

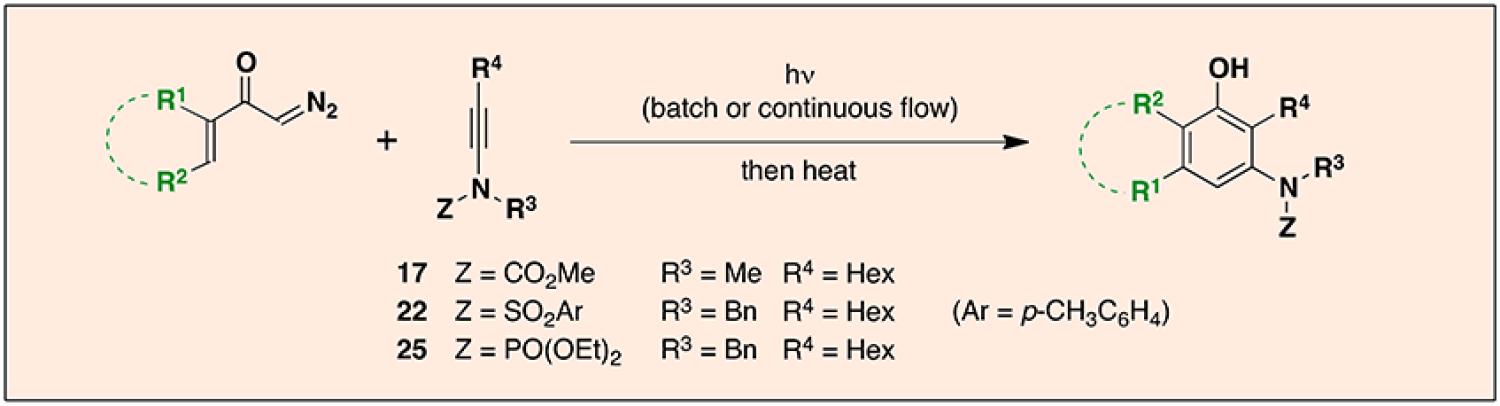
Abstract

Highly substituted polycyclic aromatic and heteroaromatic compounds are produced via a two-stage tandem benzannulation/cyclization strategy. The initial benzannulation step proceeds via a pericyclic cascade mechanism triggered by thermal or photochemical Wolff rearrangement of a diazo ketone. The photochemical process can be performed using a continuous flow reactor which facilitates carrying out reactions on a large scale and minimizes the time required for photolysis. Carbomethoxy ynamides as well as more ketenophilic bissilyl ynamines and N-sulfonyl and N-phosphoryl ynamides serve as the reaction partner in the benzannulation step. In the second stage of the strategy, RCM generates benzofused nitrogen heterocycles, and various heterocyclization processes furnish highly substituted and polycyclic indoles of types that were not available by using the previous cyclobutenone-based version of the tandem strategy.
INTRODUCTION
The design and invention of improved methods for the synthesis of benzofused nitrogen heterocycles continues to command a great deal of attention from laboratories in synthetic organic chemistry. Driving this interest are the numerous natural products and pharmaceutical compounds whose structures incorporate these heterocyclic systems. The great majority of synthetic approaches to benzofused nitrogen heterocycles involve cyclization of ortho-substituted aniline derivatives, but the application of these strategies to the synthesis of systems bearing a high level of substitution on the benzenoid ring is often problematic due to the challenges associated with the regioselective preparation of the requisite polysubstituted anilines.
Convergent benzannulation strategies1 provide the most attractive approach for the regiocontrolled construction of multiply substituted benzenoid aromatic systems. Our laboratory has developed two complementary benzannulation strategies based on the reaction of alkynes and vinylketenes2,3,4 and we have applied these methods to the synthesis of a number of bioactive natural products.5,6 Recently we extended the cyclobutenone-based version of this benzannulation strategy to include ynamine derivatives, demonstrating that the use of certain ynamides as alkyne reaction partners suppresses side reactions that are observed in reactions with simple ynamines. When employed in conjunction with ring closing metathesis, this version of our benzannulation method provides an efficient “tandem” strategy for the synthesis of highly substituted dihydroquinolines, benzazepines, and benzazocines, including a key intermediate in a formal total synthesis of (+)-FR900482.7 In the first stage of the tandem strategy, benzannulations using ynamine derivatives 2 (Z = electron withdrawing group) and cyclobutenones 1 furnish anilines having general structure 3 (Scheme 1). Application of ring closing metathesis (or other cyclization methods) to 3 then leads to benzofused heterocycles of type 4 or type 5, depending on the substitution of the aniline derivative and the cyclization method employed. Further research in our laboratory has focused on the application of various heterocyclization processes in an extension of this “tandem strategy” to the synthesis of indoles bearing a high level of substitution on the six-membered ring.8
Scheme 1.

Tandem Benzannulation-Cyclization Strategies Based on Cyclobutenones as Vinylketene Precursors
While the cyclobutenone-based benzannulation depicted in Scheme 1 is effective for the synthesis of a wide range of substituted anilines, polycyclic systems of type 6 present a challenge. As outlined in Scheme 2, the synthesis of polycyclic aniline derivatives with this general structure would require benzannulation with a cyclobutenone of type 7 as the vinylketene precursor. The synthesis of fused-ring cyclobutenones of this type generally requires lengthy and inefficient routes,9 and some potential substrates of interest are expected to be prohibitively strained compounds.
Scheme 2.
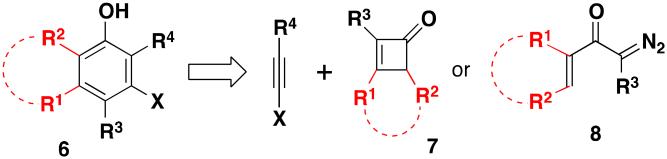
Benzannulation Disconnection for Polycyclic Compounds
In order to extend the benzannulation to include the synthesis of polycyclic systems, we previously developed a “second generation” variant of our strategy in which α-diazo ketones are employed as vinylketene precursors in place of cyclobutenones.2b Diazo ketones of type 8 are readily available (vide infra) by a number of straightforward routes and provide efficient access to the requisite ketenes via Wolff rearrangement.10 In this paper we report the application of this “second generation” version of our strategy for the first time to reactions in which ynamine derivatives function as the alkyne benzannulation partners. Based on our previous studies, the proposed benzannulation was expected to proceed via the “pericyclic cascade” mechanism outlined in Scheme 3. Irradiation (or thermolysis) of diazo ketones of type 8 would generate the vinylketene 9, which would be intercepted by the ynamide reaction partner 2 in a regioselective [2 + 2] cycloaddition to furnish vinylcyclobutenone 10. Reversible electrocyclic cleavage of 10 under the influence of heat or light would then lead to the dienylketene 11, and rapid 6-π electrocyclic ring closure and tautomerization would finally afford the polysubstituted aniline derivative 12. In principle, this version of the benzannulation should proceed smoothly not only with cyclic α,β-unsaturated diazo ketones of type 8, but also with a variety of aryl and hetaryl diazo ketones. For the synthesis of benzofused nitrogen heterocycles, the polycyclic aniline derivatives generated in this fashion would be subjected to various heterocyclization processes providing access to a wide variety of heterocyclic systems that were not available via our previous cyclobutenone-based strategy.
Scheme 3.

Pericyclic Cascade Mechanism of Benzannulations Based on Diazo Ketones
RESULTS AND DISCUSSION
Synthesis of Benzannulation Reaction Partners
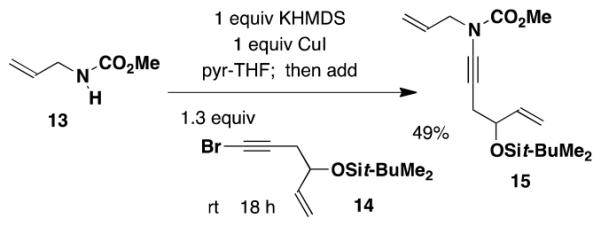
The benzannulation reaction partners employed in this investigation were easily prepared by using standard methods. For the synthesis of ynamides,11,12 we employed the closely related copper-promoted N-alkynylation methods previously developed in our laboratory13 and that of Hsung.14 Both methods involve the reaction of amine derivatives with alkynyl bromides, which are themselves readily obtained from terminal alkynes or silyl alkynes by reaction with NBS in the presence of catalytic silver nitrate.15 The Hsung N-alkynylation protocol utilizes catalytic CuCN or CuSO4 in conjunction with diamine ligands and requires elevated temperatures, while our complementary procedure employs 1 equiv of CuI but has the advantage of proceeding at room temperature and thus being applicable to thermally sensitive systems.16 All of the ynamides required for this study were known compounds with the exception of 15 which was prepared by our N-alkynylation method as outlined in eq 1.
 |
(1) |
The most popular synthetic routes to α-diazo ketones17 involve either the reaction of carboxylic acid derivatives with diazo compounds or the reaction of ketones with sulfonyl azides (“diazo transfer”). For the synthesis of the α,β-unsaturated diazo ketone benzannulation partners used in this study, we employed the detrifluoroacetylative diazo transfer protocol previously developed in our laboratory.18
Initial Photochemical Benzannulation Studies
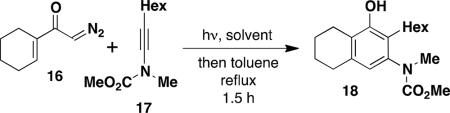
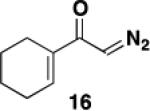
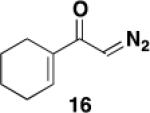
We initially examined the reaction of the diazo ketone 16 derived from acetylcyclohexene to determine the feasibility of employing ynamides in the photo-Wolff variant of the vinylketene-based benzannulation strategy. Table 1 summarizes our results. Irradiation was carried out in quartz tubes positioned ca. 20 cm from a Hanovia 450 W medium-pressure mercury lamp in a water-cooled quartz immersion well and was continued until TLC analysis indicated complete consumption of the diazo ketone. As we have noted in some prior photochemical benzannulations, at that point the reaction mixture consists of varying amounts of the desired phenolic annulation product and the intermediate vinylcyclobutenone (i.e., 10 in Scheme 3). The build up of colored polymers on the walls of the reaction tube apparently impedes the complete conversion of vinylcyclobutenone to product even on prolonged irradiation. Consequently, the crude reaction products were generally heated to complete the conversion of vinylcyclobutenone to phenol.
Table 1.
Optimization of the Photochemical Benzannulation
|
| entry | diazo ketone (equiv) |
solvent | irradiationa
time (h) |
yield (%)b |
|---|---|---|---|---|
| 1 | 1.0 | CH2Cl2 | 8 | 48 |
| 2 | 2.5 | CH2Cl2 | 5.5 | 67 |
| 3 | 1.5 | CH2Cl2 | 4.5 | 65c |
| 4 | 2.5 | CH2Cl2 | 10 | 85c |
| 5 | 2.5 | ClCH2CH2Cl | 9 | 73cd |
Irradiation with a 450 W medium-pressure mercury lamp (220-600 nm).
Isolated yields of products purified by column chromatography.
A solution of diazo ketone (0.4 M) was added to the ynamide solution via syringe pump (ca. 0.009 mL/min).
Heated in DCE (reflux, 24 h) after irradiation.
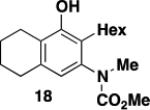
The desired benzannulation product 18 was obtained in 48% yield when 1.0 equiv of diazo ketone was used (Table 1, entry 1). We hypothesized that competing processes involving the intermediate vinylketene such as dimerization were limiting the yield, so we explored employing an excess of the diazo ketone for the reaction. In fact, when 2.5 equiv of diazo ketone was used, the yield improved to 67% (entry 2), and the yield also increased dramatically when the reaction was performed using slow addition of the diazo ketone (entry 3). Finally, the desired benzannulation product was generated in 85% yield when both slow addition was employed and the amount of diazo ketone was increased from 1.5 to 2.5 equiv (entry 4).
Dichloromethane was found to be the optimal solvent for the photochemical Wolff rearrangement. In our standard protocol, after irradiation for the indicated time the solution was concentrated and the solvent replaced with toluene. Heating at reflux then resulted in rapid conversion of any remaining intermediate vinylcyclobutenone to the desired phenolic product. For convenience, the benzannulation can be carried out without the need for solvent exchange by employing 1,2-dichloroethane (bp 83 °C), but in this case heating for 24 h is required to complete the reaction (entry 5).
For most benzannulations we found that optimal yields were obtained employing a Hanovia 450 W medium-pressure mercury lamp for irradiation. However, in some cases comparable results were obtained by employing a standard Rayonet photochemical reactor equipped with a circular array of 16 254-nm low-pressure mercury lamps. In the case of the prototypical benzannulation described in Table 1, irradiation for 1 h in a Rayonet reactor using 1 equiv of diazo ketone 16 produced 18 in somewhat lower yield (40%) as compared to reaction with the standard Hanovia protocol (entry 1).
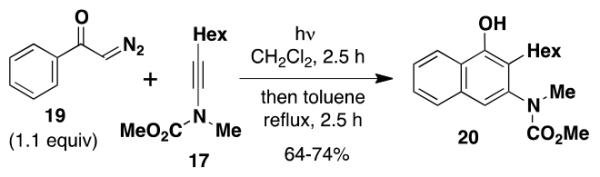
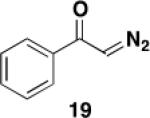
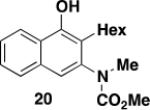
Not unexpectedly, the optimal conditions for effecting benzannulations with aryl diazo ketones differed from the protocol described above for α,β-alkenyl derivatives. In the case of α-diazoacetophenone (19), benzannulation proceeded in good yield using only 1.1 equiv of the diazo ketone and without the need for slow addition (eq 2). Wolff rearrangement of 19 generates phenylketene, which has less tendency to undergo dimerization and other unproductive side reactions as compared to the more reactive vinylketene derived from 16. Consequently, the desired [2 + 2] cycloaddition with ynamide competes more efficiently and the desired naphthylamine 20 is obtained in good yield without the need for slow addition to minimize ketene concentration.
 |
(2) |
Reactivity of Ynamine Derivatives in Ketene [2 + 2] Cycloadditions and Benzannulation
The competitive dimerization of vinylketenes presents a particular problem in the “second-generation” photo-Wolff version of the benzannulation due to the fact that the ketene intermediates are generated more rapidly and in higher concentration as compared to generation via thermolysis of cyclobutenones. Although this problem can be mitigated by the use of slow addition and by employing an excess of ketene precursor, these measures are less than ideal and we therefore became interested in replacing carbomethoxy ynamides such as 17 with ynamides that would exhibit greater ketenophilicity in the [2 + 2] cycloaddition step of the pericyclic cascade.
Previously it has been shown that N-carbonyl substituents reduce the electron-donating ability of nitrogen atoms to a greater extent than N-sulfonyl groups, a consequence of the more significant π-delocalization of the nitrogen lone pair present in the carbonyl derivatives.19,20 To confirm our prediction that N-sulfonyl ynamides would therefore exhibit increased reactivity in ketene [2 + 2] cycloadditions, we carried out the competition experiment outlined in eq 3. Reaction of an equimolar mixture of ynamides 2113a and 2214b with excess ketene in the presence of an internal standard indicated that the N-sulfonyl derivative indeed reacts at a rate twice that of the carbomethoxy ynamide.21
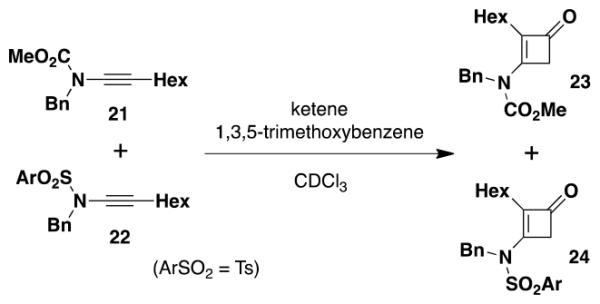 |
(3) |
As expected, ynamide 2514e with the even less electron-withdrawing phosphoryl group displays an even higher level of ketenophilicity. In the competition experiment shown in eq 4, we observed that the N-phosphoryl ynamide undergoes [2 + 2] cycloaddition at a rate five times faster than the sulfonamide.
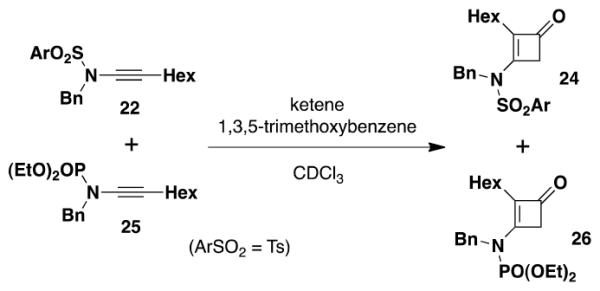 |
(4) |
Finally, a third competition experiment comparing the reactivity of the carbomethoxy ynamide and the phosphoryl derivative indicated that the rate of reaction of the phosphoryl ynamide is ca. 8 times that of the carbamate (eq 5).
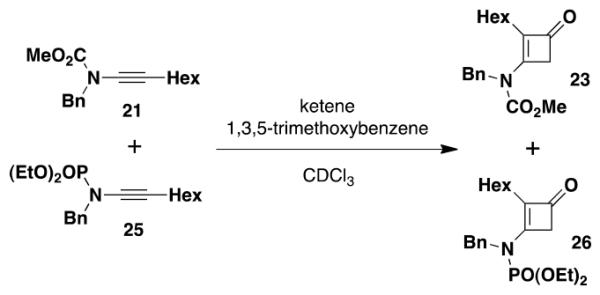 |
(5) |
N,N-Bis(trialkylsilyl) ynamines such as 27 were another class of ynamine derivatives that we expected to exhibit increased ketenophilicity compared to carbomethoxy derivatives. We anticipated that these electron-rich alkynes would prove highly reactive as benzannulation partners, furnishing aniline derivatives that would also undergo deprotection under exceptionally mild conditions. Of concern to us, however, was the known sensitivity of this class of ynamines to acid and the possibility that they might be unstable in the presence of the phenolic products of the benzannulation.
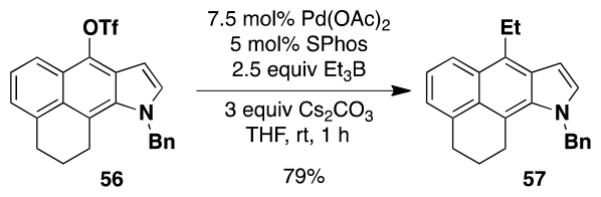
The feasibility of employing silyl ynamines in the benzannulation was investigated using the known ynamine 27 which we prepared by electrophilic amination of phenylacetylene as described by Würthwein.22 A stratagem for avoiding the potential destruction of the ynamine by phenolic product was devised based on our prior observation that the electrocyclic ring opening of vinylcyclobutenones does not take place when irradiation is conducted through a uranium filter. Uranium filters block wavelengths below 340 nm, allowing photo-Wolff rearrangement to take place but halting the pericyclic cascade at the stage of intermediate 10 (Scheme 3). Heating can then be employed to complete the benzannulation. In the event, application of this protocol to the reaction of the bis(silyl) ynamine 27 with 1.0 equiv of diazo ketone 19 afforded the desired aminonaphthol 28 in 61% overall yield (eq 6).
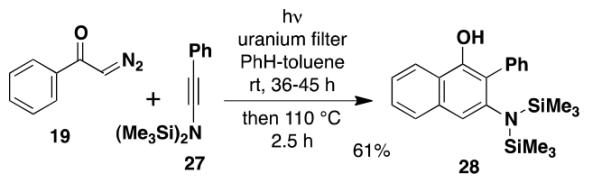 |
(6) |
Unfortunately, attempts to extend the silyl ynamine benzannulation to include heteroaryl or α,β-alkenyl diazo ketones have not been fruitful, with reactions proceeding in low (<30%) and variable yield. In view of the limited scope of this variant of the benzannulation, the use of silyl ynamines have not been investigated further.23
Scope of Photochemical Benzannulations Performed in Batch and Continuous Flow
In planning our systematic investigation of the photo-Wolff based ynamide benzannulation, we decided to examine reactions in which irradiation was performed both in batch and under conditions of continuous flow. The advantages of continuous-flow photochemical reactors are now well established.24,25 Due to their higher surface-to-volume ratio, flow reactors allow for more uniform and efficient irradiation as compared to conventional batch reactors where light does not penetrate a significant distance into the solution. Conducting photochemical reactions in flow also minimizes any secondary photo processes leading to the decomposition of the desired products. Finally, photochemical reactions conducted in continuous flow are conveniently scaled up, a significant problem and longstanding constraint when conducting photochemistry using standard batch reactors.
