent solvent lines. Line 1 was used for the sample gradient and controlled by the binary solvent manager of the nanoAcquity UPLC. Line 2 was used to deliver ESI-L low concentration tuning mix (Agilent G1969–85000) independent of line 1 during the 20 min sample injection delay period to allow for internal mass calibration of each sample. Internal calibration performed in this manner yielded better and more consistent mass accuracy across multiple samples than lock-mass calibration. Batch-processing of samples was controlled with Hystar 3.2 software (Bruker Daltonics).
Protein Identification
All peaklists were generated with Mascot Daemon and Mascot Distiller (versions 2.2.2 and 2.4.3, respectively, Matrixscience. Ltd.) using default parameters. For protein identification Mascot 2.2.7 (Matrixscience, Ltd.) (31) and Phenyx 2.6. (Geneva Bioinformatics, SA) (32) search engines were used and search results combined in Proteinscape 3.1 (Bruker Daltonics). The following parameters were used: enzyme specificity = trypsin, missed cleavages permitted = 1, fixed modification = Cys carbamidomethylation, variable modifications = Met oxidation and Pro hydroxylation, precursor ion mass tolerance = 20 ppm, fragment ion mass tolerance = 0.1 Da. A threshold score of 5% probability that a protein identification is incorrect was used for accepting individual MS/MS spectra. A database containing 28,020 protein sequences, including the complete predicted Oreochromis niloticus proteome and all available Oreochromis mossambicus sequences, was downloaded from http://www.uniprot.org/on July 28, 2012. Automatic annotation of all Uniprot ACs in this database was done with BLAST2GO (33) against the complete curated SwissProt database on July 28, 2012. In addition, annotations for all proteins identified in this study were manually confirmed by individual BLAST searches. An expanded version of the Oreochromis spec. protein database, which contains a randomly scrambled decoy sequence for each entry, was generated using PEAKS 6 (Bioinformatics Solutions, Inc.) (34). This expanded decoy database (containing 56,040 total sequences) was used for all protein identification searches to allow for consistent assessment of the protein ID false discovery rate. Redundancy in assigning peptides to protein identifications and ambiguity in protein identifications were eliminated by ProteinExtractor (Proteinscape 3.1). All data and metadata were exported from Proteinscape 3.1 to PRIDE xml using PRIDE converter (35) integrated into Proteinscape 3.1 (36). In cases where multiple proteins matched the exact same set of peptides (protein ambiguity) their corresponding UniProt accession numbers are indicated as alternative matches in the records deposited in the PRIDE database (PRIDE accession numbers 28622–28631) (29). Additional details, including mass error distribution, missed cleavage frequency, charge distribution, and number of peptides identified per protein, can be obtained by viewing PRIDE accession numbers 28622–28631 with the PRIDE Inspector tool (28, 37).
Global Quantitative Profiling
Global quantitative profiling was performed using ProfileAnalysis 2.0 and Proteinscape 3.1 (Bruker Daltonics) according to the “accurate mass and time tag” approach (38). Quantitative global profiling ratios (salinity stress versus handling controls) for identified proteins have been deposited along with the corresponding protein identification and mass spectrometry data in the PRIDE repository (29) and are available under PRIDE accession numbers 28622–28631. ProfileAnalysis 2.0 (Bruker Daltonics) was used to align and integrate chromatographic peak areas for all precursor ions exceeding an intensity of 1000 counts, being detected in at least 10 consecutive MS scans, and occurring in at least five of six biological replicates of either treated or control samples. For aligning all MS spectra from treated (n = 6) and corresponding control samples (n = 6) the retention time reproducibility limit for ProfileAnalysis was set to 1 min and the mass accuracy threshold was 30 ppm. Statistical analysis of treatment effects was carried out within ProfileAnalysis 2.0 and included determination of p values for t test, family-wise error rate (FWER), and false discovery rate (FDR) for each peptide quantified. In addition, a principal component analysis was performed using ProfileAnalysis 2.0 to visualize the overall effect of salinity stress on peptide abundances in all biological replicates. Peptides quantified with ProfileAnalysis 2.0 were imported into Proteinscape 3.1 and mapped to protein IDs based on matching m/z and retention time (Rt) combinations (deviation threshold = 0.03 Da for m/z and 1 min for Rt).
Targeted Quantitation
The initial global LFQ profiling was followed by a targeted approach for simultaneous identification and quantitation of the proteins of interest (39). Scheduled precursor lists (SPLs) were generated with Proteinscape and used to target desired ions for MS/MS in all samples. Peptides unambiguously assigned to protein NDRG1, mitochondrial proteins, molecular chaperones, and ion transport proteins were targeted for validation and more precise quantitation. These proteins were chosen because the focus of this study was on analyzing salinity effects on mitochondrial proteins and on identifying the specific isoforms of ion transport proteins and molecular chaperones that are regulated by environmental salinity. In addition, they consisted the great majority of proteins for which the global LFQ profiling analysis yielded multiple significantly regulated peptides (ProfileAnalysis, p < 0.05). Protein NDRG1 was chosen for targeted LFQ because global LFQ profiling identified it as most strongly down-regulated at elevated salinity. Targeted LFQ was performed using DataAnalysis 4.0 (Bruker Daltonics) on peptides meeting all of the following validation criteria to minimize possible sources of error (21): (1) No modification other than cysteine carbamidomethylation; (2) No missed or unspecific cleavage; (3) No alternative sequence assignment, (4) No ambiguity regarding protein ID assignment. Because gill samples were highly complex all EICs of peptides used for quantitation were visually inspected for overlap with neighboring peaks. If an adjacent peak was observed within Rt ± 0.7 min for any sample or if there were multiple missing values per group then the corresponding peptides were excluded from targeted LFQ (less than 20% overall). Visual inspection also served to verify that for each peptide all 12 EIC peaks (six treated and six controls) fall within the range of mean Rt ± 0.7 min (mean Rt was calculated by Proteinscape 3.1). These EICs were then automatically integrated using DataAnalysis 4.0 (Bruker Daltonics, m/z tolerance = 0.02 Da and Rt tolerance = 0.7 min, background subtraction algorithm version 2.1) and the quantity of peptides during a given treatment calculated as the ratio of the corresponding EIC integral relative to the mean EIC integral for the six control samples. This procedure was also performed for the base peak chromatogram (BPC) integral of each sample, which served for normalization of peptide quantities across all samples within an experiment. A two-factor ANOVA with replication was performed to discern whether treatment effects are significant, considering all peptide abundances per each protein (based on normalized peptide quantities). The two factors in the ANOVA were treatment and technical replicate and the proportions of variation in protein abundance explained by salinity treatment, biological replication, and technical replication were calculated based on the corresponding ANOVA mean square values.
RESULTS
LCMS Conditions and Protein Identifications
Because the quality of quantitative proteomics is extremely dependent on technical optimization of all steps, the entire procedure is carefully tuned to highly complex samples. A very robust initial sample preparation step uses protein extraction in ice-cold solution of 10% TCA/90% Acetone/0.2% DTT rather than UT buffer or other commonly used buffers. Using this extraction solution results in excellent recovery of not only cytosolic but also organelle and membrane proteins (e.g. mitochondrial and ion transporter proteins, see below). In addition, proteolysis is very effectively limited by the very low pH and acetone-based denaturation/precipitation (protein recovery is higher than using UT buffer or other buffers for extraction as evidenced by gel- and spectroscopy based assays). Using this method, protein extraction is highly reproducible across all samples yielding very similar amounts. Digestion of proteins with immobilized trypsin keeps trypsin contamination of samples very low and optimization of LCMS conditions ensures maximal peak capacity, Rt reproducibility, and signal intensity while avoiding that signal becomes saturated for any peak (Fig. 2). Rt reproducibility is kept within limits of ±0.7 min and isotope resolution is also very high allowing for confident charge deconvolution. In addition to high mass accuracy (threshold set to max. 30 ppm) these are critical parameters for global LFQ profiling as well as targeted LFQ. To achieve high mass accuracy, internal calibration using a mass calibrant mix is applied for each sample. A custom-fitted dual pico-emitter tip nano-spray arrangement, which delivers calibrant mix independent of the analyte and only during the injection delay window is used. This arrangement gives better results than using a single lock mass and minimizes interference of calibrant ions with analyte during sample elution. The effective Rt range for protein identification and quantitation is from 24 to 64 min (Fig. 2). Using this approach >500 proteins are identified under any condition (FDR < 2%, PRIDE ACs 28622–28627, 28629–28631). Protein and peptide identification data are summarized in supplemental Tables S1 and S2. Moreover, the LCMS data quality is very high. For instance, 85% of all proteins had >5 peptides identified, which is critical for accurate quantitation (PRIDE AC 28628).