In the case of the “second generation” photo-Wolff based benzannulation, we anticipated that the use of a continuous-flow photochemical reactor would increase the efficiency of the reaction, allow for shorter photolysis times, facilitate carrying out large scale reactions, and would lead to an increase in the yield of benzannulations by minimizing side reactions involving photoreactive aniline products. We based the design of our single pass continuous flow reactor on the very practical and easily assembled system described by Booker-Milburn and coworkers.26 UV-transparent fluorinated ethylene propylene (FEP) tubing (0.76 mm i.d., 1.59 mm o.d.) was wound around a standard water-jacketed quartz immersion well into which was placed a 450 W medium pressure mercury lamp. In most cases irradiation was conducted through a Pyrex filter as this served to reduce polymer buildup on the walls of the tubing. A syringe pump was employed to drive the reaction solution (ca. 0.25 M in ynamide and diazo ketone) through the tubing at a flow rate of 0.057-0.32 mL/min (vide infra). The residence time in the irradiated section of tubing ranged from 21 to 33 min and the throughput with this reactor was typically 0.9 mmol/h.27
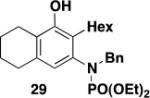
Table 2 summarizes the results of our investigation of the photochemical benzannulation reaction performed both as a batch process and using the continuous-flow reactor. The photochemical benzannulation of α,β-alkenyl diazo ketone 16 was examined first. As discussed previously, as a batch process the reaction of 16 with N-carbomethoxy ynamide 17 is best performed using slow addition of excess diazo ketone to a solution of the ynamide in order to maintain the vinylketene intermediate in low concentration and to thus minimize its dimerization. This slow addition protocol is not easily applied in a continuous flow photolysis, and so for benzannulations using α,β-alkenyl diazo ketones we have found that the more ketenophilic N-phosphoryl ynamides such as 25 are the preferred reaction partners. Using 25, benzannulation with only 1.1 equiv of diazo ketone 16 provides the desired product (29) in 66% yield when carried out as a batch process without the need for slow addition. Irradiation of the same benzannulation partners under conditions of continuous flow afforded 29 in comparable yield (entry 2). Importantly, this result established for the first time the feasibility of conducting the photochemical benzannulation under conditions of continuous flow. As discussed earlier, conducting photochemical benzannulations under flow enables the reaction to be performed on multigram scale much more easily than would be the case by employing conventional batch equipment.
Table 2.
Photochemical Benzannulations Performed in Batch and Flow
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | diazo ketone | ynamide | product | % yielda
(method)b,c |
entry | diazo ketone | ynamide | product | % yielda
(method)bc |
| 1 |
|
17 |
|
85d (batch) | 4 |
|
17 |
|
40 (batch) 37 (flow) |
| 2 |
|
25 |
|
66 (batch) 53 (flow) |
5 |
|
17 |
|
57e (batch) 47-50e (flow) |
| 3 |
|
17 |
|
64-76 (batch) 66 (flow) |
6 |
|
22 |
|
52e (batch) 47e (flow) |
| 7 |
|
25 |
|
78 (batch) 79 (flow) |
|||||
Isolated yield of products purified by column chromatography
Batch reactions: 1.1 equiv of diazo ketone, irradiation (220-600 nm) for 3-8 h in CH2CI2; then reflux, toluene, 1.5-2.5 h. For entry 6, irradiation at >340 nm (uranium filter) for 50 h.
Continuous-flow reactions: 1.1 equiv of diazo ketone, irradiation at >280 nm (Pyrex filter) in DCE, residence time = ca. 21 min; then reflux, DCE, 40-48 h (entries 2, 3, 6, and 7) or reflux, toluene, 2 h (entries 4 and 5). For entry 6, irradiation at >340 nm (uranium filter), residence time = ca. 33 min.
Slow addition of 2.5 equiv of diazo ketone.
Overall yield for 2 steps. Benzannulation product was treated with 1.2 equiv Tf2O, 2 equiv 4-DMAP, CH2Cl2, 0 °C to rt, 2-4 h.
It is worth noting with regard to the synthetic utility of N-phosphoryl benzannulation products such as 29 that cleavage of the protective group can be achieved either under acidic conditions or by reaction with hydride reagents.28 Deprotection of 29, for example, is readily accomplished upon heating with Red-Al in toluene and furnishes 37 in 82% yield (eq 7).
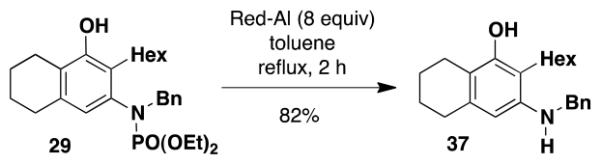 |
(7) |
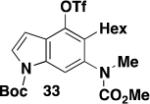
As shown in Table 2, benzannulations involving aryl and hetaryl diazo ketones proceed smoothly with all three classes of ynamides when conducted using either a batch process or in continuous flow. Particularly notable are the syntheses of indoles 31 and 33 (entries 4 and 5), demonstrating the ability of this version of the benzannulation to provide access to indoles bearing multiple substituents on the six-membered ring. In the case of benzannulation with diazo ketone 32, purification was carried out after converting the air-sensitive phenolic product to the corresponding triflate derivative. This measure facilitated purification and in principle the triflate should serve as a useful handle for the further synthetic elaboration of the product via metal-catalyzed coupling reactions (vide infra).
Benzannulation using the N-sulfonyl ynamide 22 worked well, but this benzannulation requires irradiation through a uranium filter (>340 nm) in order to prevent photochemical decomposition of the sulfonyl ynamide which most likely takes place via a photo-Fries type process.29 In the case of the batch process, irradiation at this longer wavelength necessitated an increase in the photolysis time up to 50 h from the usual 3 to 8 h employed in reactions of other ynamides. The alternative continuous flow protocol proved to be highly advantageous in this case, as complete conversion could be achieved simply by increasing the residence time from the usual 21 min to 33 min.
Feasibility of Triggering Benzannulations via Thermal Wolff Rearrangement
The next goal in our investigation of the scope of the diazo ketone version of the benzannulation was to determine the feasibility of effecting the initial Wolff rearrangement step under thermal rather than photochemical conditions.10 If successful, this might allow us to extend this version of the benzannulation to include photochemically unstable ynamides and could also lead to improved yields in the case of benzannulations that generate photochemically unstable products.
Initial experiments using cyclohexenyl diazo ketone 16 were not encouraging. Once again, side reactions involving the intermediate vinylketene competed with the desired [2 + 2] cycloaddition with ynamide, and useful yields were only obtained when the reaction was performed by slow addition of a 2-fold excess of diazo ketone (eq 8).
 |
(8) |
Fortunately, both slow addition and the use of excess diazo ketone were not necessary when the more ketenophilic N-phosphoryl ynamide 25 was employed in the reaction. As shown in eq 9, heating a solution of diazo ketone 16 and 25 at reflux in toluene affords benzannulation product 29 in essentially the same yield as obtained in the photochemical process (Table 2, entry 2). The application of this thermal Wolff triggered benzannulation to reactions involving photochemically unstable ynamides and aniline products is described in the next section.
 |
(9) |
Tandem Ynamide Benzannulation/RCM Strategies
The successful extension of our “second generation” strategy to include ynamides as the alkyne benzannulation partners provides access to polycyclic anilines that were not available via the cyclobutenone-based version of the method. In combination with ring closing metathesis, this new variant of the benzannulation provides an attractive route to a variety of polycyclic benzofused nitrogen heterocyclic systems. For example, as illustrated in Scheme 4, benzannulation using ynamide 15 and α-diazoacetophenone (19) leads to the formation of the highly substituted naphthol derivative 38 in good yield. Exposure of 38 to the Grubbs II catalyst 3930 then affords naphthazocine derivative 40.
Scheme 4.

Tandem Photochemical Benzannulation-RCM
The advantages associated with using the thermal Wolff rearrangement to initiate the benzannulation cascade are illustrated in the tandem benzannulation/RCM sequence shown in Scheme 5. Prior experiments had taught us that C-allyl ynamides such as 41 do not participate in photochemical benzannulations in good yield due to the facile cyclization of the o-allylphenol products that takes place on irradiation.31 This destructive side reaction is avoided when the benzannulation is effected thermally, and ring closing metathesis then provides the benzazepine 44 in excellent yield.32
Scheme 5.

Tandem Thermal Benzannulation-RCM
Synthesis of Indoles via Tandem Ynamide Benzannulation/Cyclization Strategies
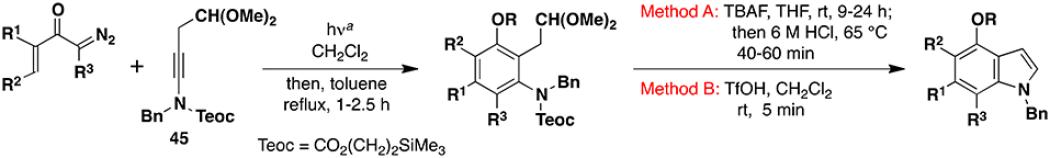
Recently we demonstrated that the combination of the cyclobutenone-based benzannulation with various heterocyclization and annulation processes provides an attractive route to indoles bearing multiple substituents on the benzenoid ring.8 A limitation of this tandem strategy, however, is that it cannot provide access to indoles with certain substitution patterns and to indoles fused to additional ring systems. As demonstrated below, the “second generation” variant of the benzannulation strategy addresses these limitations and extends the scope of this chemistry to include the construction of a number of highly substituted polycyclic indoles.
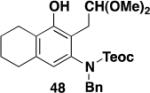
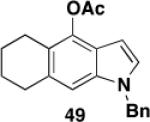
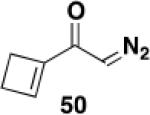
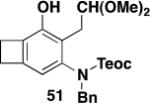
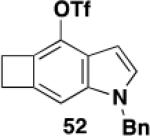
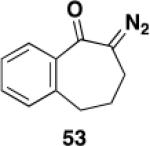
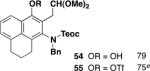
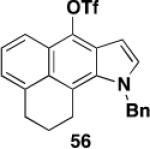
Table 3 illustrates one version of this tandem strategy. Previously we have shown that benzannulations with ynamide 458 afford products that undergo cyclization to indoles upon exposure in one flask to TBAF and then acid. As shown in Table 3, irradiation of 45 with a variety of α,β-unsaturated diazo ketones proceeds smoothly to afford benzannulation products that in most cases would not have been available via the cyclobutenone-based method. Treatment in one pot with TBAF and then HCl provides the desired indoles in good to excellent yield. Due to its limited stability, the benzannulation product 54 (entry 4) was best transformed to the corresponding triflate 55 prior to indole formation. In this case, conversion to the indole was accomplished in one step by brief exposure to excess TfOH at room temperature.33 Particularly notable is the efficient formation of the indole 52, incorporating an interesting cyclobutarene system.34 Due to their sensitivity to air oxidation, the indole products in entries 2 and 3 were best isolated as their acetate or triflate derivatives. The utility of the triflates as synthetic intermediates for the further elaboration of the indole products is illustrated by the Suzuki-Miyaura coupling shown in eq 10 which we found is best accomplished by using Buchwald’s SPhos ligand.35
 |
(10) |
Table 3.
Tandem Benzannulation/Cyclization Strategy for the Synthesis of Indoles
| ||||||
|---|---|---|---|---|---|---|
| entry | diazo ketone | benzannulation product | yield (%)b | cyclization method |
indole | yield (%)b |
| 1 |
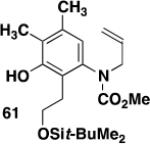
|
|
59 | A |
|
82 |
| 2 |
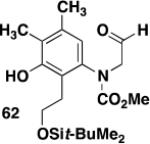
|
|
60 | A |
|
57c |
| 3 |
|
|
59 | A |
|
63d |
| 4 |
|
|
79 75e |
B |
|
76 |
Irradiation (220-600 nm) of 45 and 2.4 equiv of diazo ketone (1.5 equiv in entry 4). In entries 1-3 diazo ketone was added over 3.5-5 h followed by 1.0-6.5 h of further irradiation. In entry 4 a solution of 45 and 53 was irradiated for 2.5 h.
Isolated yield of products purified by column chromatography.
Overall yield from 48 for cyclization and acetylation (1.2 equiv AcCl, 2.2 equiv Et3N, CH2Cl2, 0 °C to rt, 2.5 h). In this case reaction with HCl was conducted at rt for 41 h.
Overall yield from 51 for cyclization and trifluoromethylsulfonylation (1.7 equiv NaH, 1.6 equiv PhNTf2, THF, 0 °C to rt, 1 h).
Trifluoromethylsulfonylation of 54 by reaction with 1.4 equiv NaH, 1.4 equiv PhNTf2, THF (0 °C to rt, 15 min).
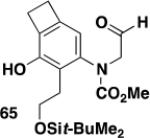
N-Allyl ynamides serve as another useful building block in our tandem benzannulation/heterocyclization strategy for the synthesis of highly substituted indoles.8 Oxidative cleavage of the allyl double bond following benzannulation affords an aldehyde that undergoes cyclization to generate the desired indole upon treatment with K2CO3 or DBU followed by brief exposure to HCl.
Table 4 illustrates the application of this cyclization protocol in conjunction with the “second generation” benzannulation to provide access to very highly substituted indoles whose synthesis would be difficult to achieve by alternative methods. In these experiments to establish the scope of the method the benzannulations were carried out as batch reactions, but application of the continuous flow reactor with N-phosphoryl ynamides should allow the benzannulation to be accomplished on multigram scale since the outcome of flow processes is expected to be independent of scale.
Table 4.
Tandem Benzannulation-Aromatic Substitution Strategy for the Synthesis of Indoles
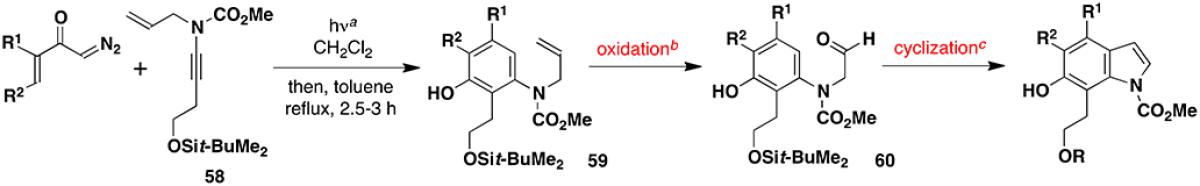
| |||||||
|---|---|---|---|---|---|---|---|
| entry | diazo ketone | benzannulation product | yield (%)d | aldehyde | yield (%)d | indole | yield (%)d |
| 1 |
|
|
76-78 |
|
89-97 |
|
59-62 |
| 2 |
|
|
62-75 |
|
88-92 |
|
48-62 |
Irradiation (220-600 nm) of 58 and 2.6 equiv of diazo ketone. Diazo ketone was added over 5-7 h followed by 0.3-5 h of further irradiation.
Oxidation conditions: 2-3 mol % OsO4, 1.3-1.5 equiv NMO, THF/H2O (rt, 24-26 h); then 2.0-2.3 equiv NaIO4-SiO2, CH2CI2 (rt, 10-30 min).
Cyclization conditions: 1.0 equiv K2CO3, i-PrOH, 75-80 °C, 2.5 h (entry 1) or 10 h (entry 2), then 1 M HCl to pH 1 (entry 1) or neutralize with 0.6 M HCl (entry 2).
Isolated yield of products purified by column chromatography.
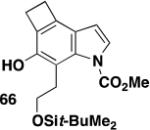
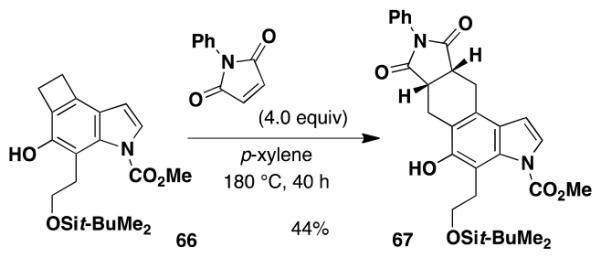
The availability of cyclobutarenes such as 66 via this methodology is of particular interest. Thermolysis of cyclobutarenes provides access to o-quinodimethanes, whose utility as highly reactive dienes in Diels-Alder cycloadditions is well documented.36 Eq 11 and Scheme 6 illustrate the application of the o-quinodimethanes derived from 66 in [4 + 2] cycloadditions with N-phenylmaleimide and ethyl propiolate, respectively. Not unexpectedly, little regioselectivity was observed in the Diels-Alder reaction of the unsymmetrical dienophile.
 |
(11) |
Scheme 6.

Tricyclic Indoles via Diels-Alder Reactions of o-Quinodimethanes
CONCLUSION
A wide range of benzofused nitrogen heterocyclic compounds are available via the application of our “second generation” benzannulation strategy employed in conjunction with various heterocyclization reactions. The triggering step for the pericyclic cascade involved in the benzannulation is the Wolff rearrangement of an α,β-unsaturated diazo ketone that can be effected photochemically or by heating in refluxing toluene. The photochemical process can be performed by irradiation as a batch process or by using a simple photochemical continuous flow reactor. The benzannulation has also been extended to include several new classes of ynamides that exhibit enhanced reactivity in the key [2 + 2] cycloaddition step of the pericyclic cascade. Ring closing metathesis and several types of nucleophilic cyclization processes are employed to convert the benzannulation products to benzazepines, benzacines, and in particular, to highly substituted indoles and polycyclic indoles that were not available by using previous versions of our tandem benzannulation/cyclization strategy.
EXPERIMENTAL SECTION
General Procedures
All reactions were performed in flame-dried or oven-dried glassware under a positive pressure of argon. Reaction mixtures were stirred magnetically unless otherwise indicated. Air- and moisture-sensitive liquids and solutions were transferred by syringe or cannula and introduced into reaction vessels through rubber septa. Reaction product solutions and chromatography fractions were concentrated by rotary evaporation at ca. 20 mmHg and then at ca. 0.1 mmHg (vacuum pump) unless otherwise indicated. Thin layer chromatography was performed on precoated glass-backed silica gel 60 F-254 0.25 mm plates. Column chromatography was performed on silica gel 60 (230-400 mesh) or basic alumina (80-325 mesh). Slow additions were performed with a syringe pump.
Materials
Commercial grade reagents and solvents were used without further purification except as indicated below. Dichloromethane, diethyl ether, and tetrahydrofuran were purified by pressure filtration through activated alumina. Toluene was purified by pressure filtration through activated alumina and Cu(II) oxide. Acetonitrile, 1,2-dichloroethane, 1,1,1,3,3,3-hexamethyldisilazane, pyridine, triethylamine, 2,2,2-trifluoroethyl trifluoroacetate (TFETFA), and p-xylene were distilled under argon from calcium hydride. Triflic anhydride was distilled under argon from P2O5. N-Bromosuccinimide was recrystallized from boiling water. 4-DMAP was recrystallized from boiling toluene. Copper (I) iodide was extracted with THF for 24 h in a Soxhlet extractor and then dried under vacuum (0.1 mmHg). Sodium periodate supported on silica gel was prepared according to the procedure of Zhong and Shing.37 Methanesulfonyl azide was prepared by reaction of methanesulfonyl chloride with sodium azide.18an-Butyllithium was titrated using menthol in THF with 1,10-phenanthroline as an indicator.38 Deactivated alumina was prepared by first generating Brockman I grade by heating 150 g of alumina at 250 °C under vacuum (0.050 mmHg) for 12-18 h and then cooling under Ar. The alumina was then transferred to a 500-mL Erlenmeyer with a screw cap and the requisite amount of water was added to obtain the desired grade (Brockman grade II: 3%; grade III: 6%; grade IV: 10%; grade V: 15% w/w water). The alumina was then vigorously shaken for 10-15 min until a free flowing powder was obtained. The powder was allowed to stand at rt for 24 h with occasional mixing prior to use.
Instrumentation
Melting points are uncorrected. 1H NMR chemical shifts are expressed in parts per million (δ) downfield from tetramethylsilane (with the CHCl3 peak at 7.27 ppm used as a standard). 13C NMR chemical shifts are expressed in parts per million (δ) downfield from tetramethylsilane (with the central peak of CHCl3 at 77.23 ppm used as a standard). 31P NMR chemical shifts are expressed in parts per million (δ) downfield from an external 85% H3PO4 standard (0.0 ppm). High-resolution mass spectrometry (HRMS) was performed with a Fourier transform ion cyclotron resonance mass spectrometer.
Construction of the Continuous Flow Reactor
The continuous flow reactor employed in our experiments was constructed based on the design of Booker-Milburn et al.26 Fluorinated ethylene propylene (FEP) tubing, 1.59 mm o.d., 0.76 mm i.d., length = 440 cm (Reactor A) or 1520 cm (Reactor B) was wrapped around a quartz immersion well in tightly packed coils leaving 90 cm of tubing free at each end. The length of tubing wrapped around the well was 260 cm in the case of Reactor A (internal volume ca. 1.2 mL) and 1340 cm in the case of Reactor B (internal volume ca. 6.1 mL). The ends of the tubing were secured to the well with Teflon tape. The top end of the tubing was fitted with a nut, ferrule, and a thread to a female Luer adapter for attachment of a syringe. The bottom end of the tubing was connected through a rubber septum to a 25-mL or 50-mL pear flask equipped with an argon inlet needle and a needle vent. The flask was wrapped in aluminium foil. The immersion well was connected to recirculating tap water via PVC tubing. A Pyrex or uranium filter and a 450 W Hanovia medium-pressure mercury lamp were placed inside the immersion well as shown in the diagram.

Procedures for Preparation of Benzannulation Reaction Partners
1-Bromo-4-(tert-butyldimethylsilanyloxy)-hex-5-en-1-yne (14)
A 250-mL round-bottomed flask equipped with a rubber septum and argon inlet needle was charged with 4-(tert-butyldimethylsiloxy)-hex-5-en-1-yne39 (2.40 g, 11.4 mmol, 1.0 equiv) and 50 mL of acetone. N-bromosuccinimide (2.23 g, 12.6 mmol, 1.1 equiv) was added and the mixture was stirred until all of the NBS dissolved. AgNO3 (0.194 g, 1.14 mmol, 0.1 equiv) was added and then the reaction mixture was stirred at rt for 16 h. The resulting grey slurry was diluted with 50 mL of H2O and 80 mL of Et2O. The aqueous layer was extracted with two 50-mL portions of Et2O, and the combined organic layers were washed with two 50-mL portions of satd Na2S2O3 solution and 50 mL of brine, dried over MgSO4, filtered, and concentrated to afford ca. 3.9 g of green oil. Column chromatography on 20 g of silica (elution with hexanes) afforded 2.60 g (81%) of alkynyl bromide 14 as a colorless oil: IR (thin film) 2957, 2858, 2228, 1473, 1362, 1255, and 1090 cm±1; 1H NMR (400 MHz, CDCl3) δ 5.88 (ddd, J = 5.6, 10.4, 17.1 Hz, 1 H), 5.25 (dt, J = 1.5, 17.1 Hz, 1H), 5.11 (dt, J = 1.5, 10.4 Hz, 1H), 4.24-4.29 (m, 1H), 2.38 (app qd, J = 6.8, 16.5 Hz, 2H), 0.91 (s, 9H), 0.10 (s, 3H), and 0.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 140.1, 115.0, 77.6, 72.3, 39.8, 29.7, 26.0, 18.4, −4.4, and −4.7; HRMS (ESI) (m/z) [M + Na]+ calcd for C12H21BrNaOSi, 311.0443, found: 311.0438.
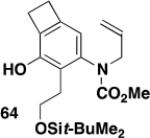
N-(Methoxycarbonyl)-N-(2-propenyl)-hex-5-en-4-(tert-butyl-dimethylsilanyloxy)-1-ynylamine (15)
A 50-mL, three-necked, round-bottomed flask fitted with a rubber septum, argon inlet adapter and glass stopper was charged with allyl carbamate 13 (0.160 g, 1.39 mmol, 1.0 equiv) and 6 mL of THF. The solution was cooled to 0 °C, and a solution of KHMDS (1.5 mL, 0.91 M in THF, 1.0 equiv) was then added rapidly dropwise over ca. 4 min. The resulting pale yellow slurry was stirred at 0 °C for 15 min and 3.8 mL of pyridine was then added, followed by CuI (0.265 g, 1.39 mmol, 1.0 equiv) in one portion. The reaction mixture was allowed to warm to rt over 2 h, and a solution of the alkynyl bromide 14 (0.528 g, 1.83 mmol, 1.3 equiv) in 5 mL of THF was then added dropwise via cannula over 25 min. The reaction mixture was stirred at rt for 18 h, diluted with 25 mL of Et2O, and washed with three 20-mL portions of a 2:1 mixture of brine and concd NH4OH solution. The combined aqueous layers were extracted with two 15-mL portions of Et2O and the combined organic phases were washed with 30 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.871 g of red-brown oil. Purification by column chromatography on 35 g of silica gel (elution with 3% EtOAc-hexanes) provided 0.222 g (49%) of 15 as a pale yellow oil: IR (thin film) 3084, 2956, 2858, 2265, 1732, 1646, and 1388 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.81-5.94 (m, 2H), 5.2105.28 (m, 3H), 5.09 (app dt, J = 1.5, 10.4 Hz, 1H), 4.20-4.25 (m, 1H), 3.98-4.08 (m, 2H), 3.79 (s, 3H), 2.40-2.55 (app qd, J = 6.0, 16.3 Hz, 2 H), 0.90 (s, 9H), 0.08 (s, 3H), and 0.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.2, 140.4, 132.0, 118.4, 114.6, 75.2, 72.8, 66.9, 54.1, 52.8, 28.5, 26.0, 18.4, −4.4, and −4.7; HRMS (ESI) (m/z) [M + Na]+ calcd for C17H29NNaO3Si: 346.1809, found: 346.1807.
1-(tert-Butoxycarbonyl)-3-acetylpyrrole
A 50-mL, pear flask equipped with a rubber septum and argon inlet needle was charged with 3-acetylpyrrole40 (0.510 g, 4.67 mmol, 1.0 equiv) and 9 mL of CH2Cl2. The resulting solution was cooled to 0 °C and a solution of Boc2O (1.05 g, 4.79 mmol, 1.1 equiv) in 7.5 mL of CH2Cl2 was added via cannula over 15 min. After 10 min, a solution of 4-DMAP (0.085 g, 0.70 mmol, 0.15 equiv) and Et3N (0.67 mL, 4.79 equiv, 1.1 equiv) in 6.5 mL of CH2Cl2 was added dropwise via cannula over 20 min. The reaction mixture was stirred at 0 °C for 2.5 h and then washed with two 25-mL portions of water, dried over MgSO4, filtered, and concentrated to yield 1.07 g of a yellow oil. Column chromatography on 50 g of silica gel (elution with 7% EtOAc-hexanes) furnished 0.952 g (97%) of 1-(tert-butoxycarbonyl)-3-acetylpyrrole as a white solid: mp 69-71 °C; IR (neat) 2977, 1752, 1673, 1494, 1396, 1280, 1150 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.80-7.83 (m, 1H), 7.21-7.24 (m, 1H), 6.62-6.65 (m, 1H), 2.45 (s, 3H), 1.63 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 193.7, 148.4, 128.6, 124.7, 121.5, 111.1, 85.4, 28.1, 27.4; HRMS (ESI) (m/z) [M + Na]+ calculated for C11H15NNaO3: 232.0944, found: 232.0945.
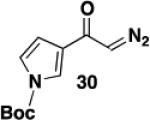
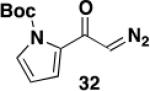
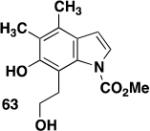
1-(tert-Butoxycarbonyl)-3-diazoacetylpyrrole (30)
A 100-mL, three-necked, round-bottomed flask equipped with two rubber septa, an argon inlet adapter, and a thermocouple was charged with a solution of 1,1,1,3,3,3-hexamethyldisilazane (1.15 mL, 5.45 mmol, 1.2 equiv) in 12 mL of THF and then cooled at 0 °C while n-BuLi (2.64 M in hexane, 1.9 mL, 5.02 mmol, 1.1 equiv) was added dropwise over 5 min. After 10 min, the resulting solution was cooled at −78 °C while a solution of 1-(tert-butoxycarbonyl)-3-acetylpyrrole (0.951 g, 4.55 mmol, 1.0 equiv) in 12 mL of THF was added dropwise via cannula over 35 min (0.8 mL THF wash). After 45 min, 2,2,2-trifluoroethyl trifluoroacetate (0.73 mL, 5.45 mmol, 1.2 equiv) was added rapidly by syringe. After 10 min, the reaction mixture was diluted with 25 mL of 1 M aq HCl solution and 30 mL of Et2O. The aqueous phase was extracted with two 15-mL portions of Et2O, and the combined organic phases were washed with 30 mL of brine, dried over Na2SO4, filtered, and concentrated to give 1.62 g of an orange oil which was immediately dissolved in 12 mL of CH3CN and transferred to a 100-mL pear flask equipped with a rubber septum and argon inlet needle. Water (0.08 mL, 4.43 mmol, 1.0 equiv) and Et3N (0.95 mL, 6.82 mmol, 1.5 equiv) were added, and a solution of methanesulfonyl azide (0.880 g, 7.27 mmol, 1.6 equiv) in 14 mL of CH3CN was added dropwise via cannula over 30 min (1 mL CH3CN wash). The resulting solution was stirred at room temperature for 3 h and then diluted with 50 mL of Et2O and washed with three 20-mL portions of 10% aq NaOH solution and 50 mL of brine, dried over Na2SO4, filtered, and concentrated to afford 1.199 g of a yellow oil. Column chromatography on 53 g of silica gel (gradient elution with 10-20% EtOAc-hexanes) furnished 1.004 g (94%) of diazo ketone 30 as a yellow solid: mp 58-59 °C; IR (neat) 3124, 2982, 2103, 1752, 1617, 1496, 1396, 1336, 1255, 1154 cm−1; UV (CH3CN) λmax, nm (ε) 294 (20642), 245 (19339), 229 (22817); 1H NMR (500 MHz, CDCl3) δ 7.70-7.72 (m, 1H), 7.23-7.25 (m, 1H), 6.51-6.52 (m, 1H), 5.63 (s, 1H), 1.62 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 181.6, 148.3, 126.9, 122.0, 121.4, 110.3, 85.4, 54.2, 28.1; HRMS (ESI) (m/z) [M + Na]+ calculated for C11H13N3NaO3: 258.0849, found: 258.0848.
Ynamide Reactivity Comparison Study
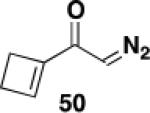
2-Hexyl-3-(N-benzyl-N-(methoxycarbonyl))-2-cyclobuten-1-one (23)
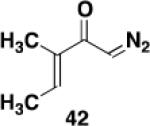
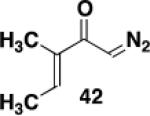
Ketene was generated by pyrolysis of acetone over an electrically heated metal filament using the apparatus described by Williams and Hurd.41 A 12x75 mm test tube fitted with a rubber septum was charged with ynamide 2113a (0.098 g, 0.358 mmol, 1.0 equiv) and 3.0 mL of toluene. The septum was fitted with an inlet needle connected to the ketene generator and an outlet needle connected via tubing to a column of calcium sulfate leading into a trap of water. Ketene was bubbled into the solution at rt over a period of 6 h. The reaction mixture was then concentrated to afford 0.127 g of an orange oil. Column chromatography on 12 g of silica gel (elution with 15% EtOAc-hexane) yielded 0.051 g of unreacted ynamide 21 as a colorless oil and 0.045 g (40%) of cyclobutenone 2342 as a yellow oil: IR (thin film) 2956, 2929, 2857, 1737, 1611, 1453, 1393, 1364, 1233 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.37 (t, J = 7.0 Hz, 2H), 7.30 (t, J = 7.0 Hz, 1H), 7.19 (d, J = 7.5 Hz, 2H), 4.99 (s, 2H), 3.84 (s, 3H), 3.54 (t, J = 1.5 Hz, 2H), 1.97 (t, J = 7.5 Hz, 2H), 1.30-1.38 (m, 2H), 1.20-1.30 (m, 2H), 1.10-1.20 (m, 4H), 0.84 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 188.1, 159.6, 153.7, 136.5, 129.1, 127.9, 127.7, 125.9, 54.4, 51.2, 50.9, 31.6, 29.4, 28.5, 24.0, 22.7, 14.2; HRMS (ESI) (m/z) [M + H]+ calcd for C19H26NO3, 316.1907, found 316.1911.
Ketene [2 + 2] Cycloaddition Competition Experiment:N-Carbomethoxy vs.N-Tosyl Ynamides
Ketene was generated by pyrolysis of acetone over an electrically heated metal filament using the apparatus described by Williams and Hurd. A 5-mL conical vial fitted with a rubber septum was charged with ynamide 21 (0.103 g, 0.377 mmol), ynamide 2214b (0.140 g, 0.379 mmol) and 1.5 mL of CDCl3 containing 1,3,5-trimethoxybenzene (0.063 g, 0.375 mmol) as an internal standard. The septum was fitted with an inlet needle connected to the ketene generator and an outlet needle connected via tubing to a column of calcium sulfate leading into a trap of water. Ketene was bubbled into the solution at rt over a period of 4 h. Aliquots (ca. 0.1 mL) of the reaction mixture were taken at intervals, diluted with CDCl3, and examined by 1H NMR (500 MHz) with a relaxation time d1 = 20 s to ensure accurate integration and auto phasing or manual phasing to ensure a level baseline. For cyclobutenone 23 the resonance at 4.96 ppm was integrated; for cyclobutenone 2442 the resonance at 5.04 ppm was integrated. For ynamide 21 the resonance at 4.61 ppm was integrated; for ynamide 22 the resonance at 4.45 ppm was integrated.
2-Hexyl-3-(N-benzyl-N-(diethylphosphoryl))-2-cyclobuten-1-one (26)
Ketene was generated by pyrolysis of acetone over an electrically heated metal filament using the apparatus described by Williams and Hurd. A 10x75 mm test tube fitted with a rubber septum was charged with ynamide 2514e (0.061 g, 0.174 mmol, 1.0 equiv) and 1.5 mL of toluene. The septum was fitted with an inlet needle connected to the ketene generator and an outlet needle connected via tubing to a column of calcium sulfate leading into a trap of water. Ketene was bubbled into the solution at rt over a period of 40 min. The reaction mixture was then concentrated to afford 0.082 g of an orange oil. Column chromatography on 7 g of silica gel (elution with 30% EtOAc-hexane) yielded 0.062 g (90%) of cyclobutenone 26 as a yellow oil: IR (thin film) 2929, 2857, 1751, 1602, 1455, 1384, 1351, 1270, 1020 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.37 (t, J = 7.5 Hz, 2H), 7.25-7.34 (m, 3H), 4.90 (d, J = 11.5 Hz, 2H), 3.90-4.20 (m, 4H), 3.37 (t, J = 1.5 Hz, 2H), 1.90 (t, J = 7.5 Hz, 2H), 1.29-1.39 (m, 8H), 1.18-1.29 (m, 2H), 1.04-1.18 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 187.1, 161.6 (d, J = 5.1 Hz), 137.4, 129.0, 127.9, 126.3, 125.6 (d, J = 9.8 Hz), 64.2 (d, J = 5.8 Hz), 52.2 (d, J = 5.0 Hz), 49.7, 31.7, 29.5, 28.6, 23.9, 22.7, 16.2 (d, J = 6.9 Hz) 14.2; 31P NMR (121 MHz, CDCl3) δ 2.17; HRMS (ESI) (m/z) [M + H]+ calcd for C21H33NO4P, 394.2142, found 394.2141.
Ketene [2 + 2] Cycloaddition Competition Experiment: N-Diethylphosphoryl vs. N-Tosyl Ynamides
Ketene was generated by pyrolysis of acetone over an electrically heated metal filament using the apparatus described by Williams and Hurd. A 13x100 mm test tube equipped with a rubber septum was charged with 3.5 mL CDCl3 and then fitted with an inlet needle connected to the ketene generator and an outlet needle connected via tubing to a column of calcium sulfate leading into a trap of water. Ketene was bubbled into the solution at rt for 5 min, then a solution of ynamide 25 (0.124 g, 0.353 mmol), ynamide 22 (0.130 g, 0.352 mmol), and 1,3,5-trimethoxybenzene (0.059 g, 0.351 mmol) as an internal standard in 0.5 mL of CDCl3 was added via syringe, and the addition of ketene continued over a period of 2 h. Aliquots (ca. 0.1 mL) of the reaction mixture were taken at intervals, diluted with CDCl3, and examined by 1H NMR (500 MHz) with a relaxation time d1 = 20 s to ensure accurate integration and auto phasing or manual phasing to ensure a level baseline. For cyclobutenone 26 the resonance at 4.90 ppm was integrated; for cyclobutenone 24 the resonance at 5.04 ppm was integrated. For ynamide 25 the resonance at 4.40 ppm was integrated; for ynamide 22 the resonance at 4.45 ppm was integrated.
Ketene [2 + 2] Cycloaddition Competition Experiment:N-Carbomethoxy vs.N-Diethylphosphoryl Ynamides
Ketene was generated by pyrolysis of acetone over an electrically heated metal filament using the apparatus described by Williams and Hurd. A 12x75 mm test tube fitted with a rubber septum was charged with 3.5 mL CDCl3 then fitted with an inlet needle connected to the ketene generator and an outlet needle connected via tubing to a column of calcium sulfate leading into a trap of water. Ketene was bubbled into the solution at rt for 10 min, then a solution of ynamide 21 (0.110 g, 0.404 mmol), ynamide 25 (0.142 g, 0.404 mmol), and 1,3,5-trimethoxybenzene (0.068 g, 0.404 mmol) as an internal standard in 0.5 mL of CDCl3 was added via syringe, and the addition of ketene continued over a period of 2 h. Aliquots (ca. 0.1 mL) of the reaction mixture were taken at intervals, diluted with CDCl3, and examined by 1H NMR (500 MHz) with a relaxation time d1 = 20 s to ensure accurate integration and auto phasing or manual phasing to ensure a level baseline. For cyclobutenone 23 the resonance at 4.96 ppm was integrated; for cyclobutenone 26 the resonance at 4.90 ppm was integrated. For ynamide 21 the resonance at 4.61 ppm was integrated; for ynamide 25 the resonance at 4.40 ppm was integrated.