Fig. 2.
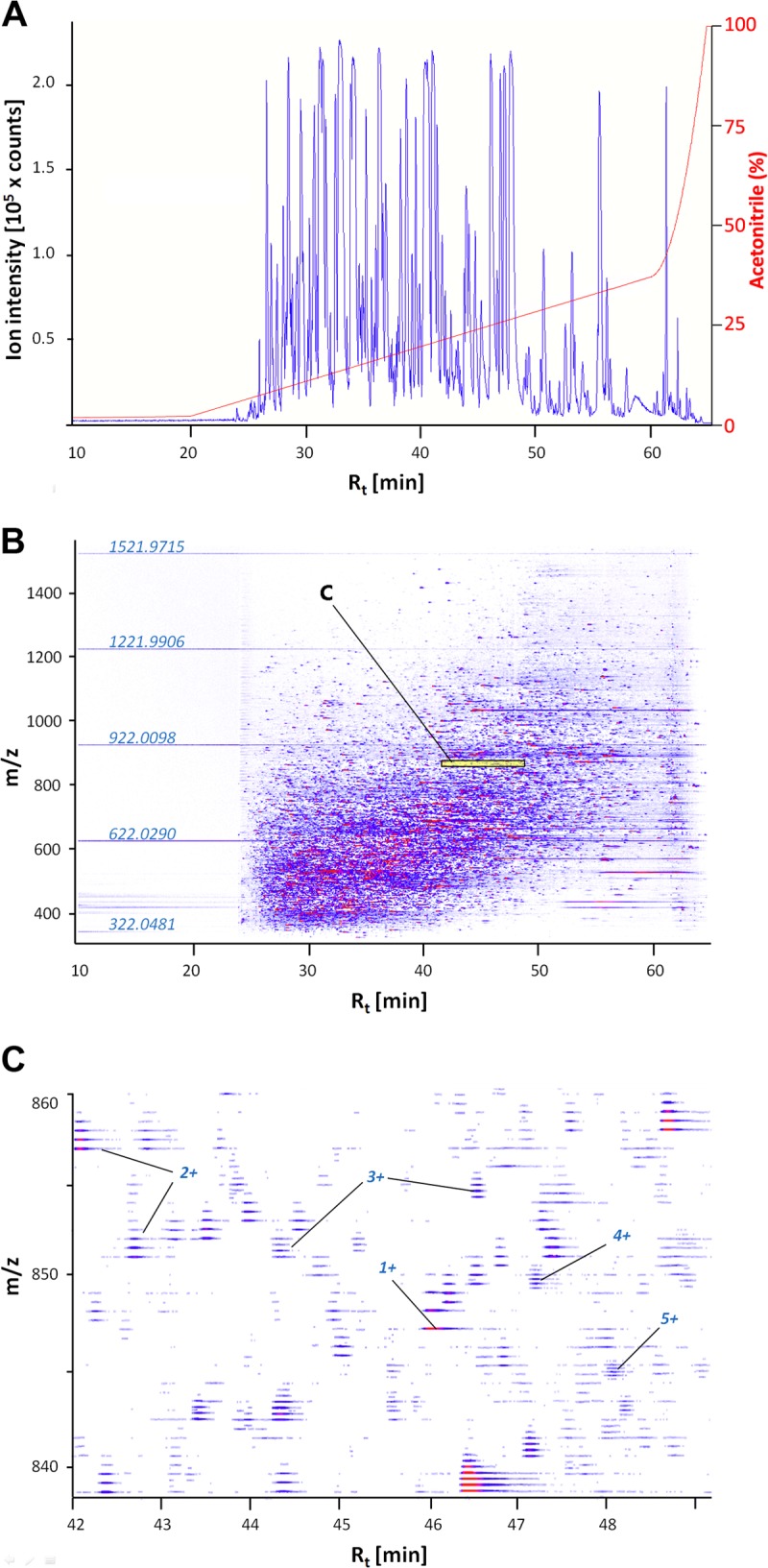
Representative LCMS data for a tryptic peptide mix obtained from a freshwater handling control sample. A, Base peak chromatogram illustrating effective separating range and LC gradient (3 - 35% ACN before rapid increase to 95%). Note that the amount of peptide loaded on the column was optimized to maximize BPC amplitude (max. = 2.55 × 105 counts) while avoiding saturation of signal for any peptide. Total BPC integral (AUC) was used for normalization during targeted LFQ (see text). B, Corresponding two-dimensional chromatogram depicting the elution profile of peptides (Rt in min) relative to m/z values. Note the appearance of mass standards used for internal calibration during the 20 min injection delay. Ion intensity increases by color intensity and from blue to red. Note that very low intensity ions are not visible because the dynamic range of ion intensities detected spans three orders of magnitude; C, Magnification of the area shaded in yellow and labeled “C” in panel B. High mass resolution allows confident deconvolution of charge states (examples for 1+ to 5+ peptides are labeled) and even very highly abundant ions (red center) elute over a narrow (ca. 30 s.) Rt range.
Global Quantitative Peptide and Protein Profiling
Even when using stringent criteria and a high intensity threshold for selecting peptides suitable for quantitative profiling (see Experimental Procedures) a large number of peptides is included in the LFQ analysis. On average 7026 ± 136 (mean ± S.E., n = 12) peptides are quantified by global LFQ profiling in each experiment (Fig. 3). Based on PCA of the quantities of these peptides in each sample the overall effect of salinity stress, i.e. the severity of salinity effects on the gill proteome, can be deduced. The PCA plots indicate that treated samples are much further separated from the controls in the 90 ppt salinity acclimation experiment (Fig. 3C) than in the other two experiments (Figs. 3A, 3B). Therefore, the overall effect of 90 ppt gradual salinity acclimation on the composition of the gill proteome (Fig. 3C) is much stronger than 70 ppt gradual acclimation (Fig. 3B) or 34 ppt acute acclimation (Fig. 3A). The overall extent of gill proteome abundance changes is similar for the 34 ppt and 70 ppt salinity acclimations. PCA plots also reveal that the technical reproducibility of the analysis is very high and that the same conclusions on overall extent of salinity effects on the gill proteome can be reached from every technical replicate. In addition, PCA plots allow an assessment of responses of individual fish. For instance, in the 70 ppt acclimation experiment one salinity stressed fish clearly responded differently from all others (orange circle in lower left corner, Fig. 3B). Although the cause for this difference is not known for the present study the existence of such individual variation and their importance for evolutionary processes is increasingly being emphasized (40).
Fig. 3.

Salinity effects on global peptide abundance patterns. ProfileAnalysis 2.0 (Bruker Daltonics) was used to quantify peptides in each experiment and derive principal component analysis PCA plots to visualize the corresponding data (see Experimental Procedures). The number of quantified peptides for each experiment is depicted in the upper right corner of each PCA plot. Three technical replicates (= separate LCMS runs of the same set of samples) are shown in each row for fish exposed to A 34 ppt, B 70 ppt, and C 90 ppt (all salinity stress samples depicted as orange circles compared with FW handling controls depicted as blue triangles).
The overall effect of salinity stress revealed by PCA at the peptide level is also observed when depicting the results of global quantitative profiling at the protein level (Fig. 4). The extent of regulation of protein abundances is greatest in samples collected from fish gradually acclimated to 90 ppt salinity and this result is highly reproducible in all three technical replicates. In each experiment and technical replicate 533 ± 2 unique proteins are identified by Proteinscape 3.1 (using Mascot and Phenyx search engines, FDR < 2%, Fig. 4). All global profiling LFQ ratios for both peptide- and protein-level analyses have been submitted to the PRIDE repository along with the corresponding MS/MS spectra, protein identification scores, and pertinent metadata. This information can be accessed for all proteins identified in each experiment and technical replicate under PRIDE ACs 28622–28627, 28629–28631. The total number of unambiguous, nonredundant gill proteins identified in all experiments together is 628 (FDR< 2%, PRIDE AC 28628).