Scope of Benzannulation Reactions
3-[N-(Carbomethoxy)-N-(methyl)amino]-2-hexyl-5,6,7,8-tetrahydronaphthalen-1-ol (18) by Batch Photochemical Benzannulation
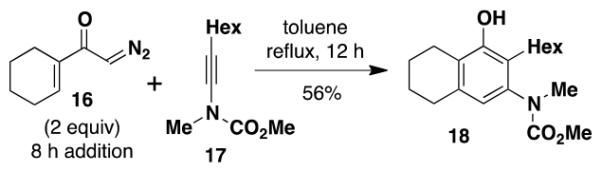
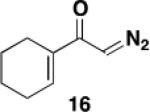
A 20-cm quartz tube (7 mm i.d., 10 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with ynamide 1742 (0.106 g, 0.537 mmol, 1.0 equiv) and 1.7 mL of CH2Cl2. A 10-mL Pyrex tube fitted with a rubber septum and argon inlet needle was charged with diazo ketone 1618a (0.202 g, 1.34 mmol, 2.5 equiv) and 3.3 mL of CH2Cl2. Both solutions were degassed via three cycles of freeze-pump-thaw (−196 °C, 0.05 mmHg). The solution of diazo ketone was taken up in a 5-mL glass syringe wrapped in aluminum foil and fitted with a 20-gauge, 20 cm long steel needle. The upper portion (ca. 5-7 cm length) of the quartz tube was wrapped in aluminum foil. The quartz tube was positioned ca. 20 cm proximity to the Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by recirculating tap water). The diazo ketone solution was added to the reaction tube via syringe pump during irradiation under argon at 28 °C. After the addition was completed (ca. 6 h), the Pyrex tube was rinsed with 0.5 mL CH2Cl2 and added with the same syringe in one portion to the reaction mixture. Irradiation was continued for 4 h. The resulting reaction mixture was concentrated to 0.320 g of orange oil and transferred to a 10-ml Pyrex tube with the aid of 3.5 mL of toluene. The solution was heated at reflux for 1.5 h and then cooled to rt and concentrated to afford 0.347 g of dark orange oil. Purification by column chromatography on 13 g of silica gel (elution with 10% EtOAc-hexanes) afforded 0.146 g (85%) of the aniline 18 as a viscous yellow oil: IR (thin film) 3408, 2960, 2929, 1688, 1455, 1261, and 1101 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.53 (s, minor rotamer), 6.49 (s, 1H), 4.95 (s, 1H), 3.79 (s, minor rotamer), 3.64 (s, 3H), 3.19 (s, 3H), 2.73-2.67 (m, 2H), 2.60 (t, J = 6.3 Hz, 2H), 2.49-2.45 (m, 2H), 1.88-1.83 (m, 2H), 1.79-1.72 (m, 2H), 1.53-1.47 (m, 2H), 1.41-1.35 (m, 2H), 1.33-1.27 (m, 4H), and 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) 156.9, 152.4, 139.6, 136.6, 123.4, 122.4, 120.1, 53.1, 38.9, 31.9, 30.1, 29.4, 29.3, 25.9, 23.1, 22.8, 22.7, and 14.3; HRMS (ESI) (m/z) [M + H]+ calcd for C19H30NO3; 320.2220, found 320.2229.
3-[N-(Carbomethoxy)-N-(methyl)amino]-2-hexyl-5,6,7,8-tetrahydronaphthalen-1-ol (18) by Thermal Benzannulation
A 10-mL round-bottomed flask equipped with a reflux condenser, rubber septum, and argon inlet needle was charged with ynamide 17 (0.060 g, 0.306 mmol, 1.0 equiv) and 1.5 mL of toluene. A 10-mL pear flask fitted with a rubber septum and argon inlet needle was charged with diazo ketone 16 (0.092 g, 0.612 mmol, 2.0 equiv) and 3 mL of toluene. Both solutions were degassed with a stream of argon for 15 min. The solution of diazo ketone was taken up in a 5-mL glass syringe wrapped in aluminum foil and fitted with a 20-gauge, 20-cm long steel needle. The reaction mixture was heated at reflux and the diazo ketone solution was added through the condenser via syringe pump. After the addition was completed (ca. 8 h), the pear flask was rinsed with 0.5 mL of toluene and added with the same syringe in one portion to the reaction mixture. Heating was continued for 4 h. After cooling to rt, the reaction mixture was concentrated to afford 0.148 g of orange oil. Purification by column chromatography on 20 g of silica gel (elution with 5% EtOAc-10% benzene-85% hexanes) afforded 0.074 g of a yellow oil that was further purified by column chromatography on 16 g of silica gel (elution with 5% EtOAc-10% benzene-85% hexanes) to afford 0.055 g (56%) of phenol 18 as a yellow oil with spectral data consistent with that reported in the procedure for the batch process given above.
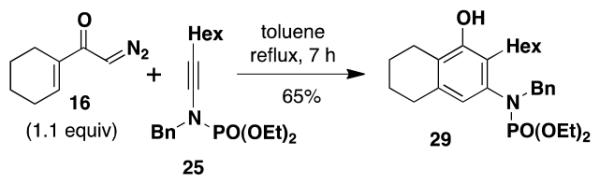
3-[N-(Diethylphosphoryl)-N-(benzyl)amino]-2-hexyl-5,6,7,8-tetrahydronaphthalen-1-ol (29) – Batch Reactor
A 20-cm quartz tube (7 mm i.d., 10 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with diazo ketone 16 (0.073 g, 0.486 mmol, 1.1 equiv), ynamide 2514e (0.151 g, 0.430 mmol, 1.0 equiv), and 2.0 mL of CH2Cl2. The yellow solution was degassed for 10 min with a stream of argon. The quartz tube was positioned ca. 20 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by recirculating tap water) and irradiated under argon at 25 °C for 6 h. The reaction mixture was transferred to a 25-mL pear flask with the aid of two 4-mL portions of CH2Cl2 and concentrated to afford 0.222 g of orange oil. This material was dissolved in 4.4 mL of toluene, the flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 2 h. The resulting mixture was concentrated to afford 0.234 g of orange oil. Column chromatography on 30 g of silica gel (gradient elution with 20-40% EtOAc-hexanes) furnished 0.149 g of an yellow oil which was further purified by column chromatography on 25 g of silica gel (elution with 1% MeOH-CH2Cl2) to afford 0.135 g (66%) of phenol 29 as a yellow oil: IR (neat) 3296 (broad), 2929, 2858, 1572, 1439, 1421, 1211, 1125, 1097, 1055, 1029, 966 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.20-7.26 (m, 3H), 7.14-7.20 (m, 2H), 6.32 (s, 1H), 4.80 (s, 1H), 4.50-4.66 (m, 1H), 4.21-4.35 (m, 1H), 3.96-4.16 (m, 4H), 2.37-2.67 (m, 6H), 1.75-1.84 (m, 2H), 1.65-1.75 (m, 2H), 1.54-1.65 (m, 1H), 1.22-1.38 (m, 12H), 1.06-1.21 (m, 1H), 0.90 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 152.7, 138.4 (d, J = 2.5 Hz), 138.1 (d, J = 3.8 Hz), 135.7 (d, J = 1.8 Hz), 129.9, 128.2, 127.6, 125.3 (d, J = 4.0 Hz), 123.5, 122.1 (d, J = 1.8 Hz), 62.9 (d, J = 5.6 Hz), 56.0 (d, J = 5.1 Hz), 32.0, 30.5, 29.5, 29.1, 26.4, 23.2, 23.0, 22.9, 16.4 (d, J = 6.9 Hz), 14.2; 31P NMR (121 MHz, CDCl3) δ 6.65; HRMS (ESI) (m/z) [M + H]+ calculated for C27H41NO4P, 474.2768; found 474.2780.
3-[N-(Diethylphosphoryl)-N-(benzyl)amino]-2-hexyl-5,6,7,8-tetrahydronaphthalen-1-ol (29) – Flow Reactor
Reactor A with a Pyrex filter was employed for this reaction. A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with diazo ketone 16 (0.115 g, 0.766 mmol, 1.1 equiv), ynamide 25 (0.240 g, 0.683 mmol, 1.0 equiv), and 2.9 mL of DCE, and the yellow solution was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The FEP tubing of the reactor was flushed with 3 mL of degassed DCE, the mercury lamp was turned on, and degassed DCE was pumped through the tubing for 5 min at a rate of 0.057 mL/min by syringe pump. The solution of 16 and 25 was transferred to a 5-mL glass syringe and pumped through the tubing at a rate of 0.057 mL/min. Once the addition was complete, the tubing was flushed with three portions of degassed DCE (0.4, 0.4, and then 4.0 mL). The 25-mL pear-shaped collection flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 40 h and then concentrated to afford 0.386 g of orange oil. This material was dissolved in 5 mL of CH2Cl2 and concentrated onto 5 g of silica gel. The free-flowing powder was deposited on a column of 55 g of silica gel and eluted with 35% EtOAc-hexanes to furnish 0.204 g of an orange oil which was further purified by column chromatography on 33 g of silica gel (gradient elution with 0-1% MeOH-CH2Cl2) to afford 0.176 g (53%) of phenol 29 as an orange oil with spectral data consistent with that reported in the procedure for the batch process given above.
3-[N-(Diethylphosphoryl)-N-(benzyl)amino]-2-hexyl-5,6,7,8-tetrahydronaphthalen-1-ol (29) – Thermal Benzannulation
A 25-mL round-bottomed flask equipped with a rubber septum and argon inlet needle was charged with ynamide 25 (0.154 g, 0.438 mmol, 1.0 equiv), diazo ketone 16 (0.070 g, 0.466 mmol, 1.1 equiv), and 5.5 mL of toluene. The septum was replaced with a condenser fitted with an argon inlet adapter and the yellow reaction mixture was stirred at reflux for 7 h. The resulting orange mixture was concentrated to yield 0.220 g of orange oil. Column chromatography on 42 g of silica gel (elution with 20% EtOAc-hexanes + 5% Et3N) provided 0.154 g of an orange oil that was further purified by column chromatography on 21 g of silica gel (elution with 20% EtOAc-hexanes) to afford 0.137 g (65%) of phenol 29 as a pale yellow oil with spectral data consistent with that reported in the procedure for the batch process given above.
3-[N-(Carbomethoxy)-N-(methyl)amino]-2-hexylnaphthalen-1-ol (20) – Batch Reactor
A 25-cm quartz tube (5 mm i.d., 7 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with diazo ketone 1918a (0.081 g, 0.552 mmol, 1.1 equiv), ynamide 17 (0.099 g, 0.502 mmol, 1.0 equiv), and 1.7 mL of CH2Cl2. The yellow reaction mixture was degassed via three cycles of freeze-pump-thaw (−196 °C, 0.05 mmHg). The quartz tube was positioned ca. 20 cm proximity to the Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by recirculating tap water) and irradiated under argon at 25 °C for 2.5 h. The reaction mixture was concentrated to 0.244 g of yellow oil, which was transferred with 2 mL of toluene to a 10-mL reaction tube. The solution was heated at reflux for 2.5 h, cooled to rt, and concentrated to give 0.262 g orange oil, which was dissolved in 1 ml of CH2Cl2 and concentrated on 0.5 g of silica gel. The resulting free flowing powder was deposited onto a column of 12 g of silica gel and eluted with 20% EtOAc-hexanes to afford 0.102 g (64%) of 20 as an orange wax: IR (thin film) 3385, 2955, 2928, 2857, 1684, 1573, 1461, 1374, 1194, and 1164 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 9 Hz, 1 H), 7.95 (bs, minor rotamer), 7.74 (d, J = 9 Hz, 1H), 7.60 (bs, minor rotamer), 7.46 (app dt, J = 3.8, 7.0 Hz, 2H), 7.34 (bs, minor rotamer), 7.26 (s, 1H), 7.24 (s, minor rotamer), 6.19 (s, minor rotamer), 5.85 (s, 1H), 3.88 (s, minor rotamer), 3.64 (s, 3H), 3.30 (s, 3H), 3.28 (s, minor rotamer), 2.67 (t, J = 7.1 Hz, 2H), 1.61-1.55 (m, 2H), 1.43-1.39 (m, 2H), 1.33-1.27 (m, 4H), and 0.90 (t, J = 6.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 157.0, 150.1, 140.6, 132.8, 127.7, 126.3, 125.8, 124.0, 121.3, 121.0, 118.8, 53.1, 39.1, 31.9, 30.0, 29.5, 26.0, 22.8, and 14.3; HRMS (EI) (m/z) M+ calcd for C19H25NO3: 315.1829, found 315.1823.
3-[N-(Carbomethoxy)-N-(methyl)amino]-2-hexylnaphthalen-1-ol (20) – Flow Reactor
Reactor A with a Pyrex filter was employed for this reaction. A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with diazo ketone 19 (0.088 g, 0.602 mmol, 1.1 equiv), ynamide 17 (0.108 g, 0.548 mmol, 1.0 equiv), and 2.2 mL of DCE and the yellow solution was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The FEP tubing of the reactor was flushed with 3 mL of degassed DCE, the mercury lamp was turned on, and degassed DCE was pumped through the tubing for 5 min at a rate of 0.057 mL/min by syringe pump. The solution of 19 and 17 was transferred to a 5-mL glass syringe and pumped through the tubing at a rate of 0.057 mL/min. Once the addition was complete, the tubing was flushed with three portions of degassed DCE (0.4, 0.4, and then 4.0 mL). The 25-mL pear-shaped collection flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 48 h and then concentrated to afford 0.188 g of orange oil. This material was dissolved in 3 mL of CH2Cl2 and concentrated onto 2 g of silica gel. The free-flowing powder was deposited on a column of 25 g of silica gel and eluted with 10% EtOAc-10% benzene-80% hexanes to furnish 0.114 g (66%) of phenol 20 as a yellow oil with spectral data consistent with that reported in the procedure for the batch process given above.
3-[N-(Carbomethoxy)-N-(methyl)amino]-2-hexylnaphthalen-1-ol (20) – Large Scale Flow Reactor
Reactor B with a Pyrex filter was employed for this reaction. A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with diazo ketone 19 (0.184 g, 1.26 mmol, 1.1 equiv), ynamide 17 (0.223 g, 1.13 mmol, 1.0 equiv), and 4.8 mL of DCE. and the yellow solution was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The FEP tubing of the reactor was flushed with 3 mL of degassed DCE, the mercury lamp was turned on, and degassed DCE was pumped through the tubing for 5 min at a rate of 0.32 mL/min by syringe pump. The solution of 19 and 17 was transferred to a 10-mL glass syringe and pumped through the tubing at a rate of 0.32 mL/min. Once the addition was complete, the tubing was flushed with three portions of degassed DCE (0.6, 0.6, and then 9.0 mL). The 50-mL pear-shaped collection flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 40 h and then concentrated to afford 0.500 g of orange oil. This material was dissolved in 5 mL of CH2Cl2 and concentrated onto 5 g of silica gel. The free-flowing powder was deposited on a column of 50 g of silica gel and eluted with 10% EtOAc-10% benzene-80% hexanes to furnish 0.220 g (63%) of phenol 20 as a yellow oil with spectral data consistent with that reported in the procedure for the batch process given above.
3-[Bis(trimethylsilyl)amino]-2-phenylnaphthalen-1-ol (28)
A 20-cm quartz tube (7 mm i.d., 10 mm o.d.) equipped with a stir bar, rubber septum, and argon inlet needle was charged with diazo ketone 19 (0.048 g, 0.325 mmol, 1.0 equiv), ynamine 2722 (0.65 mL, 0.5 M in benzene, 0.325 mmol, 1.0 equiv), and 0.7 mL of toluene. The yellow solution was degassed (three freeze-pump-thaw cycles at −196 °C, 0.05 mmHg) and backfilled with argon. The quartz tube was positioned ca. 20 cm from a Hanovia 450W medium pressure mercury lamp equipped with a uranium filter (quartz immersion well, cooled by recirculating tap water) and irradiated under argon with stirring at 25 °C for 45 h. The reaction mixture was transferred to a 15-mL threaded Pyrex tube (20 mm o.d.) with the aid of two 0.8-mL portions of toluene. The reaction tube was sealed with a threaded Teflon cap and the reaction mixture was heated at 110 °C (bath temperature) for 2.5 h. The resulting mixture was concentrated to afford 0.116 g of a dark yellow oil. Column chromatography on 20 g of silica gel (elution with 94:5:1 hexanes-EtOAc-Et3N) furnished 0.075 g (61%) of phenol 28 as a yellow solid: mp 79–81 °C; IR (neat) 3540, 3054, 2954, 1630, 1590, 1494, 1435, 1385, 1251, 1127, 1075, 942, 876, 839, and 822 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.18 (app d, J = 8.4 Hz, 1H), 7.70 (app d, J = 8.2 Hz, 1H), 7.52–7.56 (m, 2H), 7.38–7.49 (m, 5H), 7.10 (s, 1H), 5.47 (s, 1H), and −0.06 (s, 18H); 13C NMR (125 MHz, CDCl3) δ 149.1, 145.9, 135.2, 134.0, 132.1, 129.5, 128.4, 126.8, 126.7, 124.2, 122.9, 122.6, 121.8, 119.2, and 2.3; HRMS (ESI) (m/z) [M + H]+ calculated for C22H30NOSi2, 380.1860; found 380.1889.
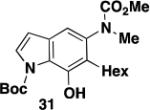
1-(tert-Butoxycarbonyl)-6-hexyl-7-hydroxy-5-[N-(methoxycarbonyl)-N-(methyl)amino]indole (31) – Batch Reactor
A 20-cm quartz tube (7 mm i.d., 10 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with diazo ketone 30 (0.135 g, 0.574 mmol, 1.1 equiv), ynamide 17 (0.103 g, 0.522 mmol, 1.0 equiv), and 2.2 mL of CH2Cl2. The solution was degassed for 10 min with a stream of argon. The quartz tube was positioned ca. 20 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by recirculating tap water) and irradiated under argon at 25 °C for 8 h. The reaction mixture was transferred to a 25-mL pear flask with the aid of two 4-mL portions of CH2Cl2 and concentrated to afford 0.222 g of an orange oil. This material was dissolved in 5.2 mL of toluene, the flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 2 h. The resulting mixture was concentrated to afford 0.234 g of an orange oil. Column chromatography on 25 g of silica gel (gradient elution with 10-20% EtOAc-hexanes) furnished 0.084 g (40%) of phenol 31 as a yellow oil: IR (neat) 3162 (broad), 2930, 2857, 1708, 1688, 1584, 1452, 1380, 1353, 1242, 1156, 1120 cm−1; 1H NMR (500 MHz, CDCl3) δ 11.13 (s, 1H), 11.09 (s, minor rotamer), 7.36-7.42 (m, 1H), 6.88 (s, minor rotamer), 6.82 (s, 1H), 6.43-6.49 (m, 1H), 3.80 (s, minor rotamer), 3.62 (s, 3H), 3.24 (s, 3H), 3.22 (s, minor rotamers), 2.53-2.68 (m, 2H), 1.65 (s, 9H), 1.50-1.60 (m, 2H), 1.36-1.45 (m, 2H), 1.27-1.36 (m, 4H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 157.0, 152.7, 143.6, 139.5, 131.1, 126.7, 124.8, 123.3, 110.7, 109.4, 86.3, 53.0, 38.9, 32.0, 30.2, 29.4, 28.2, 26.6, 22.8, 14.3; HRMS (ESI) (m/z) [M + Na]+ calculated for C22H32N2NaO5, 427.2203; found 427.2208.
1-(tert-Butoxycarbonyl)-6-hexyl-7-hydroxy-5-[N-(methoxycarbonyl)-N-(methyl)amino]indole (31) – Flow Reactor
Reactor A with a Pyrex filter was employed for this reaction. A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with diazo ketone 30 (0.134 g, 0.570 mmol, 1.1 equiv), ynamide 17 (0.100 g, 0.507 mmol, 1.0 equiv), and 2.2 mL of DCE and the yellow solution was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The FEP tubing of the reactor was flushed with 3 mL of degassed DCE, the mercury lamp was turned on, and degassed DCE was pumped through the tubing for 5 min at a rate of 0.057 mL/min by syringe pump. The solution of 30 and 17 was transferred to a 5-mL glass syringe and pumped through the tubing at a rate of 0.057 mL/min. Once the addition was complete, the tubing was flushed with three portions of degassed DCE (0.4, 0.4, and then 4.0 mL). The reaction mixture was then concentrated to afford 0.209 g of orange oil. This material was dissolved in 5.0 mL of toluene in the same 25-mL pear-shaped collection flask. The flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 2 h. The resulting mixture was concentrated to afford 0.189 g of a dark orange oil. Column chromatography on 25 g of silica gel (elution with 12% EtOAc-hexanes) furnished 0.075 g (37%) of phenol 31 as a yellow oil with spectral data consistent with that reported in the procedure for the batch process given above.
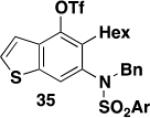
5-Hexyl-4-(trifluoromethylsulfonyloxy)-6-[N-(diethylphosphoryl)-N-(benzyl)amino]indole (33) – Batch Reactor
A 20-cm quartz tube (7 mm i.d., 10 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with diazo ketone 322b (0.137 g, 0.582 mmol, 1.1 equiv), ynamide 17 (0.103 g, 0.522 mmol, 1.0 equiv), and 2.2 mL of CH2Cl2. The yellow reaction mixture was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The quartz tube was positioned ca. 20 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by recirculating tap water) and irradiated under argon at 25 °C for 5.5 h. The reaction mixture was transferred to a 25-mL pear flask with the aid of two 4-mL portions of CH2Cl2 and concentrated to afford 0.249 g of a dark orange oil. This material was dissolved in 5.2 mL of toluene, the flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 2 h. The resulting mixture was concentrated to afford 0.262 g of a dark orange oil. Column chromatography on 30 g of silica gel (elution with 15% EtOAc-hexanes) furnished 0.152 g of crude benzannulation product as an orange oil.
A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with the crude benzannulation product (0.152 g, 1.0 equiv), 4-DMAP (0.092 g, 0.753 mmol, 2.0 equiv), and 2.7 mL of CH2Cl2. The orange solution was cooled to 0 °C and triflic anhydride (0.07 mL, 0.416 mmol, 1.1 equiv) was added dropwise by syringe over ca. 2 min. The reaction mixture was allowed to warm to rt, stirred for 2 h, and then diluted with 20 mL of CH2Cl2 and washed with two 10-mL portions of 1 M HCl solution. The combined aqueous layers were extracted with 10 mL of CH2Cl2, and the combined organic layers were washed with 20 mL of satd aq NaHCO3 solution and 20 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.187 g of orange oil. Column chromatography on 28 g of silica gel (elution with 10% EtOAc-hexanes) afforded 0.160 g (57% over two steps from the ynamide) of triflate 33 as an orange oil: IR (neat) 2958, 2932, 2860, 1744, 1716, 1529, 1449, 1409, 1373, 1347, 1304, 1249, 1215, 1154 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.12 (minor rotamer), 8.06 (s, 1H), 7.57-7.67 (m, 1H), 6.65-6.72 (m, 1H), 3.81 (minor rotamer), 3.63 (s, 3H), 3.29 (s, 3H), 2.61-2.76 (m, 2H), 1.66 (s, 9H), 1.46-1.57 (m, 2H), 1.32-1.40 (m, 2H), 1.25-1.32 (m, 4H), 0.89 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 156.7, 149.2, 139.7, 139.5, 135.0, 129.0, 128.1, 124.0, 118.9 (q, J = 318 Hz), 115.6, 103.9, 85.0, 53.2, 39.1, 31.8, 29.8, 29.8, 28.3, 26.6, 22.7, 14.2; HRMS (ESI) (m/z) [M + Na]+ calculated for C23H31F3N2NaO7S, 559.1696; found 559.1703.
5-Hexyl-4-(trifluoromethylsulfonyloxy)-6-[N-(diethylphosphoryl)-N-(benzyl)amino]indole (33) – Flow Reactor
Reactor A with a Pyrex filter was employed for this reaction. A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with diazo ketone 32 (0.570 g, 0.134 mmol, 1.1 equiv), ynamide 17 (0.100 g, 0.507 mmol, 1.0 equiv), and 2.2 mL of DCE and the yellow solution was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The FEP tubing of the reactor was flushed with 3 mL of degassed DCE, the mercury lamp was turned on, and degassed DCE was pumped through the tubing for 5 min at a rate of 0.057 mL/min by syringe pump. The solution of 32 and 17 was transferred to a 5-mL glass syringe and pumped through the tubing at a rate of 0.057 mL/min. Once the addition was complete, the tubing was flushed with three portions of degassed DCE (0.4, 0.4, and then 4.0 mL). The reaction mixture was concentrated to afford 0.202 g of orange oil. This material was dissolved in 5.0 mL of toluene in the same 25-mL pear-shaped collection flask. The flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 2 h. The resulting mixture was concentrated to afford 0.232 g of a dark orange oil. Column chromatography on 25 g of silica gel (elution with 12% EtOAc-hexanes) furnished 0.113 g of ca. 85-90% pure benzannulation product as an orange oil.
A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with the impure benzannulation product (0.113 g, 1.0 equiv), 4-DMAP (0.070 g, 0.573 mmol, 2.0 equiv), and 3.0 mL of CH2Cl2. The orange solution was cooled to 0 °C and triflic anhydride (0.06 mL, 0.357 mmol, 1.3 equiv) was added dropwise by syringe over ca. 2 min. The reaction mixture was allowed to warm to rt, stirred for 2 h, and then diluted with 20 mL of CH2Cl2 and washed with two 10-mL portions of 1 M HCl solution. The combined aqueous layers were extracted with 10 mL of CH2Cl2, and the combined organic layers were washed with 20 mL of satd aq NaHCO3 solution and 20 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.151 g of orange oil. Column chromatography on 18 g of silica gel (elution with 10% EtOAc-hexanes) afforded 0.127 g (47% over two steps from the ynamide) of triflate 33 as an orange oil with spectral data consistent with that reported in the procedure for the batch process given above.
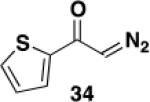
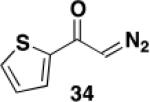
5-Hexyl-4-(trifluoromethylsulfonyloxy)-6-[N-(p-tolylsulfonyl)-N-(benzyl)amino]benzo[b]thiophene (35) – Flow Reactor
Reactor A with a uranium filter was employed for this reaction. A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with diazo ketone 34 (0.062 g, 0.407 mmol, 1.1 equiv), ynamide 22 (0.134 g, 0.363 mmol, 1.0 equiv), and 1.6 mL of DCE and the yellow solution was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The FEP tubing of the reactor was flushed with 3 mL of degassed DCE, the mercury lamp was turned on, and degassed DCE was pumped through the tubing for 5 min at a rate of 0.036 mL/min by syringe pump. The solution of 34 and 22 was transferred to a 5-mL glass syringe and pumped through the tubing at a rate of 0.036 mL/min. Once the addition was complete, the tubing was flushed with three portions of degassed DCE (0.4, 0.4, and then 4.0 mL). The 25-mL pear-shaped collection flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 42 h and then concentrated to afford 0.195 g of red oil. Column chromatography on 20 g of silica gel (elution with 10% EtOAc-hexanes) furnished 0.102 g of ca. 85-90% pure benzannulation product as an orange oil.
A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with the impure benzannulation product (0.102 g, 1.0 equiv), 4-DMAP (0.050 g, 0.409 mmol, 2.0 equiv), and 1.6 mL of CH2Cl2. The orange solution was cooled to 0 °C and triflic anhydride (0.04 mL, 0.238 mmol, 1.2 equiv) was added dropwise by syringe over ca. 2 min. The reaction mixture was allowed to warm to rt, stirred for 4 h, and then diluted with 10 mL of CH2Cl2 and washed with two 5-mL portions of 1 M HCl solution. The combined aqueous layers were extracted with 5 mL of CH2Cl2, and the combined organic layers were washed with 15 mL of satd aq NaHCO3 solution and 15 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.125 g of a red oil. Column chromatography on 13 g of silica gel (elution with 7% EtOAc-hexanes) afforded 0.106 g (47% over two steps from the ynamide) of triflate 35 as an off-white solid with spectral data consistent with that reported in the procedure for the batch process given above.
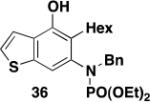
5-Hexyl-4-hydroxy-6-[N-(diethylphosphoryl)-N-(benzyl)amino]benzo[b]thiophene (36) – Batch Reactor
A 20-cm quartz tube (5 mm i.d., 7 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with diazo ketone 34 (0.049 g, 0.322 mmol, 1.1 equiv), ynamide 25 (0.099 g, 0.282 mmol, 1.0 equiv), and 1.3 mL of CH2Cl2. The yellow reaction mixture was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The quartz tube was positioned ca. 20 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by recirculating tap water) and irradiated under argon at 25 °C for 5.5 h. The reaction mixture was transferred to a 25-mL pear flask with the aid of two 4-mL portions of CH2Cl2 and concentrated to afford 0.143 g of an orange oil. This material was dissolved in 3 mL of toluene, the flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 1.5 h. The resulting mixture was concentrated to afford 0.159 g of a dark orange oil. Column chromatography on 15 g of silica gel (elution with 30% EtOAc-hexanes) furnished 0.115 g of an orange oil which was further purified by column chromatography on 15 g of silica gel (elution with 0.5% MeOH-CH2Cl2) to afford 0.105 g (78%) of phenol 36 as an orange oil: IR (neat) 3225 (broad), 2924, 2856, 1605, 1552, 1495, 1424, 1392, 1330, 1215, 1126, 1051, 970 cm−1; 1H NMR (500 MHz, CDCl3) 1H NMR (500 MHz, CDCl3) δ 7.32 (dd, J = 0.8 Hz, J = 5.5 Hz, 1H) 7.18-7.24 (m, 3H), 7.10-7.14 (m, 2H), 7.09 (d, J = 5.5 Hz), 6.95-6.97 (m, 1H), 6.74 (s, 1H), 4.59-4.67 (m, 1H), 4.02-4.32 (m, 5H), 2.58-2.68 (m, 2H), 2.45-2.55 (m, 2H), 1.57-1.70 (m, 1H), 1.21-1.42 (m, 12H), 0.99-1.11 (m, 1H), 0.90 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 150.4, 137.9 (d, J = 1.4 Hz), 137.7 (d, J = 3.4), 137.6 (d, J = 2.9 Hz), 129.9, 129.1 (d, J = 1.1 Hz), 128.3, 127.7, 124.2, 123.4 (d, J = 4.0 Hz), 120.3, 116.3, 63.2 (m), 55.9 (d, J = 4.5 Hz), 31.9, 30.5, 29.2, 26.2, 23.0, 16.4 (m), 14.4; 31P NMR (121 MHz, CDCl3) δ 6.18; HRMS (ESI) (m/z) [M + H]+ calculated for C25H35NO4PS, 476.2019; found 476.2034.
5-Hexyl-4-hydroxy-6-[N-(diethylphosphoryl)-N-(benzyl)amino]benzo[b]thiophene (36) – Flow Reactor
Reactor A with a Pyrex filter was employed for this reaction. A 25-mL pear flask equipped with a rubber septum and argon inlet needle was charged with diazo ketone 34 (0.069 g, 0.448 mmol, 1.1 equiv), ynamide 25 (0.143 g, 0.407 mmol, 1.0 equiv), and 1.7 mL of DCE and the yellow solution was degassed via three freeze-pump-thaw (−196 °C, 0.05 mmHg) cycles. The FEP tubing of the reactor was flushed with 3 mL of degassed DCE, the mercury lamp was turned on, and degassed DCE was pumped through the tubing for 5 min at a rate of 0.057 mL/min by syringe pump. The solution of 34 and 25 was transferred to a 5-mL glass syringe and pumped through the tubing at a rate of 0.057 mL/min. Once the addition was complete, the tubing was flushed with three portions of degassed DCE (0.4, 0.4, and then 4.0 mL). The 25-mL pear-shaped collection flask was equipped with a stir bar and cold-finger condenser, and the reaction mixture was heated at reflux for 42 h and then concentrated to afford 0.199 g of red oil. This material was dissolved in 2 mL of CH2Cl2 and concentrated onto 2 g of silica gel. The free-flowing powder was deposited on a column of 20 g of silica gel and eluted with 30% EtOAc-hexanes to furnish 0.159 g of an orange oil which was further purified by column chromatography on 20 g of silica gel (elution with 1% MeOH-CH2Cl2) to afford 0.154 g (79%) of phenol 36 as an orange oil with spectral data consistent with that reported in the procedure for the batch process given above.
Reductive Cleavage of N-Phosphorylaniline 29. 3-[N-(Benzyl)amino]-2-hexyl-5,6,7,8-tetrahydronaphthalen-1-ol (37)
A 25-mL, pear flask equipped with a rubber septum and argon inlet needle was charged with phosphoramide 29 (0.132 g, 2.79 mmol, 1.0 equiv) and 4 mL of toluene. Red-Al (≥65 wt% in toluene, 0.65 mL, 2.13 mmol, 8 equiv) was added and the resulting solution was heated at 85 °C for 2 h. The reaction mixture was quenched at room temperature by the addition of 10 mL of satd aq K2CO3 solution, diluted with 10 mL of ether, and the organic layer was washed with 10 mL of satd aq K2CO3 solution. The combined aqueous layers were extracted with three 10-mL portions of Et2O. The combined organic layers were washed with 20 mL of brine, dried over Na2SO4, filtered, and concentrated to give 0.122 g of red oil. Column chromatography on 11 g of silica gel (elution with 2% EtOAc-hexanes) provided 0.077 g (84%) of amine 37 as a red solid: mp 58-60 °C; IR (neat) 3556 (broad), 3452 (broad) 2926, 2856, 1621, 1582, 154, 1438, 1352, 1317, 1198, 1143 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.36-7.45 (m, 4H), 7.31 (tt, J = 7.0 Hz, J = 1.5 Hz, 1H), 6.09 (s, 1H), 4.62 (s, 1H), 4.36 (s, 2H), 3.87 (s, 1H), 2.68 (t, J = 6.3 Hz, 2H), 2.52-2.60 (m, 4H), 1.83-1.89 (m, 2H), 1.73-1.79 (m, 2H), 1.52-1.60 (m, 2H), 1.38-1.47 (m, 2H), 1.29-1.38 (m, 4H), 0.91 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.8, 144.5, 140.1, 136.2,128.8, 127.7, 127.3, 112.1, 110.7, 104.1, 48.9, 32.0, 30.0, 29.9, 28.8, 24.1, 23.3, 23.2, 22.9, 22.6, 14.3; HRMS (ESI) (m/z) [M + H]+ calculated for C23H32NO, 338.2478; found 338.2470.
Tandem Benzannulation/Ring Closing Metathesis
3-[N-(Carbomethoxy)-N-(allyl)amino]-2-[but-3-en-2-(tert-butyldimethylsilanyloxy)yl]naphthalen-1-ol (38)
A 25-cm quartz tube (5 mm i.d., 7 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with diazo ketone 19 (0.075 g, 0.510 mmol, 1.1 equiv), ynamide 15 (0.150 g, 0.469 mmol, 1.0 equiv), and 1.8 mL of CH2Cl2. The yellow reaction mixture was degassed via three cycles of freeze-pump-thaw (−196 °C, 0.05 mmHg). The quartz tube was positioned ca. 20 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by recirculating tap water) and irradiated under argon at 25 °C for 2.5 h. The reaction mixture was concentrated to afford 0.260 g of yellow oil, which was transferred with 2 mL of toluene to a 10-mL reaction tube. The solution was heated at reflux for 2.5 h, allowed to cool to rt, and then concentrated to give 0.291 g orange oil. Purification by column chromatography on 16 g of silica gel (elution with 10% EtOAc-hexanes) afforded 0.036 g (24%) of unreacted ynamide 15 and 0.129 g (62%) of 38 as a viscous yellow oil: IR (thin film) 3238, 2955, 2859, 1716, 1636, 1597, 1574, 1448, 1386, and 1256 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.16 (s, minor rotamer), 9.01 (s, 1H), 8.28-8.30 (m, 1H), 7.71-7.74 (m, 1H), 7.45-7.47 (m, 2H), 7.20-7.23 (m, 1H), 5.84-6.08 (m, 2H), 5.11-5.30 (m, 4H), 4.47-4.54 (m, 2H), 3.81-3.95 (m, 2H), 3.66 (s, 3H), 3.60 (s, minor rotamer), 3.03-3.11 (m, 1H), 2.82-2.89 (m, 1H), 0.92 (s, 9H), 0.08 (s, minor rotamer), 0.05 (s, 3H), -0.05 (s, minor rotamer), and -0.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.5, 152.9, 139.8, 139.1, 139.0, 133.5, 133.1, 127.4, 126.6, 124.6, 125.3, 122.8, 119.5, 118.7, 117.3, 115.7, 54.4, 53.0, 35.5, 35.0, 25.9, 18.2, -4.3, and -5.0, (Peaks corresponding to the minor rotamer: δ 153.2, 133.2, 127.3, 122.9, 119.2, 118.5, 117.0, 54.4, 53.1, 26.0, 18.3, -4.2, and -4.8); HRMS (ESI) (m/z) [M + Na]+ calcd for C25H35NNaO4Si: 464.2228, found: 464.2217.
Methyl 5-(tert-Butyldimethylsilanyloxy)-7-hydroxy-5,6-dihydro-2H-napthaleno[b]azocine-1-carboxylate (40)
A 100-mL, round-bottomed flask fitted with a rubber septum and argon inlet needle was charged with a solution of the Grubbs catalyst 39 (0.012 g, 0.014 mmol, 0.05 equiv) in 20 mL of CH2Cl2. Naphthol 38 (0.124 g, 0.281 mmol, 1.0 equiv) in 8 mL of CH2Cl2 was added to the reaction flask via cannula in one portion. The rubber septum was replaced with a reflux condenser fitted with an argon inlet adapter. The pale pink reaction mixture was heated at reflux for 6 h, allowed to cool to rt, and then concentrated to afford ca. 0.134 g of yellow-brown foam. Column chromatography on 12 g of silica gel (elution with 15% EtOAc-hexane) yielded 0.088 g (76%) of azocine 40 as an off-white solid: mp 74-76°C; IR (thin film) 3351, 2955, 2857, 1709, 1684, 1572, 1450, 1388, 1250 and 1065 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.24-8.26 (m, 1H), 7.70 (s, 1H), 7.72-7.75 (m, 1H), 7.44 (app dd, J = 3.1, 6.2 Hz, 2H), 7.26 (bs, 1H), 5.57 (app d, J = 11.6 Hz, 1H), 5.34-5.41 (m, 1H), 5.14 (app s, 1H), 4.44 (bs, 1H), 4.20 (dd, J = 7.9, 14.4 Hz, 1H), 3.72 (bs, 3H), 3.24 (app d, J = 2.7 Hz, 2H), 1.01 (s, 9H), 0.26 (s, 3H), and 0.19 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.3, 150.6, 140.6, 137.7, 133.1, 127.5, 127.3, 126.3, 125.3, 124.7, 122.5, 118.9, 117.7, 72.6, 53.3, 48.0, 35.9, 26.0, 18.4, -4.5, and -4.8; HRMS (EI) (m/z) [M + Na]+ calcd for C23H31NNaO4Si, 436.1915; found: 436.1922.
N-(Methoxycarbonyl)-N-allyl-(2-allyl-4,5-dimethyl-3-hydroxyphenyl)amine (43)
A 25-mL recovery flask equipped with a condenser, rubber septum and argon inlet needle was charged with ynamide 417 (0.146 g, 0.815 mmol, 1.0 equiv) and 6.0 mL of toluene. A 25-mL pear flask fitted with a rubber septum and argon inlet needle was charged with diazo ketone 4218a (0.244 g, 1.96 mmol, 2.4 equiv) and 8 mL of toluene. Both solutions were degassed with a stream of argon for 15 min. The solution of diazo ketone was taken up in a 10-mL glass syringe wrapped in aluminum foil and fitted with a 20-gauge, 20-cm long steel needle. The reaction mixture was heated at reflux and the diazo ketone solution was added through the condenser via syringe pump. After the addition was completed (ca. 13.5 h), the pear flask was rinsed with 0.5 mL of toluene and added with the same syringe in one portion to the reaction mixture. Heating was continued for 5 h. After cooling to rt, the reaction mixture was concentrated to afford 0.450 g of orange oil that was diluted with 5 mL of MeOH and 3 mL of 5 M KOH solution. After heating at 60-70 °C for 2 h, the resulting mixture was cooled to rt, quenched with 16 mL of 1 M HCl solution, and diluted with 20 mL of Et2O. The aqueous layer was extracted with two 20-mL portions of Et2O and the combined organic layers were washed with 40 mL of satd aq NaHCO3 solution and 40 mL of brine, dried over MgSO4, filtered, and concentrated to provide 0.308 g of orange oil. Purification by column chromatography on 39 g of silica gel (gradient elution with 15-20% EtOAc-hexanes) afforded 0.146 g of aniline 43 (65%) as a yellow solid: mp 70-72 °C; IR (neat) 3412, 3078, 2925, 2857, 1686, 1575, 1455, 1389, 1320, 1257, 1196 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.58 (s, 1 H), 5.85-6.02 (m, 2 H), 5.18-5.30 (m, 3 H), 5.07-5.15 (m, 2 H), 4.21-4.32 (m, 1 H), 3.85-4.03 (m, 1 H), 3.78 (s, minor rotamer), 3.62 (s, 3 H), 3.26-3.39 (m, 2 H), 2.24 (s, 3 H), 2.15 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 156.4, 153.5, 137.8, 136.8, 135.9, 133.4, 123.4, 122.1, 120.2, 118.4, 117.3, 54.1, 53.1, 30.9, 20.2, 12.0; HRMS (ESI) (m/z) [M + H]+ calculated for C16H22NO3, 276.1594; found 276.1598.