Fig. 4.

Global profile of salinity effects on the gill proteome. Each graph depicts the protein LFQ ratio (= mean protein abundance in treated/mean protein abundance in controls) on the Y axis and the protein identification (ID) score obtained with Proteinscape 3.1 on the X axis for every identified protein. The number of identified proteins is depicted in the upper right corner of each graph. Note that all axis scales are plotted as Log2. Three technical replicates (= separate LCMS runs of the same set of samples) are shown for fish exposed to A 34 ppt, B 70 ppt, and C 90 ppt (relative to the corresponding FW handling controls).
Targeted Quantitation of Protein NDRG1
Protein NDRG1 was chosen for targeted LFQ because global LFQ profiling indicated that this protein is strongly down-regulated in gills when tilapia are acclimated to high salinity (70 and 90 ppt). Three NDRG1 peptides fulfill all validation criteria for targeted quantitation. The EICs for those peptides show very clear differences in amplitude between samples from 90 ppt acclimated versus FW handling control fish (Fig. 5A). To visualize a greater amount of quantitative peptide-level data, EIC integrals are compacted into heat maps and depicted for all three experiments (34, 70, and 90 ppt acclimations) in Fig. 5B. These heat maps clearly show that all NDRG1 peptides are very strongly down-regulated in all biological and technical replicates during gradual acclimation of fish to 70 and 90 ppt. In contrast, no effect of salinity on NDRG1 peptide abundances is discernible from the heat maps for the 34 ppt acute salinity stress experiment (Fig. 5B). Peptide-level data were compacted to protein abundance by averaging the abundances of all three peptides in each sample (biological replicate). Fig. 5C depicts the means ± S.E. of protein abundances for six biological replicates from treated samples and corresponding handling controls for all three salinity acclimation experiments. From these data it is evident that gill NDRG1 protein abundance is significantly reduced by >80% in response to gradual acclimation of fish to 70 and 90 ppt but unchanged during 34 ppt acute salinity stress (Fig. 5C). More than 99.5% of the variability in NDRG1 protein abundance can be explained by the effect of salinity treatment for 70 and 90 ppt gradual acclimation (Table I). In addition, Table I shows that for every experiment the portion of NDRG1 protein abundance variability resulting from biological replication (variability among individual fish) is approximately an order of magnitude greater than that resulting from technical replication.
Fig. 5.

Effects of salinity stress on NDRG1 protein abundance. NDRG1 (n-myc downstream regulated gene 1) protein abundance was quantified by targeted proteomics. A, Extracted ion chromatograms (EICs) of three NDRG1 peptides (m/z 518.274, m/z 736.654, m/z 912.417, gray traces = six controls, blue traces = six samples from fish acclimated to 90 ppt). Three technical replicates are depicted (from top to bottom). B, Condensation of targeted EIC integrals into heat maps for all three experiments (34 ppt, 70 ppt, 90 ppt acclimations). Heat maps were generated with Gene-E (BROAD Institute). Peptide abundances are shown as arbitrary units (AU) corresponding to a FW handling control mean of 1. Three technical replicates are depicted (from top to bottom). C, Further condensation of targeted EIC integrals at the protein level. Means ± S.E. of NDRG1 protein abundances in 6 controls (blue triangles) and 6 samples from salinity acclimated fish (orange circles) are shown for all three experiments (34 ppt, 70 ppt, and 90 ppt salinity acclimations). Protein abundance of each biological replicate was calculated as the average of the three corresponding peptide abundances (see panel B). Three technical replicates are depicted (from left to right). An asterisk indicates a significant effect of salinity.
Table I. Detailed two-factorial ANOVA results for targeted quantitation of salinity effects on N-myc downstream regulated gene 1 (NDRG1) protein.
| NDRG1 |
ANOVA |
Treatment |
Control |
Variation resulting from: |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment | Treatment p value | Tech. Rep. p value | Average | Variance | Average | Variance | Treatment effect | Technical replication | Biological replication |
| 34 ppt | 9.28E-01 | 0.90 | 0.99 | 0.096 | 1.00 | 0.180 | 0.74% | 9.71% | 89.55% |
| 70 ppt | 6.83E-18 | 1.00 | 0.18 | 0.002 | 1.00 | 0.029 | 99.71% | 0.00% | 0.29% |
| 90 ppt | 1.62E-16 | 0.93 | 0.16 | 0.002 | 1.00 | 0.040 | 99.60% | 0.02% | 0.37% |
Targeted Quantitation of Proteins Involved in Transepithelial Ion Transport
All nine proteins identified and known to be key proteins for transepithelial ion transport across gill epithelium have been analyzed by targeted quantitation. They include two isoforms of carbonic anhydrase (CA1 and CA2), Na+-K+-2Cl− cotransporter (NKCC), three isoforms of the Na+/K+-ATPase alpha subunit (NKA-α1a, NKA-α1b, NKA-α1c), the Na+/K+-ATPase beta subunit 233 (NKA-β233), the V-type H+-ATPase catalytic subunit A (HA-a), and the V-type H+-ATPase subunit B (HA-b). The UniprotKB accession numbers and information about peptides used for targeted LFQ of these proteins are shown in supplemental Fig. S1A. The heat map in supplemental Fig. S1A demonstrates the overall high technical reproducibility and consistency of peptide-level data for targeted LFQ of these proteins. When compacted at the protein level, targeted LFQ reveals that one of the two carbonic anhydrase isoforms (CA1) is significantly increased at all salinities, the largest increase (fourfold) being observed at 90 ppt (Fig. 6A). The other carbonic anhydrase isoform (CA2) does not change much although a slight decrease is evident at 34 ppt and a slight increase at 90 ppt. NKCC is significantly increased at 70 ppt (fourfold) and 90 ppt (sevenfold) but not at 34 ppt. NKA-α1b is significantly (fourfold) increased at 34 and 90 ppt but unchanged at 70 ppt whereas NKA-β233 is significantly increased at all salinities (twofold at 34 ppt, 1.5-fold at 70 ppt, fourfold at 90 ppt). Interestingly, NKA-α1c is only significantly increased at 34ppt (twofold) and NKA-α1a is down-regulated after gradual salinity acclimation to 70 ppt and 90 ppt. A general trend toward down-regulation is also seen for both subunits of the V-type H+-ATPase at all salinities but this trend is only statistically significant for the B subunit at 70 and 90 ppt (Fig. 6A).
Fig. 6.

Salinity-dependent regulation of A transepithelial ion transport proteins, B molecular chaperones, and C mitochondrial proteins. Means ± S.E. of three technical replicates are depicted. A filled circle indicates a statistically significant effect of salinity on the abundance of the corresponding protein whereas open circles indicate that there is no significant difference in the abundance of the corresponding protein relative to fresh water handling controls. For abundances of all diagnostic peptides representing each protein in each biological replicate (n = 6 for each experimental group) please consult the heat maps in supplemental Fig. S2. Detailed results of two-factor ANOVA analyses including p values for treatment effects and technical replicates and the proportions of variation in protein abundances that are attributable to treatment effects, technical replication, and biological (individual) variability are provided in supplemental Table S3.
Targeted Quantitation of Molecular Chaperones
All ten molecular chaperones confidently identified have been analyzed by targeted LFQ. They include heat shock protein 10 (HSP10), HSP60, five distinct isoforms of HSP70, HSP90-beta, and the beta and zeta subunits of T-complex protein 1. At least three peptides are valid for targeted LFQ of each of these molecular chaperones and the level of consistency of the abundance ratio of these peptides (treated versus control) within a single protein is high (supplemental Fig. S1B). All molecular chaperones increase significantly in abundance during 34 ppt acute salinity stress (Fig. 6B). In addition, three molecular chaperones - HSP10, HSP60, and one HSP70 isoform (UniProtKB AC I3J8F4) are strongly and significantly (twofold) more abundant at 90 ppt compared with FW handling controls. At 70 ppt only a single molecular chaperone (HSP70 isoform I3J8F4) is significantly (1.5-fold) up-regulated whereas most other molecular chaperones are slightly but significantly down-regulated (Fig. 6B).