Methyl 6-Hydroxy-7,8-dimethyl-2,5-dihydro-benzo[b]azepine-1-carboxylate (44)
A 100-mL, round-bottomed flask equipped with a rubber septum and argon inlet needle was charged with a solution of the Grubbs catalyst 39 (0.014 g, 0.016 mmol, 0.05 equiv) in 30 mL of CH2Cl2. The aniline 43 (0.094 g, 0.341 mmol, 1.0 equiv) in 6 mL of CH2Cl2 was added to the reaction flask via cannula in one portion and the rubber septum was replaced with a reflux condenser fitted with an argon inlet adapter. The pale pink reaction mixture was heated at reflux for 40 min, allowed to cool to rt, and then concentrated onto 2 g of silica gel. The free-flowing powder was deposited on a column of 20 g of silica gel and eluted with 25% EtOAc-hexanes to afford 0.081 g (96%) of dihydrobenzoazepine 44 as a tan wax: IR (neat) 3399, 3024, 2955, 1685, 1615, 1579, 1456, 1395, 1333, 1262, 1193, 1088 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.70 (s, minor rotamer), 6.64 s, 1 H), 5.75-5.84 (m, 1 H), 5.43-5.52 (m, 1 H), 4.72-5.16 (m, 1 H), 4.69 (s, 1H), 4.22 (s, minor rotamer), 3.79 (s, minor rotamer) 3.67 (s, 3 H), 3.34-3.66 (m, 3 H), 2.26 (s, 3 H), 2.24 (s, minor rotamer), 2.15 (s, 3 H), 2.12 (s, minor rotamer); 13C NMR (125 MHz, CDCl3 mixture of two rotamers) for major rotamer: δ 156.4, 150.4, 139.5, 135.8, 127.3, 126.0, 125.2, 123.8, 121.5, 53.4, 48.3, 23.3, 20.4, 12.3; additional resonances appeared for the minor rotamer at δ: 126.9, 121.4, 48.1, 23.1; HRMS (ESI) (m/z) [M + H]+ calculated for C14H18NO3, 248.1281; found 248.1272.
Synthesis of Indoles via Tandem Ynamide Benzannulation/Cyclization Strategies
2-(Trimethylsilyl)ethyl Benzyl[2-(2,2-dimethoxyethyl)-3-hydroxy-4,5-dimethylphenyl]carbamate (46)
A solution of ynamide 458 (0.340 g, 0.936 mmol, 1.0 equiv) in 3.5 mL of CH2Cl2 was distributed evenly between two 25-cm quartz reaction tubes (13 mm i.d., 15 mm o.d.) fitted with rubber septa and argon inlet needles. A 15-mL Pyrex tube fitted with a rubber septum and argon inlet needle was charged with a solution of diazo ketone 42 (0.310 g, 2.07 mmol, 2.4 equiv) in 7 mL of CH2Cl2. All solutions were purged with argon for 10 min. The upper portions (ca. 10 cm) of the quartz tubes were wrapped in aluminum foil and the tubes were positioned ca. 4 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by tap water). The quartz reaction tubes and the immersion well were partially submerged in a room-temperature water bath contained in a 2-L beaker wrapped in aluminum foil. The diazo ketone solution was taken up in two 5-mL glass syringes fitted with 20 gauge, 20-cm steel needles and wrapped in aluminum foil, and the solution was added to the reaction mixture using a syringe pump over 5 h (0.5 mL CH2Cl2 rinse). Irradiation was continued for 1 h, and then the reaction mixtures were combined in a 25-mL round-bottomed flask, concentrated, and the residue was diluted with 5 mL of toluene. The flask was fitted with a cold-finger condenser with an argon inlet sidearm, and the solution was heated at reflux for 1 h, allowed to cool to rt, and then concentrated to give 0.625 g of an orange oil. Column chromatography on 20 g of grade II neutral alumina (gradient elution with 0–5% EtOAc–hexanes) gave 0.292 g of a mixture of unreacted ynamide 45 and phenol 46. Mixed fractions (0.057 g) were purified by chromatography on 5 g of neutral alumina (gradient elution with 5–10% EtOAc–hexanes) to give additional 0.034 g of the mixture of 45 and 46. The combined mixtures were then further purified on 25 g of silica gel (elution with 10:10:80 Et2O:benzene:CH2Cl2) to give 0.255 g (59%) of phenol 46 as a yellow oil: IR (film) 3314, 2952, 1698, 1619, 1573, 1454, 1407, 1311, 1251, 1116, 1093, 1074, 1041 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.03 (br s, 1H), 7.20–7.28 (m, 5H), 6.43 (br s, minor rotamer), 6.35 (br s, 1H), 4.68 (s, 2H), 4.00–4.36 (br m, 3H), 3.32 (s, 3H), 3.23 (s, 3H), 2.52 (d, J = 5.6 Hz, 2H), 2.13 (s, 3H), 2.12 (s, 3H), 1.08 (br m, minor rotamer), 0.86 (br m, 2H), 0.04 (s, minor rotamer), -0.10 (s, 9H); 13C NMR (100 MHz, CDCl3), (mixture of two rotamers) for major rotamer: δ 156.6, 154.5, 138.3, 137.7, 137.1, 129.5, 128.5, 127.7 124.6, 121.8, 118.7 105.7, 64.2, 55.1, 54.8, 53.6, 30.8, 20.1, 18.1, 12.3, -1.5; additional resonances appeared for the minor rotamer at δ 129.0; HRMS (ESI) [M + Na]+ calcd for C25H37NNaO5Si: 482.2333, found 482.2336.
N-Benzyl-N-2-(trimethylsilyl)ethoxycarbonyl-[1-hydroxy-2-(2,2-dimethoxyethyl)-5,6,7,8-tetrahydronaphthalen-3-yl]amine (48)
A solution of ynamide 45 (0.313 g, 0.86 mmol, 1.0 equiv) in 2.8 mL of CH2Cl2 was distributed equally between two 25-cm quartz reaction tubes (13 mm i.d., 15 mm o.d.) fitted with rubber septa and argon inlet needles. A 15-mL Pyrex tube fitted with a rubber septum and argon inlet needle was charged with diazo ketone 16 (0.310 g, 2.07 mmol, 2.4 equiv) and 5.8 mL of CH2Cl2. Both solutions were degassed by three freeze-pump-thaw cycles at -196 °C, 0.05 mmHg. The solution of diazo ketone was taken up in two 5-mL glass syringes wrapped in aluminum foil and fitted with 20-gauge, 20-cm steel needles. The upper portions (ca. 18-cm length) of the quartz tubes were wrapped in aluminum foil and the tubes were positioned ca. 3 cm from a Hanovia 450W medium pressure mercury lamp cooled in a quartz immersion well. The quartz tubes and the immersion well were partially submerged in a room temperature water bath contained in a 2-L beaker wrapped in aluminum foil. The diazo ketone solution was added to the reaction tubes via syringe pump over ca. 5 h (0.5 mL CH2Cl2 wash) while the reaction mixture was irradiated and then irradiation was continued for 90 min. The combined reaction mixture was concentrated to provide 0.583 g of an orange oil which was transferred with the aid of 5 mL of toluene to a 25-mL, one-necked, round-bottomed flask equipped with a cold-finger condenser with argon inlet side arm. The solution was heated at reflux for 1 h, allowed to cool to room temperature, and then concentrated to give 0.550 g of an orange oil. This material was dissolved in ca. 10 mL of CH2Cl2 and concentrated onto 3.5 g of silica gel. The free-flowing powder was added to the top of a column of 55 g of silica gel and eluted with 15% EtOAc-hexanes to afford 0.224 g of acetal 48 as a viscous yellow oil and 0.059 g of a mixture of phenol 48 and unreacted ynamide 45. This mixture was dissolved in ca. 5 mL of CH2Cl2 and concentrated onto 0.5 g of silica gel. The free flowing powder was added to the top of a column of 6 g of silica gel and eluted with 15% EtOAc-hexanes to afford 0.027 g of phenol 48 as a viscous yellow oil. The combined yield of phenol 48 was 0.251 g (60%): IR (film) 3315, 2935, 2858, 2838, 1698, 1617, 1572, 1496, 1436, 1406, 1333, 1299, 1249, 1116, and 1072 cm−1; 1H NMR (500 MHz, CDCl3, ca. 68:32 mixture of rotamers) for major rotamer: δ 8.13 (s, minor rotamer), 8.09 (s, 1H), 7.18-7.35 (m, 5H), 6.38 (s, minor rotamer), 6.28 (s, 1H), 4.75 (d, J = 14.4 Hz, 1H), 4.66 (d, J = 14.4 Hz, 1H), 4.05-4.47 (m, 3H), 3.40 (s, minor rotamer), 3.37 (s, 3H), 3.31 (s, minor rotamer), 3.28 (s, 3H), 2.51-2.71 (m, 6H), 1.68-1.84 (m, 4H), 1.05-1.19 (m, minor rotamer), 0.83-1.01 (m, 2H), 0.07 (s, minor rotamer), -0.07 (s, 9H); 13C NMR (125 MHz, CDCl3, mixture of two rotamers) for major rotamer: δ 156.5, 154.1, 138.3, 137.7, 137.6, 129.4, 128.4, 127.5, 125.3, 120.8, 117.6, 105.6, 64.1, 55.0, 54.7, 53.5, 30.9, 30.6, 29.4, 23.5, 22.8, 17.9, and -1.5; additional resonances appeared for the minor rotamer at: δ 156.0, 139.0, 128.8, 120.4, 117.8, 55.2, 54.9, and -1.4; HRMS (ESI) [M+Na]+ calcd for C27H39NNaO5Si: 508.2490, found 508.2497.
2-(Trimethylsilyl)ethyl Benzyl[5-(2,2-dimethoxyethyl)-6-hydroxy-2,3-dihydro-1H-phenalen-4-yl]carbamate (54)
Two 25-cm quartz tubes (13 mm i.d., 15 mm o.d.) were each charged with ynamide 45 (0.300 g, 0.825 mmol, 1.0 equiv) and diazo ketone 532b (0.231 g, 1.24 mmol, 1.5 equiv). A stir bar was added to each reaction tube, the tubes were fitted with rubber septa and argon inlet needles, and purged with argon. Dichloromethane (8.5 mL) was then added to each tube and the reaction tubes were positioned ca. 3 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by tap water) in a room-temperature water bath contained in a 2-L beaker wrapped in aluminum foil. The reaction solutions were irradiated for 2.5 h and then combined and concentrated to give 1.129 g of an orange oil. A solution of this material in 20 mL of toluene was transferred to a flask fitted with a cold-finger condenser. The solution was heated at reflux for 2 h, allowed to cool to rt, and concentrated to give 1.15 g of an orange oil. Column chromatography on 50 g of grade II neutral alumina (elution with 10% EtOAc–hexanes) gave 0.680 g (79%) of phenol 54 as a yellow oil: IR (film) 3291, 2949, 2835, 1698, 1620, 1583, 1496, 1398, 1357, 1318, 1250, 1208, 1184, 1141, 1115 cm−1; 1H NMR (400 MHz, CDCl3, ca. 80:20 mixture of rotamers) for major rotamer: δ 8.50 (s, 1H), 8.48 (s, minor rotamer), 8.14 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 8.3 Hz, minor rotamer), 7.41–7.36 (m, 1H), 7.31-7.10 (m, 6H), 4.84 (d, J = 13.9 Hz, 1H), 4.58 (d, J = 13.9 Hz, 1H), 4.48 (d, J = 14.2 Hz, minor rotamer), 4.44–4.04 (m, 3H), 3.39 (s, minor rotamer), 3.37 (s, 3H), 3.28 (s, minor rotamer), 3.27 (s, 3H), 3.01 (t, J = 5.9 Hz, 2H), 2.73 (d, J = 6.9 Hz, minor rotamer), 2.69 (d, J = 6.9 Hz, 2H), 2.84–2.42 (m, 2H), 1.89 (pq, J = 6.1 Hz, minor rotamer), 1.81 (pq, J = 6.0 Hz, 2H), 1.26–1.18 (m, minor rotamer), 0.95–0.75 (m, 2H), 0.12 (s, minor rotamer), and 0.10 (s, 9H); 13C NMR (100 MHz, CDCl3, mixture of two rotamers) for major rotamer: δ 156.7, 150.8, 137.0, 136.3, 135.0, 130.1, 129.9, 128.5, 128.0, 125.7, 125.5, 125.2, 124.8, 120.6, 114.9, 106.0, 64.2, 55.1, 54.4, 53.0, 31.36, 31.34, 26.8, 22.8, 18.1, −1.5; additional resonances appeared for the minor rotamer at δ: 155.7, 136.9, 135.5, 130.4, 130.2, 128.0, 125.1, 124.6, 115.4, 106.2, 64.3, 55.6, 54.3, 52.9, 31.5, 22.9, 18.2, −1.2; HRMS (ESI) [M + Na]+ calcd for C30H39NNaO5Si: 544.2490, found 544.2502.
4-[Benzyl([2-(trimethylsilyl)ethoxy]carbonyl)amino]-5-(2,2-dimethoxyethyl)-2,3-dihydro-1H-phenalen-6-yl Trifluoromethanesulfonate (55)
A 25-mL recovery flask was charged with phenol 54 (0.233 g, 0.447 mmol, 1 equiv) and 4 mL of THF and cooled to 0 °C. Sodium hydride (60% wt oil disp., 0.025 g, 0.63 mmol, 1.4 equiv) was then added and the reaction mixture was stirred for ca. 5 min until gas evolution ceased. A solution of N-phenyltriflimide (0.224 g, 0.627 mmol, 1.4 equiv) in 2 mL of THF was added, and the resulting brown mixture was allowed to warm to rt over 15 min. The reaction mixture was cooled to 0 °C, and 3 mL of satd aq NaHCO3 solution was added. The resulting mixture was diluted with 15 mL of Et2O, and the aqueous layer was separated and extracted with two 15-mL portions of Et2O. The combined organic phases were washed with two 20-mL portions of 2.5 M NaOH solution and 20 mL of brine, dried over MgSO4, filtered and concentrated to give 0.329 g of a yellow oil. Column chromatography on 15 g of silica gel (elution with 10% EtOAc–hexanes) gave 0.219 g (75%) of trifluoromethanesulfonate 55 as a viscous colorless oil: IR (film) 2953, 2835, 1703, 1597, 1403, 1318, 1248, 1215, 1138, 1121, 1074, 943 and 729 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.00 (d, J = 8.5 Hz, 1H), 7.59–7.60 (m, 1H), 7.38–7.14 (m, 6H), 5.35 (d, J = 14.3 Hz, 1H), 5.19 (d, J = 14 Hz, minor rotamer), 4.76–4.78 (m, 1H), 4.70–4.72 (m, minor rotamer), 4.16–4.56 (m, 3H), 3.35–3.46 (m, 3H), 3.29–3.35 (m, 3H), 2.96–3.13 (m, 3H), 2.54–2.60 (m, 1H), 2.14–2.27(m, 1H), 1.27–1.96 (m, 4H), 0.87–1.00 (m, 2H), 0.20 (s, minor rotamer), 0.00 (s, 9H); 13C NMR (150 MHz, CDCl3, mixture of two rotamers) for major rotamer: δ 156.4, 142.4, 137.3, 137.1, 136.6, 134.7, 130.4, 130.3, 128.6, 127.6, 127.2, 126.87, 125.7, 119.6, 103.6, 64.5, 54.5, 54.1, 53.2, 34.8, 32.1, 31.1, 27.5, 22.1, 18.2, −1.5; additional resonances appeared for the minor rotamer at δ: 155.2, 142.5, 137.6, 137.5, 136.4, 134.9, 130.0, 128.7, 127.4, 126.93, 125.6, 104.3, 64.7, 55.2, 54.4, 52.2, 32.6, 31.7, 27.3, 22.8, 18.3, −1.3; HRMS (ESI) [M + Na]+ calcd for C31H38F3NNaO7SSi: 676.1983, found 676.1986.
Methyl Allyl(2-[2-(tert-butyldimethylsilyloxy)ethyl]-3-hydroxy-4,5-dimethylphenyl)carbamate (61)
Two 25-cm quartz tubes (13 mm i.d., 15 mm o.d.) fitted with rubber septa and argon inlet needles were charged with solutions of ynamide 588 (flask A: 0.168 g, 0.565 mmol, 1.0 equiv; flask B: 0.151 g, 0.508 mmol, 1.0 equiv, each in 2 mL of CH2Cl2). A 25-mL recovery flask was charged with a solution of diazo ketone 42 (0.340 g, 2.74 mmol, 2.6 equiv) in 7 mL of CH2Cl2. All solutions were purged with argon for 15 min. The upper portions (ca. 10 cm) of the quartz reaction tubes were wrapped in aluminum foil and the tubes were positioned ca. 5 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by tap water). The reaction tubes and the immersion well were partially submerged in a room-temperature water bath contained in a 2-L beaker wrapped in aluminum foil.44 The diazo ketone solution was taken up into two 5-mL gastight syringes wrapped in aluminum foil and fitted with 20 gauge, 20-cm steel needles and added to the reaction tubes using a syringe pump over 7 h (1 mL CH2 Cl2 rinse).45 Irradiation was continued for 20 min, and then the reaction mixtures were combined and concentrated in a 25-mL round-bottomed flask. The residue was diluted with 7 mL of toluene, the flask was fitted with a cold-finger condenser with an argon inlet sidearm, and the solution was heated at reflux for 3 h. The reaction mixture was allowed to cool to rt and concentrated to give 0.7 g of a dark orange oil. Column chromatography on 50 g of silica gel (elution with 10% EtOAc–hexanes) gave 0.328 g (78%) of phenol 61 as a yellow oil which crystallized upon refrigeration: mp 48–49 °C; IR (neat) 3273, 2859, 2930, 1709, 1620, 1574, 1449, 1388, 1313, 1259, 1214, 1192, 1147, 1092, 1039 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 6.51 (s, 1H), 5.85–5.95 (m, 1H), 5.12 (m, 2 H), 3.70– 4.30 (m, 4H), 3.62 (s, 3H), 2.78 (m, 2H), 2.23 (s, 3H), 2.17 (s, 3H), 0.92 (s, 9H), 0.08 (d, J = 5.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 156.5, 154.7, 137.6, 136.7, 133.3, 124.7, 122.4, 121.2, 118.3, 65.3, 54.3, 53.0, 29.5, 25.9, 20.2, 18.4, 12.3, −5.5; HRMS (ESI) [M + Na]+ calcd for C21H35NNaO4Si: 416.2228, found 416.2218.
Methyl Allyl[4-(2-[(tert-butyldimethylsilyl)oxy]ethyl)-3-hydroxybenzocyclobutene]-5-carbamate (64)
A 25-cm quartz tube (13 mm i.d., 15 mm o.d.) equipped with two stir bars, a rubber septum, and argon inlet needle was charged with ynamide 58 (0.310 g, 1.04 mmol, 1.0 equiv) and 3 mL of CH2Cl2. An 18x160 mm test tube was charged with a solution of diazo ketone 50 (0.328 g, 2.69 mmol, 2.6 equiv) in 8 mL of CH2Cl2. Both solutions were purged with argon for 15 min. The upper portion (ca. 10 cm) of the quartz reaction tube was wrapped in aluminum foil and the tube was positioned ca. 15 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by tap water). The diazo ketone solution was taken up into a 10-mL gastight syringe wrapped in aluminum foil and fitted with a 20 gauge, 20-cm steel needle and added to the reaction tubes using a syringe pump over 5 h (1 mL CH2Cl2 rinse). Irradiation was continued for 5 h and the reaction mixture was concentrated in a 50-mL round-bottomed flask. The residue was diluted with 15 mL of toluene, the flask was fitted with a reflux condenser with an argon inlet, and the solution was heated at reflux for 2.5 h. The resulting red-brown solution was allowed to cool to rt and then concentrated to give 0.74 g of a red-brown oil. Column chromatography on 15 g of silica gel (elution with 10% EtOAc–hexanes) yielded 0.099 g (32%) of unreacted ynamide 58 as a colorless oil and 0.251 g (62%; 91% based on recovered ynamide) of carbamate 64 as a white solid: mp 129–130 °C; IR (KBr pellet) 3317, 2928, 1679, 1593, 1457, 1432, 1310, 1258, 1148, 1089, 982, 921, 859, 834 and 773 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 6.45 (s, 1H), 5.87–5.93 (m, 1H), 5.11–5.14 (m, 2H), 4.31 (dd, J = 19.1, 10.1 Hz, 1H), 3.59–3.91(m, 6H), 3.09–3.15 (m, 4H), 2.74–2.85 (m, 2H), 0.92 (s, 9H), 0.10 (d, J = 4.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 156.6, 151.0, 145.7, 133.2, 130.9, 124.2, 118.5, 118.4, 115.5, 65.3, 54.4, 53.0, 29.7, 29.2, 27.4, 26.0, 18.5, −5.4; Anal. calcd for C21H33NO4Si: C, 64.41; H, 8.49; N, 3.58. Found: C, 64.50; H, 8.36; N, 3.53.