Targeted Quantitation of Mitochondrial Proteins
All mitochondrial proteins identified and analyzed by targeted LFQ are up-regulated in response to 34 ppt and 90 ppt salinity acclimations, the great majority of them significantly (Fig. 6C). This concerted response of mitochondrial proteins to severe salinity stress is highly reproducible in all three technical replicates (supplemental Fig. S1C). Supplemental Fig. S1C shows peptides used for targeted quantitation, protein annotation information, and a peptide-level quantitative heat map for all identified mitochondrial proteins. From this comprehensive heat map it is evident that the majority of quantitative peptide-level ratios (treated versus control) is consistent for any given protein. Therefore, the precision of targeted LFQ quantitation is high when LCMS conditions meet the requirements for reproducibility, accuracy, and resolution, and diagnostic peptides are carefully validated using the criteria outlined above (see Experimental Procedures). Interestingly, proteins associated with amino acid metabolism are generally not as strongly up-regulated than the other mitochondrial proteins (Fig. 6C). The results shown in Fig. 6C suggest that, although the overall regulation of mitochondrial proteins during salinity stress is highly coordinated, not all mitochondrial proteins are regulated to the same extent. Even at 90 ppt there are several mitochondrial proteins whose abundance does not significantly increase. Nevertheless, the strong overall increase in mitochondrial proteins involved in energy and oxidative metabolism at 90 ppt indicates that the energetic demand for fueling ion transporters and molecular chaperones discussed above and other ATP-consuming processes involved in osmotic stress coping responses increases greatly at salinities approaching the tolerance limit of this species. This observation is consistent with the significantly reduced body mass of tilapia acclimated to 90 ppt compared with FW controls, which suggests that ATP and nutrients are shunted away from growth (and reproduction) during severe salinity stress. Acclimation of fish to 70 ppt has a much smaller effect on mitochondrial protein abundance than the 34 and 90 ppt acclimations. This observation is consistent with undisturbed growth of fish during long-term gradual acclimation to 70 ppt and indicates that the energy demands for both osmoregulation and growth/basal metabolism are readily met at this more moderate salinity.
The relative portions of variation in the abundances of ion transport proteins, molecular chaperones, and mitochondrial proteins that are resulting from salinity treatments, individual variability (biological replication), and technical replication are shown in supplemental Table S3. The average ratio of biological/technical variability is 6.41 ± 2.10 (mean ± S.E., n = 27) for ion transport proteins, 3.40 ± 1.44 (mean ± S.E., n = 30) for molecular chaperones, and 6.02 ± 1.03 (mean ± S.E., n = 219) for mitochondrial proteins.
DISCUSSION
Accurate Quantitation Via Two-tiered LFQ Proteomics
The present study uses a two-tiered LFQ workflow for investigating how salinity affects the tilapia gill proteome. This species and tissue were chosen because considerable prior knowledge on gill responses to salinity stress exists (4, 8, 13, 17). Therefore, it is possible to validate the workflow by assessing how well it reproduces previously established or expected effects on abundances of certain gill proteins. In particular, it is well known that the number and size of gill mitochondria-rich ionocytes increase significantly when FW-acclimated Mozambique tilapia are exposed to severe acute (34 ppt) or gradual (90 ppt) salinity stress (8, 41–43). The extent of increase in mitochondria-rich ionocyte quantity at the cellular level is readily reflected in the extent of increased abundance of mitochondrial proteins under comparable conditions in this study. In addition, the overall high consistency of up-regulation of mitochondrial proteins agrees with a general increase in mitochondria in gills of fish exposed to severe salinity stress (as opposed to a change in particular mitochondrial proteins). Moreover, the abundances of key proteins involved in active ion transport across gill epithelium of tilapia and other euryhaline species are altered by salinity in the same direction and to very similar extent as previously reported using other methods (6, 13, 44–49). Furthermore, significant up-regulation of HSP70 and HSP90 in gills has been previously observed with other methods in tilapia and other species of euryhaline fish exposed to severe salinity stress (3, 50).
The initial global LFQ profiling step is sufficient to identify many up- or down-regulated proteins and discern different severities of salinity stress (Figs. 3, 4). However, quantitative changes in the abundance of proteins determined by global LFQ profiling only represent estimates because all peptides identified for a particular protein are considered valid in this global screening approach. This assumption is incorrect because some of these peptides bear post-translational modifications or are incompletely or nonspecifically cleaved at variable stoichiometry. Other peptides used in global LFQ profiling belong to more than just a single protein and isoform. Furthermore, some peptides may be erroneously assigned to a protein if they co-elute with peptides of nearly identical mass and retention time but belong to a different protein (e.g. when the peptide consists of the same amino acids arranged in a different order). Other peptides produce missing values for multiple biological replicates because the intensity threshold (peak height) for global profiling is high to minimize inclusion of noisy signals. These shortcomings can be efficiently addressed by targeted LFQ of proteins of interest that are selected based on the initial global profiling step. For targeted LFQ only peptides meeting all validation criteria (see Experimental Procedures) are used and agreement among peptide quantities for a particular protein is high as illustrated by peptide-level heat maps. Such heat maps represent a very effective and rapidly comprehendible visualization and quality control tool for quantitative proteomics data (Fig. 5 and supplemental Fig. S1). They illustrate that a two-tiered workflow consisting of (a) an “accurate mass and time tag” approach for initial global profiling (20, 38, 51–53) in combination with (b) targeted LFQ of particular proteins of interest based on scheduled precursor lists and alternating MS and MS/MS acquisition (39) improves label-free quantitation accuracy. Although the additional targeted LFQ step is time-consuming when applied to a large number of proteins it offers similar advantages as other targeted approaches such as selected reaction monitoring (SRM) (54).
The present study uses advantages of ion current and chromatographic peak area integration LFQ approaches for analyzing tissues from complex organisms. An important advantage of LFQ approaches consists of avoiding difficulties in applying isotope labels to intact fish. Another advantage concerns the absence of chemical derivatization of reactive amino groups of tryptic peptides, the extent of which may vary during isotope labeling. In addition, a large number of independent previous studies have thoroughly established the suitability of LFQ approaches for quantitative proteomics (20, 25, 39). Normalization of peptide (and correspondingly protein) abundances to account for systematic differences in sample loading or instrument sensitivity represents an important aspect of quantitative proteome analyses, in particular for label-free approaches. During the initial global profiling step, quantitative peptide ratios are normalized on the basis of the assumption that the median values of peptide abundance ratios in the compared samples are equal (55, 56). However, the validity of this assumption is not certain for samples that differ substantially (e.g. 90 ppt salinity stress compared with FW handling controls). Alternative normalization procedures use certain “housekeeping” proteins whose abundances are assumed unaffected by the treatment but such normalization may not be adequate in many cases (57). Likewise, spiking of a reference standard into already highly complex gill samples would not account for variability before the spiking event and further increase sample complexity, which exacerbates ion suppression problems. Therefore, the BPC integral is used for normalization because it reflects the overall ion intensity of not just peptides from one or a few “housekeeping” protein(s) but from many different proteins at once. The results of this study clearly illustrate that the technical reproducibility is very high given the workflow, instrumentation, and parameters used. The portion of biological variability among individual fish is substantially higher than that explained by technical replication for all groups of proteins targeted. Therefore, future studies using this workflow should emphasize biological replication over technical replication for minimizing variability that is not a result of treatment effects.
Novel Insights Into Salinity Effects on Tilapia Gill Proteins
The overall effect of salinity stress on mitochondrial proteins, proteins involved in transepithelial ion transport, and molecular chaperones is as expected and had already been well documented using approaches other than quantitative proteomics (see above). In addition to independently confirming well-documented general trends using label-free quantitative proteomics, this study reveals differences in the extent of salinity effects on relevant tilapia proteins. Moreover, specific isoforms of proteins that are involved in salinity stress responses in tilapia gill are identified. For instance, two isoforms of carbonic anhydrase are highly expressed in gills of tilapia but only one (CA1) is regulated in response to salinity stress. This enzyme is important for acid-base balance and facilitation of CO2 secretion across fish gills (58). Because oxidative metabolism in mitochondria-rich ionocytes increases greatly at 90 ppt there is an increased demand for CO2 secretion and metabolic compensation of acidosis, which is facilitated by up-regulation of CA1. Salinity-induced up-regulation of CA activity has previously been reported for tilapia gills (6). The data presented here indicate that the isoform mostly responsible for this activity increase is CA1 and that the mechanism is an increase in CA1 abundance. The present study also identifies the specific alpha and beta subunits of NKA and the NKCC, which are key transporters that are increased by salinity stress and localized to mitochondria-rich ionocytes in gills of euryhaline fish (48, 59). The observed salinity-induced regulation of two distinct NKA alpha subunits in opposite direction is consistent with previous reports on tilapia and other species of euryhaline fish (46, 60). Thus, NKA alpha 1b is used for active salt secretion in a hyperosmotic (relative to plasma) environment whereas NKA alpha 1a contributes to active salt absorption in a hyposmotic (relative to plasma) environment. The abundances of both HA subunits either decrease or do not change during salinity stress, which is consistent with their localization in the freshwater-type ionocytes rather than the mitochondria-rich salt-secretory ionocytes (60–62).
In addition, the present study shows that a specific isoform of HSP70 (stress 70 protein) increases in all three salinity acclimation experiments. All other HSP70 isoforms are regulated differently than that particular isoform. Like the stress 70 protein isoform of HSP70, HSP10 and HSP60 are robustly up-regulated during severe acute (34 ppt) and gradual (90 ppt) salinity stress. All three of these molecular chaperones recognize unfolded proteins in mitochondria and cytosol to aid in their refolding or proteolytic processing (depending on the degree of unfolding) (63). Therefore, it is likely that the up-regulation of these molecular chaperones indicates an increase in the amount of protein unfolding, in particular in mitochondria, during severe (34 ppt acute, 90 ppt gradual) salinity stress (64). At more moderate salinity (70 ppt), the stress 70 protein isoform of HSP70 may be sufficient to recognize and reconstitute proteins damaged by milder salinity stress without a need for an increased contribution from the other HSPs. In general, knowledge about specific protein isoforms that are most responsive to salinity changes in the environment is not only useful for biomarker assessment but will also facilitate future studies aimed at identifying the mechanisms underlying transduction of environmental signals from sensors to specific effector proteins.
An intriguing observation in the present study is that salinity effects on certain mitochondrial proteins are not as pronounced as for the majority of mitochondrial proteins. In particular, proteins involved in amino acid metabolism are consistently increased to a lesser extent during salinity stress than most other mitochondrial proteins. During severe salinity stress, activation of energy metabolism may have priority to produce ATP needed for fueling active transepithelial ion transport (65–67). In addition, activation of fatty acid metabolism may be essential to repair membrane damage and adjust the lipid composition/physicochemical properties of cell membranes in the face of altered salinity (68, 69). Proteins that are marginally or not significantly up-regulated may not be rate-limiting for energy metabolism and membrane maintenance (70, 71). Taken together, the data support the notion that an increase in mitochondria-rich ionocytes and the number of mitochondria is accompanied by fine-tuning the mitochondrial proteome to maximize energetic efficiency and meet the demands for optimizing cell membrane integrity during severe salinity stress.
Regulation of Protein NDRG1 During Salinity Stress
The primary intent of this study is to apply a thoroughly optimized, two-tiered LFQ proteomics workflow to perform quantitative molecular phenotyping of mitochondrial proteins, ion transport proteins, and molecular chaperones that play an important role during salinity acclimation of euryhaline fish. However, the initial global profiling step of quantitative analysis has revealed additional candidate proteins likely to be regulated during salinity stress in tilapia gill (see PRIDE ACs 28622–28627, 28629–28631). Because most mitochondrial proteins, ion transport proteins, and molecular chaperones are up-regulated during salinity stress, a candidate for a strongly down-regulated protein was also included in the targeted LFQ approach. Protein NDRG1 has not been previously identified in fish gills or in the context of salinity stress. However, it is known from experiments on other organisms that it is a cytoplasmic tumor suppressor protein involved in the regulation of cell proliferation and differentiation (72, 73). Elevated salinity strongly decreases NDRG1 levels whereas accelerating the turnover and proliferation rates of gill cells (74, 75). This inverse relationship between NDRG1 levels and cell proliferation suggests a possible antiproliferative role of NDRG1 in tilapia gill epithelium. Mammalian NDRG1 compartmentalizes to endosomes and has a function in the recycling of certain membrane proteins (e.g. E-cadherin) via vesicular transport (76). Therefore, its down-regulation at high salinity may promote redistribution of membrane proteins and formation of the extensive tubulovesicular membrane network that is characteristic of salt-secretory mitochondria-rich ionocytes (2). Moreover, protein NDRG1 induces key intracellular stress signaling proteins in mammalian cells via the TGF-beta pathway (protein phosphatase PTEN, transcription factors GLIS3 and SMAD4, and E3 ubiquitin ligase NEDD4) whereas it suppresses PI3K/AKT, Ras/ERK, and p53 (via p21 up-regulation) signaling pathways (77–79). Because these pathways have previously been implicated in cellular responses to osmotic stress and in maintaining distinct states of cellular differentiation (30, 80–83) NDRG1 may contribute substantially to the remodeling of gill epithelium that occurs when euryhaline fish are transferred from a hyposmotic to a hyperosmotic (relative to plasma) environment. However, in contrast to rapid up-regulation in response to hypoxic stress (84), during acute salinity stress (34 ppt/2 days) NDRG1 protein abundance is not significantly altered. The lack of a rapid change in abundance may indicate that NDRG1 protein is relatively stable. Hence, changes in its abundance may serve to stabilize rather than induce particular salinity-specific gill epithelial cell phenotypes. Moreover, posttranslational modification of NDRG1 can alter its function more rapidly than abundance changes. NDRG1 is a specific substrate for serum-and glucocorticoid-induced kinase 1 (SGK1), which phosphorylates Thr 346/356/366 in mammalian NDRG1 (85). In addition, once phosphorylated by SGK1 it becomes a specific substrate for glycogen synthase kinase 3 (GSK3), which phosphorylates Ser 342/352/362 in mammalian NDRG1 (86). These six phosphorylation sites are found in the decapeptide GTRSRSHTSE arranged in triplicate in mammalian NDRG1 (86). This decapeptide triplicate near the C terminus is conserved with the corresponding Ser and Thr residues at identical positions in tilapia NDRG1 (UniProtKB AC I3J169). Considering the evolutionary conservation of these phosphorylation sites together with previous reports on potent osmotic regulation of SGK1 and GSK3 activities in other organisms (87–90), the role of phosphorylation in the regulation of tilapia NDRG1 during salinity stress represents an intriguing target for future studies.
In summary, the present study demonstrates the utility of a two-tiered LFQ proteomics workflow for phenotyping molecular changes occurring in gills of euryhaline tilapia during environmental salinity change. The quantitative data agree with known salinity effects on mitochondria-rich ionocytes, key transport proteins involved in transepithelial ion transport, and molecular chaperones. In addition, the specific protein isoforms that are involved in these responses have been quantified and it was shown that salinity effects on the mitochondrial proteome are not entirely uniform. Moreover, protein NDRG1 was identified as a novel cell differentiation protein that is strongly down-regulated during long-term salinity stress. Taken together, these data provide new insight into the molecular mechanisms that promote remodeling of gill epithelium in euryhaline fish exposed to environmental salinity change.
Supplementary Material
Acknowledgments
We thank the Bruker Daltonics technical support team, in particular Wolfgang Jabs, Peter Hufnagel, and Henri O'Connor. In addition, we thank Alex Masselot and Pierre-Alain Binz (Genebio), Richard Jacob (Matrixscience), and Attila Csordas (PRIDE team at EBI) for technical assistance.
Footnotes
* This work was supported by grants from the National Science Foundation (IOS-1049780) and the National Institutes of Health (NIEHS P42-ES004699).
 This article contains supplemental Fig. S1 and Tables S1 to S3.
This article contains supplemental Fig. S1 and Tables S1 to S3.
1 The abbreviations used are:
- FW
- fresh water
- AC
- accession number
- ACN
- acetonitrile
- BPC
- base peak chromatogram
- BW
- brackish water
- CA
- carbonic anhydrase
- EIC
- extracted ion chromatogram
- ERK
- extracellular signal-regulated kinase
- FA
- formic acid
- GSK3
- glycogen synthase kinase 3
- HA
- V-type H+-ATPase
- TIC
- total ion chromatogram
- LFQ
- label-free quantitation
- NDRG1
- N-myc downstream regulated gene 1
- NEDD4
- neural precursor cell expressed developmentally down-regulated protein 4
- NKA
- Na+/K+-ATPase
- NKCC
- Na+/K+/Cl− cotransporter
- PI3K
- phosphoinositide 3-kinase
- PCA
- principal component analysis
- ppt
- parts per thousand
- Ras
- Rat sarcoma protein
- SGK1
- serum and glucocorticoid inducible kinase 1
- SMAD
- small body size/mothers against decapentaplegic homolog
- SW
- seawater.