1-Benzyl-5,6-dimethyl-4-hydroxyindole (47)
A 25-mL recovery flask fitted with a rubber septum and argon inlet needle was charged with a solution of phenol 46 (0.325 g, 0.712 mmol, 1.0 equiv) in 3.5 mL of THF. The solution was cooled at 0 °C while TBAF solution (1 M in THF, 3.5 mL, 3.5 mmol, 5 equiv) was added via syringe over 3 min. The resulting brown solution was stirred at rt for 18 h, and then cooled at 0 °C while 6 M aqueous HCl solution (3.5 mL, 21.0 mmol, 30 equiv) was added dropwise over ca. 2 min. The resulting light yellow solution was stirred at 0 °C for 5 min, and then heated at reflux for 40 min. The resulting blue-brown mixture was allowed to cool to rt, diluted with 80 mL of Et2O, and washed with two 30 mL portions of satd aq NaHCO3 solution. The combined aqueous layers were extracted with three 15 mL portions of Et2O, and the combined organic phases were washed with 50 mL of brine, dried over Na2SO4, filtered, and concentrated to give 0.184 g of a brown oil. Column chromatography on 20 g of silica gel (elution with 10% EtOAc–hexanes) gave 0.146 g (82%) of indole 47 as a tan solid: IR (KBr pellet) 3592, 3054, 2986, 2924, 1570, 1491, 1454, 1442, 1297, 1265, 1236, 909, 705 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.25–7.32 (m, 3H), 7.09–7.11 (m, 2H), 6.98 (d, J = 3.2 Hz, 1H), 6.75 (s, 1H), 6.47 (dd, J = 3.2, 0.4 Hz, 1H), 5.27 (s, 2H), 4.85 (s, 1H), 2.35 (s, 3H), 2.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 146.2, 137.9, 136.2, 132.5, 128.9, 127.6, 126.8, 126.6, 116.6, 111.6, 103.5, 97.1, 50.1, 21.4, 11.3; HRMS (ESI) [M + H]+ calcd for C17H18NO: 252.1383, found 252.1371.
1-Benzyl-5,6,7,8-tetrahydro-1H-benz[f]indol-4-yl Acetate (49).46
A 25-mL, two-necked, round-bottomed flask fitted with a rubber septum and argon inlet adapter was charged with a solution of carbamate 48 (0.305 g, 0.63 mmol, 1.0 equiv) in 3.2 mL of THF. The solution was cooled at 0 °C while TBAF solution (1 M in THF, 3.2 mL, 3.2 mmol, 5 equiv) was added via syringe over 3 min. The resulting orange solution was stirred at rt for 8 h and then cooled at 0 °C while 6 M aq HCl solution (3.2 mL, 19.2 mmol, 30 equiv) was added dropwise over 3 min. The resulting dark orange solution was stirred at rt for 41 h. The resulting dark green mixture was diluted with 50 mL of Et2O and washed with two 25 mL portions of satd aq NaHCO3 solution. The combined aqueous layers were extracted with two 20 mL portions of Et2O, and the combined organic phases were washed with 40 mL of brine, dried over Na2SO4, filtered, and concentrated to give 0.207 g of a dark brown oil. This indole was immediately protected as the acetate derivative due to its propensity to oxidize. The unpurified indole was transferred to a 25-mL, one-necked, round-bottomed flask equipped with a rubber septum and argon inlet needle, and 6.3 mL of CH2Cl2 and Et3N (0.138 g, 0.190 mL, 1.36 mmol, 2.2 equiv) were added. The resulting brown solution was cooled at 0 °C while acetyl chloride (0.061 g, 0.055 mL, 0.77 mmol, 1.2 equiv) was added in one portion. The resulting solution was stirred at room temperature for 2.5 h and then diluted with 10 mL of CH2Cl2 and washed with 10 mL of 1 M HCl solution. The aqueous layer was extracted with 10 mL of CH2Cl2 and the combined organic phases were washed with 10 mL of satd aq NaHCO3 solution, 10 mL of H2O, and 10 mL of brine, dried over MgSO4, filtered, and concentrated to provide 0.240 g of a brown foam. This material was dissolved in ca. 10 mL of CH2Cl2 and concentrated onto 2.4 g of silica gel. The free flowing powder was added to the top of a column of 35 g of silica gel and eluted with 15% EtOAc-hexanes to give 0.115 g (57% over two steps) of acetate 49 as a pale yellow solid: mp 112-115 °C; IR (KBr pellet) 3348, 3102, 3031, 2935, 2855, 1752, 1636, 1563, 1509, 1478, 1453, 1437, 1367, and 1203 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.24-7.35 (m, 3H), 7.10-7.15 (m, 2H), 7.01 (d, J = 3.2 Hz, 1H), 6.93 (s, 1H), 6.32 (dd, J = 3.2, 0.7 Hz, 1H), 5.26 (s, 2H), 2.89 (t, J = 5.6 Hz, 2H), 2.70 (t, J = 6.0 Hz, 2H), 2.42 (s, 3H), 1.75-1.86 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 169.2, 141.1, 137.6, 136.5, 132.6, 128.8, 128.2, 127.6, 126.9, 120.5, 120.0, 107.6, 97.8, 50.1, 30.5, 23.3, 23.3, 23.1, and 20.9; Anal. Calcd for C21H21NO2: C, 78.97; H, 6.63; N, 4.39. Found: C, 78.75; H, 6.46; N, 4.34.
In a separate experiment, an impure sample of hydroxy indole S1 was isolated and observed to have the following spectroscopic properties: IR (film) 3425, 2926, 2855, 1633, 1568, 1510, 1495, 1453, 1360, 1296, 1240, and 1218 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.23-7.34 (m, 3H), 7.12 (app d, J = 6.8 Hz, 2H), 6.98 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 6.50 (d, J = 3.2 Hz, 1H), 5.26 (s, 2H), 4.91 (s, 1H), 2.87 (t, J = 6.2 Hz, 2H), 2.78 (t, J = 6.5 Hz, 2H), 1.86-1.91 (m, 2H), and 1.77-1.82 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 146.0, 137.9, 136.7, 133.1, 128.8, 127.6, 126.9, 126.8, 116.4, 112.2, 102.2, 97.2, 50.1, 30.8, 23.5, and 22.7.
N-Benzyl-5,6-dihydro-1H-cyclobuta[f]indol-4-yl Trifluoromethanesulfonate (52)
A 20-cm quartz reaction tube (13 mm i.d., 15 mm o.d.) fitted with a rubber septum and an argon inlet needle was charged with ynamide 45 (0.351 g, 0.966 mmol, 1.0 equiv) and flushed with argon. Dichloromethane (3 mL) was then added and the reaction tube was positioned ca. 15 cm from a Hanovia 450W medium pressure mercury lamp (quartz immersion well, cooled by tap water). A solution of the diazo ketone 502b (0.287 g, 2.35 mmol, 2.4 equiv) in 7 mL of CH2Cl2 was added to the vigorously stirred reaction mixture via a syringe wrapped in aluminum foil over 3.5 h. Irradiation was continued for 6.5 h and then the reaction mixture was concentrated to give 0.7 g of a dark red-orange oil. A solution of this oil in 15 mL of toluene was transferred to a flask fitted with a cold-finger condenser with argon inlet side arm and heated at reflux for 2 h. Concentration gave 0.68 g of a red-brown oil. Column chromatography on 15 g of silica gel (elution with 15% EtOAc–hexanes) afforded 0.262 g (59%) of phenol 51 as a yellow oil: IR (film) 3583, 3297, 2952, 2832, 1698, 1671, 1594, 1431, 1251, 1116, 1073, 859, 838 cm−1; 1H NMR (400 MHz, CDCl3): 8.08 (s, 1H), 6.37 (s, minor rotamer), 6.28 (s, 1H), 4.14–4.84 (m, 5H), 3.29-3.37 (m, 6H), 3.03–3.11 (m, 4H), 2.60 (d, 2H), 0.89-1.60 (m, 2H), 0.08 (s, minor rotamer), −0.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.5, 150.6, 145.8, 140.9, 137.5, 130.7, 129.4, 128.9, 128.4, 127.6, 120.4, 116.1, 105.7, 64.2, 55.1, 54.8, 53.5, 34.7, 30.8, 29.0, 27.2, 18.0, −1.56.
A 25-mL recovery flask fitted with a rubber septum and an argon inlet needle was charged with a solution of phenol 51 (0.170 g, 0.372 mmol, 1.0 equiv) in 2 mL of THF. The solution was cooled at 0 °C while TBAF solution (1 M in THF, 2 mL, 2 mmol, 5 equiv) was added via syringe over 1 min. The resulting brown solution was stirred at rt for 24 h, and then cooled to 0 °C while 6 M HCl solution (2 mL, 12 mmol, 32 equiv)47 was added dropwise over ca. 2 min. The resulting solution was stirred at 0 °C for 5 min, and then heated at reflux for 1 h. The resulting blue-brown mixture was allowed to cool to rt, diluted with 20 mL of Et2O, and washed with 20 mL of satd aq NaHCO3 solution. The combined aqueous layers were extracted with Et2O, and the combined organic phases were washed with brine, dried over MgSO4 containing a small amount of decolorizing carbon, filtered, and concentrated to give 0.191 g of hydroxy indole S2 as a brown oil used in the next step without purification: 1H NMR (400 MHz, CDCl3) δ 7.31–7.24 (m, 3H), 7.09–7.11 (m, 2H), 6.98 (d, J = 3.2 Hz, 1H), 6.64 (m, 1H), 6.61 (s, 1H), 5.26 (s, 2H), 3.54 (s, 1H) 3.11–3.15 (m, 4H).
The hydroxy indole prepared in the previous step was transferred into a 25-mL recovery flask equipped with a rubber septum and an argon inlet needle, dissolved in 3 mL of THF, and cooled to 0 °C. Sodium hydride (60% wt oil disp., 0.025 g, 0.63 mmol, 1.7 equiv) was added and the reaction mixture was stirred for 2 min until no more gas evolved. A solution of N-phenyltriflimide (0.210 g, 0.588 mmol, 1.6 equiv) in 2 mL of THF was added, and the resulting mixture was stirred at rt for 1 h. Satd aq NaHCO3 (3 mL), was added, and the resulting mixture was extracted with three 20 mL-portions of Et2O. The combined organic phases were washed with two 20-mL portions of 10% NaOH solution and 20 mL of brine, dried over MgSO4, filtered, and concentrated to give 0.225 g of a brown oil. Column chromatography on 5 g of silica gel (elution with 10% EtOAc–hexanes) gave 0.130 g of an oily white solid. Column chromatography on 5 g of silica gel (elution with 5% EtOAc–hexanes) gave 0.093 g of colorless oil. The oil was taken up in 4 mL of pentane, and the solution was cooled to −20 °C. The resulting white crystals were washed with cold pentane and dried under vacuum. A second crop was collected by concentrating the mother liquor and the washes to a volume of ca. 0.5 mL and cooling to −20 °C. Crystallization provided 0.089 g (63% from 45) of indole 52 as a white solid: mp 77–78 ºC; IR (KBr pellet) 2926, 1497, 1421, 1250, 1200, 1187, 998, 916, 839, 727, 617 and 603 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.31–7.34 (m, 3H), 7.14 (d, J = 3.2 Hz, 1H), 7.07-7.13 (m, 2 H), 7.00 (s, 1H), 6.62 (d, J = 3.2 Hz, 1H), 5.31 (s, 2H), 3.18–3.36 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 140.5, 138.6, 137.0, 135.4, 129.1, 128.3, 128.0, 126.9, 126.0, 120.6, 119.0 (q, J =320 Hz, CF3), 105.3, 98.4, 50.9, 28.5, 27.1; Anal. calcd for C21H33NO4Si: C, 64.41; H, 8.49; N, 3.58. Found: C, 64.50; H, 8.36; N, 3.53.
N-Benzyl-1,2,3,10-tetrahydronaphtho[1,8-fg]indol-7-yl Trifluoromethanesulfonate (56)
A 25-mL recovery flask was charged with carbamate 55 (0.189 g, 0.289 mmol, 1 equiv) and 5 mL of CH2Cl2. Trifluoromethanesulfonic acid (0.225 mL, 2.88 mmol, 10 equiv) was added via syringe in one portion producing a dark red-brown solution. The reaction mixture was stirred vigorously at rt for 5 min and then 2 mL of water was added. This biphasic reaction mixture turned light yellow within 30 sec. Satd aq NaHCO3 (5 mL) was then added,48 and the reaction mixture was diluted with 20 mL CH2Cl2 and separated. The organic phase was washed with 10 mL of water and brine, dried over MgSO4, filtered, and concentrated to give 0.117 g of a tan solid. Column chromatography on 10 g of silica gel (elution with 20% benzene–hexanes) followed by a column chromatography on 6 g of silica gel (elution with 20% benzene–hexanes) furnished 0.098 g (76%) of indole 56 as a white powder: mp 112–113 °C (dec); IR (film) 3055, 2987, 1422, 1266,737 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 8.7 Hz, 1H), 7.36–7.40 (m, 1H), 7.25–7.35 (m, 4H), 7.15 (dd, J = 10.0, 3.9 Hz, 1H), 6.96 (d, J = 7.3 Hz, 2H), 6.82 (d, J = 3.4 Hz, 1H), 5.69 (s, 2H), 3.41 (t, J = 6.2 Hz, 2H), 3.06 (t, J = 6.2 Hz, 2H), 2.02–1.99 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 139.0, 136.6, 136.1, 135.0, 134.4, 129.2, 127.8, 127.2, 125.6, 124.5, 123.9, 122.6, 121.7, 119.1 (q, J = 310 Hz), 118.6, 118.3, 98.1, 53.2, 31.4, 26.9, 22.7; HRMS (ESI) [M + H]+ calcd for C23H19F3NO3S: 446.1032, found 446.1020.
N-Benzyl-7-ethyl-1,2,3,10-tetrahydronaphtho[1,8-fg]indole (57)
A 25-mL pear flask was charged with Pd(OAc)2 (2.5 mg, 0.011 mmol, 5 mol %), SPhos (6.6 mg, 0.016 mmol, 7.5 mol %), and Cs2CO3 (0.208 g, 0.638 mmol, 3 equiv). The flask was equipped with a rubber septum fitted with an argon inlet needle and flushed with argon. A solution of triflate 56 (0.095 g, 0.213 mmol, 1 equiv) in 4.5 mL of THF and a solution of Et3B (1 M in THF, 0.533 mL, 0.533 mmol, 2.5 equiv) were added. The orange-yellow reaction mixture was stirred at rt for 1 h, diluted with 15 mL of Et2O, and filtered through 5 g of silica gel with the aid of two 20-mL portions of Et2O. The resulting yellow solution was concentrated to give 0.061 g of an off-white solid. This material was dissolved in 50 mL of hot pentane and crystallized at −78 °C to give 0.055 g (79%) of indole 57 as an off-white powder: mp 114–115 °C; IR (film) 3054, 2936, 1453, 1386, 1355, 1265, 739 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 9 Hz, 1H), 7.27–7.34 (m, 5H), 7.13 (d, J = 3.6 Hz, 1H), 7.01 (d, J = 7.2 Hz, 2H), 6.81 (d, J = 3.0 Hz, 1H), 5.70 (s, 2H), 3.45–3.48 (m, 4H), 3.11 (t, J = 6 Hz, 2H), 2.02–2.06 (m, 2H), 1.49 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 140.0, 136.3, 134.5, 134.1, 129.9, 129.3, 129.0, 127.5, 127.4, 126.2, 125.8, 122.2, 114.8, 100.1, 53.2, 32.1, 27.2, 23.1, 22.9, 15.5; HRMS (ESI) [M + H]+ calcd for C24H24N: 326.1903, found 326.1900.
Methyl 2-(2-[(tert-Butyldimethylsilyl)oxy]ethyl)-3-hydroxy-4,5-dimethylphenyl(2-oxoethyl)carbamate (62)
A 50-mL recovery flask fitted with a rubber septum and argon inlet needle was charged with alkene 61 (0.325 g, 0.826 mmol, 1.0 equiv), 6 mL of THF, 2 mL of H2O, and OsO4 (4 wt% in H2O, 0.11 mL, 0.11 g, 0.018 mmol, 0.02 equiv). The solution was stirred at rt for 10 min and then NMO (0.130 g, 1.11 mmol, 1.3 equiv) was added. The resulting cloudy tan mixture was stirred at rt for 24 h. Aqueous NaHSO3 solution (1.0 M, 9.0 mL, 9.0 mmol, 11 equiv) was added, and the reaction mixture was stirred at rt for 10 min, diluted with 15 mL of brine, and extracted with three 20-mL portions of EtOAc. The combined organic phases were dried over MgSO4, filtered, and concentrated to give 0.401 g of diol S3 as a pale yellow oil used in the next step without purification: IR (neat) 3281, 2955, 2931, 2884, 2860, 1685, 1619, 1573, 1455, 1391, 1321, 1260, 1197, 1164, 1092, 1039, 900, 839, 781, 732 cm−1; 1H NMR (400 MHz, CDCl3), (ca. 50:50 mixture of rotamers) δ 8.69 and 8.60 (s, 1H), 6.60 and 6.42 (s, 1H), 3.81–3.73 (m, 2H), 3.73–3.59 (m, 6H), 2.94–2.70 (m, 3H), 2.22 (d, J = 8.9 Hz, 3H), 2.17 (s, 3H), 0.92 (s, 9H), 0.10–0.08 (m, 6H).
A 25-mL recovery flask was charged with NaIO4 supported on silica gel (0.68 mmol/g, 2.5 g, 1.7 mmol, 2.0 equiv) and 5 mL of CH2Cl2. A solution of the diol prepared above in 5 mL of CH2Cl2 was added to the stirring suspension via pipette (0.5 mL CH2Cl2 rinse). The reaction mixture was stirred at rt for 0.5 h and then filtered through a sintered glass funnel with the aid of three 3-mL portions of CH2Cl2. Concentration of the filtrate gave 0.316 g (97%) of aldehyde 62 as a pale yellow oil: IR (neat) 3259, 2955, 2931, 2885, 2859, 1706, 1619, 1574, 1451, 1387, 1331, 1260, 1136, 1093, 1053, 1038, 899, 839, 782 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.71 (s, 1H), 8.65 (s, minor rotamer), 8.59 (s, 1H), 6.61 (s, 1H), 4.43 (d, J = 18 Hz, 1H), 4.34 (d, J = 18, minor rotamer), 4.03 (d, J = 18 Hz, 1H), 3.87 (m, 2H), 3.75 (s, minor rotamer), 3.68 (s, 3H), 2.88 (m, 2H), 2.23 (s, 3H), 2.17 (s, 3H), 0.92 (s, 9H), 0.09 (d, J = 6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 197.3, 156.9, 154.9, 137.8, 137.2, 125.3, 122.2, 120.6, 65.2, 61.1, 53.5, 29.3, 25.9, 20.1, 18.4, 12.2, −5.5; additional resonance appeared for the minor rotamer at δ 120.4; HRMS (ESI) [M + H]+ calcd for C20H34NO5Si: 396.2201, found 396.2216.