REFERENCES
- 1. Evans D. H. (2008) Teleost fish osmoregulation: what have we learned since August Krogh, Homer Smith, and Ancel Keys. Am. J. Physiol. 295, R704–R713 [DOI] [PubMed] [Google Scholar]
- 2. Karnaky K. J. (1986) Structure and function of the chloride cell of Fundulus heteroclitus and other teleosts. Am. Zool. 26, 209–224 [Google Scholar]
- 3. Kültz D. (1996) Plasticity and stressor specificity of osmotic and heat shock responses of Gillichthys mirabilis gill cells. Am. J. Physiol.-Cell 271, C1181–C1193 [DOI] [PubMed] [Google Scholar]
- 4. Dymowska A. K., Hwang P. P., Goss G. G. (2012) Structure and function of ionocytes in the freshwater fish gill. Resp. Physiol. Neurobiol. 184, 282–292 [DOI] [PubMed] [Google Scholar]
- 5. Galvez F., Reid S. D., Hawkings G., Goss G. G. (2002) Isolation and characterization of mitochondria-rich cell types from the gill of freshwater rainbow trout. Am. J. Physiol.-Reg. Int. Comp. Physiol. 282, R658–R668 [DOI] [PubMed] [Google Scholar]
- 6. Kültz D., Bastrop R., Jürss K., Siebers D. (1992) Mitochondria-rich (MR) Cells and the activities of the Na+/K+-ATPase and carbonic anhydrase in the gill and opercular epithelium of Oreochromis mossambicus adapted to various salinities. Comp. Biochem. Physiol. B 102, 293–301 [Google Scholar]
- 7. Foskett J. K., Bern H. A., Machen T. E., Conner M. (1983) Chloride cells and the hormonal control of teleost fish osmoregulation. J. Exp. Biol. 106, 255–281 [DOI] [PubMed] [Google Scholar]
- 8. Kültz D., Jürss K., Jonas L. (1995) Cellular and epithelial adjustments to altered salinity in the gill and opercular epithelium of a cichlid fish (Oreochromis mossambicus). Cell Tissue Res. 279, 65–73 [Google Scholar]
- 9. Stickney R. R. (1986) Tilapia tolerance of saline waters - a review. Prog. Fish Cult. 48, 161–167 [Google Scholar]
- 10. Costa-Pierce B. A. (2003) Rapid evolution of an established feral tilapia (Oreochromis spp.): The need to incorporate invasion science into regulatory structures. Biol. Invasions 5, 71–84 [Google Scholar]
- 11. Sardella B. A., Brauner C. J. (2007) Cold temperature-induced osmoregulatory failure: The physiological basis for tilapia winter mortality in the Salton Sea? Calif. Fish Game 93, 200–213 [Google Scholar]
- 12. Miles K. A., Ricca M. A., Meckstroth A., Spring S. E. (2009) Salton Sea Ecosystem Monitoring Project. U.S. Geol. Survey Open-File Rep. 1276, 1–150 [Google Scholar]
- 13. Sardella B. A., Matey V., Cooper J., Gonzalez R. J., Brauner C. J. (2004) Physiological, biochemical and morphological indicators of osmoregulatory stress in ‘California’ Mozambique tilapia (Oreochromis mossambicus x O. urolepis hornorum) exposed to hypersaline water. J. Exp. Biol. 207, 1399–1413 [DOI] [PubMed] [Google Scholar]
- 14. D'Amato M. E., Esterhuyse M. M., van der Waal B. C. W., Brink D., Volckaert F. A. M. (2007) Hybridization and phylogeography of the Mozambique tilapia Oreochromis mossambicus in southern Africa evidenced by mitochondrial and microsatellite DNA genotyping. Conserv. Genet. 8, 475–488 [Google Scholar]
- 15. Wang W. S., Hung S. W., Lin Y. H., Tu C. Y., Wong M. L., Chiou S. H., Shieh M. T. (2007) Purification and localization of nitric oxide synthases from hybrid tilapia (Nile tilapia x Mozambique tilapia). J. Aquat. Anim. Health 19, 168–178 [DOI] [PubMed] [Google Scholar]
- 16. Grau E. G., Helms L. M. (1990) The tilapia prolactin cell: twenty-five years of investigation. Prog. Clin. Biol. Res. 342, 534–540 [PubMed] [Google Scholar]
- 17. Cataldi E., Mandich A., Ozzimo A., Cataudella S. (2005) The interrelationships between stress and osmoregulation in a euryhaline fish, Oreochromis mossambicus. J. Appl. Ichthyol. 21, 229–231 [Google Scholar]
- 18. Ong S. E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nature Chem. Biol. 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 19. Bostanci N., Heywood W., Mills K., Parkar M., Nibali L., Donos N. (2010) Application of label-free absolute quantitative proteomics in human gingival crevicular fluid by LC/MS E (gingival exudatome). J. Proteome Res. 9, 2191–2199 [DOI] [PubMed] [Google Scholar]
- 20. Cutillas P. R., Vanhaesebroeck B. (2007) Quantitative profile of five murine core proteomes using label-free functional proteomics. Mol. Cell. Proteomics 6, 1560–1573 [DOI] [PubMed] [Google Scholar]
- 21. Matzke M. M., Brown J. N., Gritsenko M. A., Metz T. O., Pounds J. G., Rodland K. D., Shukla A. K., Smith R. D., Waters K. M., McDermott J. E., Webb-Robertson B. J. (2013) A comparative analysis of computational approaches to relative protein quantification using peptide peak intensities in label-free LC-MS proteomics experiments. Proteomics 13, 493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mallick P., Kuster B. (2010) Proteomics: a pragmatic perspective. Nat. Biotechnol. 28, 695–709 [DOI] [PubMed] [Google Scholar]
- 23. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 24. Cutillas P. R., Geering B., Waterfield M. D., Vanhaesebroeck B. (2005) Quantification of gel-separated proteins and their phosphorylation sites by LC-MS using unlabeled internal standards: analysis of phosphoprotein dynamics in a B cell lymphoma cell line. Mol. Cell. Proteomics 4, 1038–1051 [DOI] [PubMed] [Google Scholar]
- 25. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 26. Hofmann G. E., Buckley B. A., Airaksinen S., Keen J. E., Somero G. N. (2000) Heat shock protein expression is absent in the Antarctic fish Trematomus bernacchii (family Nototheniidae). J. Exp. Biol. 203, 2331–2339 [DOI] [PubMed] [Google Scholar]
- 27. Ojima N., Mekuchi M., Ineno T., Tamaki K., Kera A., Kinoshita S., Asakawa S., Watabe S. (2012) Differential expression of heat-shock proteins in F2 offspring from F1 hybrids produced between thermally selected and normal rainbow trout strains. Fisheries Sci. 78, 1051–1057 [Google Scholar]
- 28. Csordas A., Ovelleiro D., Wang R., Foster J. M., Rios D., Vizcaino J. A., Hermjakob H. (2012) PRIDE: quality control in a proteomics data repository. Database 2012, bas004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fiol D. F., Sanmarti E., Lim A. H., Kültz D. (2011) A novel GRAIL E3 ubiquitin ligase promotes environmental salinity tolerance in euryhaline tilapia. Biochim. Biophys. Acta 1810, 439–445 [DOI] [PubMed] [Google Scholar]
- 31. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 32. Masselot A., Binz P. A., Cambria L., Appel R. D. (2004) Phenyx: Combining high-throughput and pertinence in protein identification. Mol. Cell. Proteomics 3, S257–S257 [Google Scholar]
- 33. Götz S., Garcia-Gómez J. M., Terol J., Williams T. D., Nagaraj S. H., Nueda M. J., Robles M., Talón M., Dopazo J., Conesa A. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J., Xin L., Shan B. Z., Chen W. W., Xie M. J., Yuen D., Zhang W. M., Zhang Z. F., Lajoie G. A., Ma B. (2012) PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteomics 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Côté R. G., Griss J., Dianes J. A., Wang R., Wright J. C., van den Toorn H. W., van Breukelen B., Heck A. J., Hulstaert N., Martens L., Reisinger F., Csordas A., Ovelleiro D., Perez-Rivevol Y., Barsnes H., Hermjakob H., Vizcaino J. A. (2012) The PRoteomics IDEntification (PRIDE) Converter 2 framework: an improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Mol. Cell. Proteomics 11, 1682–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thiele H., Glandorf J., Hufnagel P. (2010) Bioinformatics strategies in life sciences: from data processing and data warehousing to biological knowledge extraction. J. Integr. Bioinformatics 7, 141. [DOI] [PubMed] [Google Scholar]
- 37. Wang R., Fabregat A., Rios D., Ovelleiro D., Foster J. M., Cote R. G., Griss J., Csordas A., Perez-Riverol Y., Reisinger F., Hermjakob H., Martens L., Vizcaino J. A. (2012) PRIDE Inspector: a tool to visualize and validate MS proteomics data. Nat. Biotechnol. 30, 135–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim Y. J., Feild B., Fitzhugh W., Heidbrink J. L., Duff J. W., Heil J., Ruben S. M., He T. (2009) Reference map for liquid chromatography-mass spectrometry-based quantitative proteomics. Anal. Biochem. 393, 155–162 [DOI] [PubMed] [Google Scholar]
- 39. Silva J. C., Denny R., Dorschel C., Gorenstein M. V., Li G. Z., Richardson K., Wall D., Geromanos S. J. (2006) Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol. Cell. Proteomics: MCP 5, 589–607 [DOI] [PubMed] [Google Scholar]
- 40. Kültz D., Clayton D. F., Robinson G. E., Albertson C., Carey H. V., Cummings M. E., Dewar K., Edwards S. V., Hofmann H. A., Gross L. J., Kingsolver J. G., Meaney M. J., Schlinger B. A., Shingleton A. W., Sokolowski M. B., Somero G. N., Stanzione D. C., Todgham A. E. (2013) New frontiers for organismal biology. Bioscience 63, 464–471 [Google Scholar]
- 41. Inokuchi M., Kaneko T. (2012) Recruitment and degeneration of mitochondrion-rich cells in the gills of Mozambique tilapia Oreochromis mossambicus during adaptation to a hyperosmotic environment. Comp. Biochem. Physiol. A 162, 245–251 [DOI] [PubMed] [Google Scholar]
- 42. Ouattara N., Bodinier C., Negre-Sadargues G., D'Cotta H., Messad S., Charmantier G., Panfili J., Baroiller J. F. (2009) Changes in gill ionocyte morphology and function following transfer from fresh to hypersaline waters in the tilapia Sarotherodon melanotheron. Aquaculture 290, 155–164 [Google Scholar]
- 43. Sardella B. A., Brauner C. J. (2008) The effect of elevated salinity on ‘California’ Mozambique tilapia (Oreochromis mossambicus x O-urolepis hornorum) metabolism. Comp. Biochem. Physiol. C 148, 430–436 [DOI] [PubMed] [Google Scholar]
- 44. Laverty G., Skadhauge E. (2012) Adaptation of teleosts to very high salinity. Comp. Biochem. Physiol. A 163, 1–6 [DOI] [PubMed] [Google Scholar]
- 45. Uchiyama M., Komiyama M., Yoshizawa H., Shimizu N., Konno N., Matsuda K. (2012) Structures and immunolocalization of Na+, K+-ATPase, Na+/H+ exchanger 3 and vacuolar-type H+-ATPase in the gills of blennies (Teleostei: Blenniidae) inhabiting rocky intertidal areas. J. Fish Biol. 80, 2236–2252 [DOI] [PubMed] [Google Scholar]
- 46. Tipsmark C. K., Breves J. P., Seale A. P., Lerner D. T., Hirano T., Grau E. G. (2011) Switching of Na+, K+ -ATPase isoforms by salinity and prolactin in the gill of a cichlid fish. J. Endocrinol. 209, 237–244 [DOI] [PubMed] [Google Scholar]
- 47. Lin C. H., Huang C. L., Yang C. H., Lee T. H., Hwang P. P. (2004) Time-course changes in the expression of Na, K-ATPase and the morphometry of mitochondrion-rich cells in gills of euryhaline tilapia (Oreochromis mossambicus) during freshwater acclimation. J. Exp. Zool. A 301, 85–96 [DOI] [PubMed] [Google Scholar]
- 48. Wu Y. C., Lin L. Y., Lee T. H. (2003) Na+,K+,2Cl--cotransporter: A novel marker for identifying freshwater- and seawater-type mitochondria-rich cells in gills of the euryhaline tilapia, Oreochromis mossambicus. Zool. Stud. 42, 186–192 [Google Scholar]
- 49. Lee T. H., Tsai J. C., Fang M. J., Yu M. J., Hwang P. P. (1998) Isoform expression of Na+-K+-ATPase alpha-subunit in gills of the teleost Oreochromis mossambicus. Am. J. Physiol. 275, R926–R932 [DOI] [PubMed] [Google Scholar]
- 50. Gonzalez R. J., Cooper J., Head D. (2005) Physiological responses to hyper-saline waters in sailfin mollies (Poecilia latipinna). Comp. Biochem. Physiol. A 142, 397–403 [DOI] [PubMed] [Google Scholar]
- 51. Smith R. D., Anderson G. A., Lipton M. S., Pasa-Tolic L., Shen Y., Conrads T. P., Veenstra T. D., Udseth H. R. (2002) An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics 2, 513–523 [DOI] [PubMed] [Google Scholar]
- 52. Stanley J. R., Adkins J. N., Slysz G. W., Monroe M. E., Purvine S. O., Karpievitch Y. V., Anderson G. A., Smith R. D., Dabney A. R. (2011) A statistical method for assessing peptide identification confidence in accurate mass and time tag proteomics. Anal. Chem. 83, 6135–6140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jaitly N., Monroe M. E., Petyuk V. A., Clauss T. R., Adkins J. N., Smith R. D. (2006) Robust algorithm for alignment of liquid chromatography-mass spectrometry analyses in an accurate mass and time tag data analysis pipeline. Anal. Chem. 78, 7397–7409 [DOI] [PubMed] [Google Scholar]
- 54. Boja E. S., Rodriguez H. (2012) Mass spectrometry-based targeted quantitative proteomics: achieving sensitive and reproducible detection of proteins. Proteomics 12, 1093–1110 [DOI] [PubMed] [Google Scholar]
- 55. Wang W., Zhou H., Lin H., Roy S., Shaler T. A., Hill L. R., Norton S., Kumar P., Anderle M., Becker C. H. (2003) Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem. 75, 4818–4826 [DOI] [PubMed] [Google Scholar]
- 56. Silva J. C., Denny R., Dorschel C. A., Gorenstein M., Kass I. J., Li G. Z., McKenna T., Nold M. J., Richardson K., Young P., Geromanos S. (2005) Quantitative proteomic analysis by accurate mass retention time pairs. Anal. Chem. 77, 2187–2200 [DOI] [PubMed] [Google Scholar]
- 57. Ralston-Hooper K. J., Turner M. E., Soderblom E. J., Villeneuve D., Ankley G. T., Moseley M. A., Hoke R. A., Ferguson P. L. (2013) Application of a label-free, gel-free quantitative proteomics method for ecotoxicological studies of small fish species. Environ. Sci. Technol. 47, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 58. Gilmour K. M., Perry S. F. (2009) Carbonic anhydrase and acid-base regulation in fish. J. Exp. Biol. 212, 1647–1661 [DOI] [PubMed] [Google Scholar]
- 59. Hiroi J., McCormick S. D. (2007) Variation in salinity tolerance, gill Na+/K+-ATPase, Na+/K+/2Cl- cotransporter and mitochondria-rich cell distribution in three salmonids Salvelinus namaycush, Salvelinus fontinalis and Salmo salar. J. Exp. Biol. 210, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 60. McCormick S. D., Regish A. M., Christensen A. K. (2009) Distinct freshwater and seawater isoforms of Na+/K+-ATPase in gill chloride cells of Atlantic salmon. J. Exp. Biol. 212, 3994–4001 [DOI] [PubMed] [Google Scholar]
- 61. Lin H., Pfeiffer D. C., Vogl A. W., Pan J., Randall D. J. (1994) Immunolocalization of H+-ATPase in the gill epithelia of rainbow trout. J. Exp. Biol. 195, 169–183 [DOI] [PubMed] [Google Scholar]
- 62. Hiroi J., McCormick S. D., Ohtani-Kaneko R., Kaneko T. (2005) Functional classification of mitochondrion-rich cells in euryhaline Mozambique tilapia (Oreochromis mossambicus) embryos, by means of triple immunofluorescence staining for Na+/K+-ATPase, Na+/K+/2Cl- cotransporter and CFTR anion channel. J. Exp. Biol. 208, 2023–2036 [DOI] [PubMed] [Google Scholar]
- 63. Roberts R. J., Agius C., Saliba C., Bossier P., Sung Y. Y. (2010) Heat shock proteins (chaperones) in fish and shellfish and their potential role in relation to fish health: a review. J. Fish Dis. 33, 789–801 [DOI] [PubMed] [Google Scholar]
- 64. Martin J. (1997) Molecular chaperones and mitochondrial protein folding. J. Bioenerget. Biomemb. 29, 35–43 [DOI] [PubMed] [Google Scholar]
- 65. Kültz D., Somero G. N. (1995) Ion transport in gills of the euryhaline fish Gillichthys mirabilis is facilitated by a phosphocreatine circuit. Am. J. Physiol. 268, R1003–R1012 [DOI] [PubMed] [Google Scholar]
- 66. Weng C. F., Chiang C. C., Gong H. Y., Chen M. H., Huang W. T., Cheng C. Y., Wu J. L. (2002) Bioenergetics of adaptation to a salinity transition in euryhaline teleost (Oreochromis mossambicus) brain. Exp. Biol. Med. 227, 45–50 [DOI] [PubMed] [Google Scholar]
- 67. Suresh N., Shivakumar K., Jayaraman J. (1983) The adaptation to salinity: protein synthesis and some aspects of energy transduction in fish gill mitochondria. J. Bioenerget. Biomemb. 15, 379–394 [DOI] [PubMed] [Google Scholar]
- 68. Shivkamat P., Roy R. (2005) Regulation of membrane lipid bilayer structure during salinity adaptation: a study with the gill epithelial cell membranes of Oreochromis niloticus. Comp. Bioch. Physiol. B 142, 28–36 [DOI] [PubMed] [Google Scholar]
- 69. Hazel J. R., Williams E. E. (1990) The role of alterations in membrane lipid composition in enabling physiological adaptation of organisms to their physical environment. Prog. Lipid Res. 29, 167–227 [DOI] [PubMed] [Google Scholar]
- 70. Collman J. P., Devaraj N. K., Decreau R. A., Yang Y., Yan Y. L., Ebina W., Eberspacher T. A., Chidsey C. E. (2007) A cytochrome C oxidase model catalyzes oxygen to water reduction under rate-limiting electron flux. Science 315, 1565–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Covian R., Trumpower B. L. (2009) The rate-limiting step in the cytochrome bc1 complex (ubiquinol-cytochrome c oxidoreductase) is not changed by inhibition of cytochrome b-dependent deprotonation: implications for the mechanism of ubiquinol oxidation at center P of the bc1 complex. J. Biol. Chem. 284, 14359–14367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McCaig C., Potter L., Abramczyk O., Murray J. T. (2011) Phosphorylation of NDRG1 is temporally and spatially controlled during the cell cycle. Biochem. Biophys. Res. Commun. 411, 227–234 [DOI] [PubMed] [Google Scholar]
- 73. Ellen T. P., Ke Q., Zhang P., Costa M. (2008) NDRG1, a growth and cancer related gene: regulation of gene expression and function in normal and disease states. Carcinogenesis 29, 2–8 [DOI] [PubMed] [Google Scholar]
- 74. Conte F. P., Lin D. H. Y. (1967) Kinetics of cellular morphogenesis in gill epithelium during sea water adaptation of Oncorhynchus (Walbaum). Comp. Biochem. Physiol. 23, 945–957 [DOI] [PubMed] [Google Scholar]
- 75. Chretien M., Pisam M. (1986) Cell renewal and differentiation in the gill epithelium of fresh water adapted or salt water adapted euryhaline fish as revealed by [H3] thymidine autoradiography. Biol. Cell 56, 137–150 [Google Scholar]
- 76. Kachhap S. K., Faith D., Qian D. Z., Shabbeer S., Galloway N. L., Pili R., Denmeade S. R., DeMarzo A. M., Carducci M. A. (2007) The N-myc down regulated gene1 (NDRG1) is a Rab4a effector involved in vesicular recycling of E-cadherin. Plos One 2, e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kovacevic Z., Chikhani S., Lui G. Y. L., Sivagurunathan S., Richardson D. R. (2013) The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid. Redox. Sign. 18, 874–887 [DOI] [PubMed] [Google Scholar]
- 78. Dixon K. M., Lui G. Y., Kovacevic Z., Zhang D., Yao M., Chen Z., Dong Q., Assinder S. J., Richardson D. R. (2013) Dp44mT targets the AKT, TGF-beta and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Brit. J. Cancer 108, 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kovacevic Z., Sivagurunathan S., Mangs H., Chikhani S., Zhang D., Richardson D. R. (2011) The metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1), upregulates p21 via p53-independent mechanisms. Carcinogenesis 32, 732–740 [DOI] [PubMed] [Google Scholar]
- 80. Kültz D., Burg M. B. (1998) Intracellular signaling in response to osmotic stress. Contrib. Nephrol. 123, 94–109 [DOI] [PubMed] [Google Scholar]
- 81. Kültz D., Avila K. (2001) Mitogen-activated protein kinases are in vivo transducers of osmosensory signals in fish gill cells. Comp. Biochem. Physiol. B 129, 821–829 [DOI] [PubMed] [Google Scholar]
- 82. Irarrazabal C. E., Burg M. B., Ward S. G., Ferraris J. D. (2006) Phosphatidylinositol 3-kinase mediates activation of ATM by high NaCl and by ionizing radiation: Role in osmoprotective transcriptional regulation. Proc. Natl. Acad. Sci. U.S.A. 103, 8882–8887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dmitrieva N., Kültz D., Michea L., Ferraris J., Burg M. (2000) Protection of renal inner medullary epithelial cells from apoptosis by hypertonic stress-induced p53 activation. J. Biol. Chem. 275, 18243–18247 [DOI] [PubMed] [Google Scholar]
- 84. Cangul H. (2004) Hypoxia upregulates the expression of the NDRG1 gene leading to its overexpression in various human cancers. BMC Genet. 5, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Inglis S. K., Gallacher M., Brown S. G., McTavish N., Getty J., Husband E. M., Murray J. T., Wilson S. M. (2009) SGK1 activity in Na+ absorbing airway epithelial cells monitored by assaying NDRG1-Thr(346/356/366) phosphorylation. Pflüg Arch Eur. J. Physiol. 457, 1287–1301 [DOI] [PubMed] [Google Scholar]
- 86. Murray J. T., Campbell D. G., Morrice N., Auld G. C., Shpiro N., Marquez R., Peggie M., Bain J., Bloomberg G. B., Grahammer F., Lang F., Wulff P., Kuhl D., Cohen P. (2004) Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 384, 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Klaus F., Palmada M., Lindner R., Laufer J., Jeyaraj S., Lang F., Boehmer C. (2008) Up-regulation of hypertonicity-activated myo-inositol transporter SMIT1 by the cell volume-sensitive protein kinase SGK1. J Physiol. 586, 1539–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen S. C., McCormick J. A., Prabaker K., Wang J., Pearce D., Gardner D. G. (2004) Sgk1 mediates osmotic induction of NPR-A gene in rat inner medullary collecting duct cells. Hypertension 43, 866–871 [DOI] [PubMed] [Google Scholar]
- 89. Perez-Pinera P., Menendez-Gonzalez M., del Valle M., Vega J. A. (2006) Hypertonicity activates GSK3 beta in tumor cells. Mol. Cell. Biochem. 291, 93–100 [DOI] [PubMed] [Google Scholar]
- 90. Richard O., Paquet N., Haudecoeur E., Charrier B. (2005) Organization and expression of the GSK3/Shaggy kinase gene family in the moss Physcomitrella patens suggest early gene multiplication in land plants and an ancestral response to osmotic stress. J. Mol. Evol. 61, 99–113 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.