4,5-Dimethyl-6-hydroxy-7-(2-hydroxyethyl)-1-methoxycarbonylindole (63)
A 50-mL recovery flask equipped with a cold finger condenser with an argon inlet sidearm was charged with aldehyde 62 (0.316 g, 0.800 mmol, 1.0 equiv), 40 mL of isopropanol, and K2CO3 (0.11 g, 0.80 mmol, 1.0 equiv). The pale yellow reaction mixture was heated at 75 °C for 2.5 h. The oil bath was removed, and the warm mixture was diluted with 15 mL of H2O and then treated with 5 mL of aq 1 M HCl to adjust the pH to 1. The resulting mixture was allowed to stir at rt for 0.5 h and then concentrated to a volume of ca. 10 mL. The resulting heterogeneous mixture was diluted with 200 mL of Et2O and washed with two 40-mL portions of H2O and 40 mL of brine. The aqueous layers were extracted with two 20-mL portions of Et2O, and the combined organic phases were dried over MgSO4, filtered, and concentrated to afford 0.209 g of a brown oil. Column chromatography on 7 g of silica gel (elution with 50% EtOAc–hexanes) afforded 0.130 g (62%) of indole 63 as a white solid: mp 116–117 °C; IR (KBr pellet) 3472, 3311, 3153, 3113, 2994, 2952, 2891, 1719, 1560, 1450, 1351, 1294, 1252, 1156, 1041, 930 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 7.42 (d, J = 4.0 Hz, 1H), 6.56 (d, J = 4.0 Hz, 1H), 4.36 (m, 2H), 3.93 (s, 3H), 3.13 (t, J = 4.8 Hz, 2H), 2.43 (br s, 1H), 2.41 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.0, 152.4, 133.7, 127.8, 125.6, 125.4, 121.5, 111.9, 107.4, 66.3, 54.0, 31.1, 15.8, 12.6; HRMS (ESI) [M + H]+ calcd for C14H18NO4: 264.1230, found 264.1231.
Methyl 2-Oxoethyl[4-(2-[(tert-butyldimethylsilyl)oxy]ethyl)-3-hydroxybenzocyclobutene]-5-carbamate (65)
A 100-mL recovery flask fitted with a rubber septum and argon inlet needle was charged with alkene 64 (1.22 g, 3.12 mmol, 1.0 equiv), 22.5 mL of THF, 7.5 mL of H2O, OsO4 (4 wt% in H2O, 0.5 mL, 0.08 mmol, 0.03 equiv), and NMO (0.552 g, 4.71 mmol, 1.5 equiv). The resulting yellow mixture was stirred at rt for 26 h. Aqueous NaHSO3 solution (2.0 M, 9.0 mL, 9.0 mmol, 9 equiv) was added, and the reaction mixture was stirred at rt for 10 min. The resulting mixture was transferred to a separatory funnel containing 60 mL of brine and extracted with three 40-mL portions of Et2O. The combined organic phases were dried over MgSO4, filtered, and concentrated to give 1.4 g of diol S4 as an off-white foam used in the next step without purification: IR (neat) 3345, 2939, 2858, 1680, 1595, 1455, 1391, 1254, 1084, 914, 836 and 777 cm−1; 1H NMR (400 MHz, CDCl3), (ca. 50:50 mixture of rotamers): δ 8.75 (s, 1H), 8.63 (s, minor rotamer), 6.55 (s, 1H), 6.38 (s, minor rotamer), 3.55–3.91 (m, 9H), 3.35–3.50 (m, 3H), 3.05–3.15 (m, 4H), 2.61–2.85 (m, 2H), 0.90 (s, 9H), 0.09 (m, 6H).
A 100-mL recovery flask was charged with NaIO4 supported on silica gel (0.68 mmol/g, 10.6 g, 7.2 mmol, 2.3 equiv) and 20 mL of CH2Cl2. A solution of the diol prepared above in 15 mL of CH2Cl2 was added to the stirring suspension via pipette (two 5 mL CH2Cl2 rinses). The reaction mixture was stirred at rt for 20 min and then filtered through 1 g of silica gel with the aid of CH2Cl2. Concentration of the filtrate gave 1.08 g (88%) of aldehyde 65 as a white powder: mp 134–135 °C; IR (KBr pellet) 3275, 3055, 2932, 1737, 1703, 1592, 1451, 1382, 1265, 1057, 914, 839 and 738 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1H), 8.71 (s, minor rotamer), 8.65 (s, 1H), 6.58 (s, 1H), 3.75–4.51 (m, 4H), 3.68 (s, 3H), 3.35 (s, minor rotamer), 3.05–3.14 (m, 4H), 2.87–2.90 (m, 2H), 0.92 (s, 9H), 0.10 (d, J = 2.8 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 197.1, 156.9, 151.2, 146.2, 140.5, 131.4, 124.0, 115.1, 65.2, 61.2, 53.5, 29.6, 29.1, 27.4, 26.0, 18.5, −5.45; Anal. calcd for C20H31NO5Si: C, 61.04; H, 7.94; N, 3.56. Found: C, 60.77; H, 7.96; N, 3.52.
N-Carbomethoxy-4-(2-[(tert-butyldimethylsilyl)oxy]ethyl)-5-hydroxy-6,7-dihydro-3H-cyclobuta[e]indole (66)
A 300-mL round-bottom flask was charged with aldehyde 65 (0.735 g, 1.87 mmol, 1.0 equiv), 100 mL of isopropanol, and K2CO3 (0.260 g, 1.89 mmol, 1.0 equiv). The reaction flask was equipped with a reflux condenser fitted with an argon inlet adapter, and the mixture was heated at 80 °C for 10 h. The resulting black mixture was allowed to cool to rt, neutralized with 3.3 mL of aq 0.6 M HCl, and concentrated to a volume of ca. 20 mL. This mixture was extracted with three 50 mL-portions of Et2O, and the combined organic phases were washed with brine, dried over MgSO4, and concentrated to afford 0.82 g of a brown oil. This oil was taken up in 200 mL of hexanes and filtered, and the filtrate was concentrated to give 0.577 g of a light brown oil. Column chromatography on 25 g of silica gel (elution with 5% EtOAc–hexanes) afforded 0.337 g (48%) of indole 66 as a white powder: mp 90–91 °C; IR (KBr pellet) 3248, 2928, 2857, 1751, 1608, 1548, 1465, 1440, 1371, 1336, 1273, 1256, 1174, 1145, 1041, 901, 786 and 719 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H), 7.45 (d, J = 4.0 Hz, 1H), 6.40 (d, J = 4.0 Hz, 1H), 4.30 (m, 2H), 3.94 (s, 3H), 3.24–3.30 (m, 4H), 3.08 (t, J = 4.4 Hz, 2H), 0.98 (s, 9H), 0.18 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 152.4, 149.2, 136.9, 136.7, 128.0, 127.1, 121.4, 114.2, 105.6, 67.2, 54.0, 32.3, 28.6, 28.0, 26.1, 18.5, −5.31; HRMS (ESI) [M + H]+ calcd for C14H18NO4: 264.1230, found 264.1231; Anal. calcd for C20H29NO4Si: C, 63.97; H, 7.78; N, 3.73. Found: C, 64.20; H, 7.50; N, 3.46.
Methyl 4-(2-[(tert-Butyldimethylsilyl)oxy]ethyl)-5-hydroxy-7,9-dioxo-8-phenyl-6a,7,8,9,9a,10-hexahydroisoindolo[5,6-e]indole-3(6H)-carboxylate (67)
A 10-cm threaded Pyrex tube (14 mm i.d., 25 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with N-phenylmaleimide (0.183 g, 1.06 mmol, 3.6 equiv) and flushed with argon. A solution of indole 66 (0.110 g, 0.293 mmol, 1 equiv) in 12 mL of p-xylene was added, the septum was replaced with a threaded Teflon cap, and reaction mixture was heated at 180 °C (bath temperature) for 40 h. The reaction mixture was allowed to cool to rt and concentrated to give 0.376 g of a dark orange oil. Column chromatography on 15 g of silica gel (elution with 20% EtOAc–hexanes) gave 0.070 g (44%) of indole 67 as a white powder: mp 172–174 °C; IR (KBr pellet) 3449, 2926, 1625, 1569, 1497, 1455, 1438, 1380, 1320, 1287, 1250, 1220, 1140, 998, 916, 840 and 727 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.24 (s, 1H), 7.19–7.50 (m, 4H), 6.97 (d, J = 7.2 Hz, 2H), 6.59 (d, J = 3.6 Hz, 1H), 4.227–4.28 (m, 2H), 3.94 (s, 3H), 2.85–3.80 (m, 8H), 0.92 (s, 9H), 0.11 (s, 3H), 0.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 179.0, 178.8, 152.22, 152.17, 134.6, 132.1, 129.4, 129.1, 128.5, 126.7, 126.5, 123.8, 119.8, 113.6, 106.2, 67.2, 54.1, 40.29, 40.26, 31.8, 26.2, 26.0, 22.7, 18.4, −5.42, −5.46; HRMS (ESI) [M + H]+ calcd for C30H37N2O6Si: 549.2415, found 549.2419.
7-Ethyl 3-Methyl 4-(2-[(tert-Butyldimethylsilyl)oxy]ethyl)-5-hydroxy-3H-benzo[e]indole-3,7(6H,9H)-dicarboxylate (69) and 8-Ethyl 3-Methyl 4-(2-[(tert-Butyldimethylsilyl)oxy]ethyl)-5-hydroxy-3H-benzo[e]indole-3,8(6H,9H)-dicarboxylate (70)
A 10-cm threaded Pyrex tube (14 mm i.d., 25 mm o.d.) equipped with a rubber septum and argon inlet needle was charged with a solution of indole 66 (0.109 g, 0.290 mmol, 1 equiv) in 10 mL of p-xylene followed by ethyl propiolate (0.108 mL, 1.07 mmol, 3.7 equiv). The septum was replaced with a threaded Teflon cap and the reaction mixture was heated at 180 °C (bath temperature) for 40 h. The reaction mixture was allowed to cool to rt and concentrated to give 0.159 g of an orange solid. Column chromatography on 15 g of silica gel (elution with 10% EtOAc–hexanes) gave 0.085 g (62%) of an inseparable ca. 60:40 mixture of isomers 69 and 70 as a white powder: IR (KBr pellet) 3250, 2956, 2931, 2884, 2858, 1750, 1714, 1667, 1440, 1337, 1292, 1257, 1230, 1144, 1038, 918, 838, 781 and 719 cm−1; 1H NMR (600 MHz, CDCl3), (ca. 60:40 mixture of regioisomers) for major isomer: δ 9.24 (s, 1H), 7.53 (d, J = 2.8 Hz, 1H), 7.34–7.35 (m, 1H), 6.67 (d, J = 2.4 Hz, 1H), 4.35–4.39 (m, 4H), 4.02 (s, 3H), 3.72–3.82 (m, 2H), 3.67–3.71(m, 2H), 3.22–3.24 (m, 2H), 1.43–1.45 (m, 3H), 1.03 (s, 9H), 0.21 (s, 6H); for the minor isomer: δ 9,23 (s, 1H), 7.52 (d, J = 2.4 Hz, 1H), 7.24–7.35 (m, 1H), 6.56 (d, J = 2.4 Hz, 1H), 4.35–4.39 (m, 4H), 4.02 (s, 3H), 3.72–3.82 (m, 2H), 3.67–3.71(m, 2H), 3.22–3.24 (m, 2H), 1.43–1.45 (m, 3H), 1.0 (s, 9H), 0.21 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 153.0, 152.6, 152.2, 137.2, 134.9, 134.1, 129.0, 127.0, 125.8, 124.7, 123.8, 123.3, 123.1, 119.1, 117.6, 112.5, 112.4, 106.5, 106.3, 67.3, 60.7, 60.6, 54.1, 31.7, 31.7, 28.4, 26.7, 26.3, 26.1, 26.0, 24.4, 18.5, 18.4, 14.6, −5.3, −5.4; HRMS (ESI) [M + H]+ calcd for C25H36NO6Si: 474.2306, found 474.2312.
Supplementary Material
Acknowledgment
We thank the National Science Foundation (CHE-1111567), the National Institutes of Health (GM 28273), Merck Research Laboratories, and Boehringer Ingelheim Pharmaceuticals for generous financial support. T. P. W. was supported in part by an AstraZeneca Graduate Fellowship and David A. Johnson, Merck, and George Büchi Summer Fellowships. T. Y. L. was supported in part by a fellowship from the C. P. Chu and Y. Lai Fund. Y.-P. Wang was supported in part by an Eli Lilly Graduate Fellowship.
Supporting Information Available: 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kotha S, Misra S, Halder S. Tetrahedron. 2008;64:10775–10790. Reviews: [Google Scholar]; (b) Saito S, Yamamoto Y. Chem. Rev. 2000;100:2901–2915. doi: 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]
- 2.(a) Danheiser RL, Gee SK. J. Org. Chem. 1984;49:1672–1674. [Google Scholar]; (b) Danheiser RL, Brisbois RG, Kowalczyk JJ, Miller RF. J. Am. Chem. Soc. 1990;112:3093–3100. [Google Scholar]
- 3.(a) Danheiser RL, Dudley GB, Austin WF. Alkenylketenes. In: Danheiser RL, editor. Science of Synthesis. Vol. 23. Thieme; Stuttgart: 2006. pp. 493–568. For reviews of the chemistry of vinylketenes, see. [Google Scholar]; (b) Tidwell TT. Ketenes, Second John Wiley & Sons; Hoboken, NJ: 2006. pp. 206–214. [Google Scholar]
- 4.(a) Liebeskind LS, Iyer S, Jewell CF. J. Org. Chem. 1986;51:3065–3067. A related, complementary strategy has been reported by Liebeskind and Moore. See ref. 3a and. [Google Scholar]; (b) Perri ST, Foland LD, Decker OHH, Moore HW. J. Org. Chem. 1986;51:3067–3068. [Google Scholar]
- 5.Dudley GB, Takaki KS, Cha DD, Danheiser RL. Org. Lett. 2000;2:3407–3410. doi: 10.1021/ol006561c. For examples, see. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kowalski CJ, Lal GS. J. Am. Chem. Soc. 1988;110:3693–3695. For additional examples of the application of this benzannulation strategy in natural product synthesis, see. [Google Scholar]; (b) Smith AB, III, Adams CM, Kozmin SA, Paone DV. J. Am. Chem. Soc. 2001;123:5925–5937. doi: 10.1021/ja0106164. [DOI] [PubMed] [Google Scholar]
- 7.Mak XY, Crombie AL, Danheiser RL. J. Org. Chem. 2011;76:1852–1873. doi: 10.1021/jo2000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam TY, Wang Y-P, Danheiser RL. J. Org. Chem. 2013;78:9396–9414. doi: 10.1021/jo401635c. (DOI: 10.1021/jo401635c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Wakamatsu T, Miyachi N, Ozaki F, Ban Y. Heterocycles. 1987;26:1445–1448. For example, see. [Google Scholar]; (b) Curini M, Epifano F, Marcotullio MC, Rosati O, Guo M, Guan Y, Wenkert E. Helv. Chim. Acta. 2005;88:330–338. [Google Scholar]
- 10.(a) Kirmse W. Eur. J. Org. Chem. 2002:2193–2256. For reviews on the Wolff rearrangement, see: [Google Scholar]; (b) Meier H, Zeller K-P. Angew. Chem. Int. Ed. 1975;14:32–43. [Google Scholar]
- 11. For the purpose of this paper, we refer to N-sulfonyl and N-phosphoryl ynamines as ynamides.
- 12.(a) Evano G, Jouvin K, Coste A. Synthesis. 2013;45:17–26. For recent reviews on the synthesis and chemistry of ynamides, see: [Google Scholar]; (b) DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem. Rev , 110. 2010:5064–5106. doi: 10.1021/cr100003s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Evano G, Coste A, Jouvin K. Angew. Chem. Int. Ed. 2010;49:2840–2859. doi: 10.1002/anie.200905817. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dunetz JR, Danheiser RL. Org. Lett. 2003;5:4011–4014. doi: 10.1021/ol035647d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohnen AL, Dunetz JR, Danheiser RL. Org. Synth. 2007;84:88–101. [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas J. Am. Chem. Soc. 2003;125:2368–2369. doi: 10.1021/ja021304j. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org. Lett. 2004;6:1151–1154. doi: 10.1021/ol049827e. [DOI] [PubMed] [Google Scholar]; (c) Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Shen L, Tracey MR. J. Org. Chem. 2006;71:4170–4177. doi: 10.1021/jo060230h. [DOI] [PubMed] [Google Scholar]; (d) Sagamanova IK, Kurtz KCM, Hsung RP. Org. Synth. 2007;84:359–367. [Google Scholar]; (e) DeKorver KA, Walton MC, North TD, Hsung RP. Org. Lett. 2011;13:4862–4865. doi: 10.1021/ol201947b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofmeister H, Annen K, Laurent H, Wiechert R. Angew. Chem. Int. Ed. 1984;23:727–729. [Google Scholar]
- 16.(a) Hamada T, Ye X, Stahl SS. J. Am. Chem. Soc. 2008;130:833–835. doi: 10.1021/ja077406x. For other useful coupling routes to ynamides, see. [DOI] [PubMed] [Google Scholar]; (b) Jouvin K, Couty F, Evano G. Org. Lett. 2010;12:3272–3275. doi: 10.1021/ol101322k. [DOI] [PubMed] [Google Scholar]; (c) Jouvin K, Coste A, Bayle A, Legrand F, Karthikeyan G, Tadiparthi K, Evano G. Organometallics. 2012;31:7933–7947. [Google Scholar]; (d) Jouvin K, Heimburger J, Evano G. Chem. Sci. 2012;3:756–760. [Google Scholar]
- 17.(a) Maas G. Angew. Chem. Int. Ed. 2009;48:8186–8195. doi: 10.1002/anie.200902785. For reviews on the synthesis and chemistry of diazo ketones, see: [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Wang J. Chem. Commun. 2009:5350–5361. doi: 10.1039/b908378b. [DOI] [PubMed] [Google Scholar]; (c) Zhang Y, Wang J. Tetrahedron. 2008;64:6577–6605. [Google Scholar]; (d) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis of Diazo Compounds: from Cyclopropanes to Ylides. Wiley & Sons; New York: 1998. Synthesis of α- Diazocarbonyl Compounds; pp. 1–60. [Google Scholar]; (e) Regitz M, Maas G. Diazo Compounds: Properties and Synthesis; Academic Press; Orlando, Fl: 1986. [Google Scholar]
- 18.(a) Danheiser RL, Miller RF, Brisbois RG, Park SZ. J. Org. Chem. 1990;55:1959–1964. [Google Scholar]; (b) Danheiser RL, Miller RF, Brisbois RG. Org. Synth. 1996;73:134–140. [Google Scholar]
- 19.Chataigner I, Panel C, Gérard H, Piettre SR. Chem. Commun. 2007:3288–3290. doi: 10.1039/b705034h. [DOI] [PubMed] [Google Scholar]
- 20.(a) Witulski B, Schweikert T, Schollmeyer D, Nemkovich NA. Chem. Commun. 2010;46:2953–2955. doi: 10.1039/b919275a. For examples involving the reactivity of ynamide derivatives, see. [DOI] [PubMed] [Google Scholar]; (b) Li H, Hsung RP, DeKorver KA, Wei Y. Org. Lett. 2010;12:3780–3783. doi: 10.1021/ol101418d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. For details, see Experimental Section.
- 22.(a) Weigmann RH, Würthwein E-U. Tetrahedron Lett. 1989;30:6147–6150. [Google Scholar]; (b) Schulte N, Möller MH, Rodewald U, Würthwein E-U. Chem. Ber. 1994;127:1287–1293. [Google Scholar]
- 23. The moisture sensitivity of N,N-bis(trimethylsilyl) ynamines also limits their utility as benzannulation partners. To date our attempts to prepare more stable bis-silyl ynamines have not been fruitful.
- 24.(a) Knowles JP, Elliott LD, Booker-Milburn KI. Beilstein J. Org. Chem. 2012;8:2025–2052. doi: 10.3762/bjoc.8.229. For reviews of continuous flow photochemical reactions, see. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Oelgemöller M, Shvydkiv O. Molecules. 2011;16:7522–7550. doi: 10.3390/molecules16097522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Nguyen JD, Reiss B, Dai C, Stephenson CRJ. Chem. Commun. 2013;49:4352–4354. doi: 10.1039/c2cc37206a. For recent examples of continuous flow photochemical reactions, see. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y, Blackman ML, Leduc AB, Jamison TF. Angew. Chem. Int. Ed. 2013;52:4251–4255. doi: 10.1002/anie.201300504. [DOI] [PubMed] [Google Scholar]; (c) Maskill KG, Knowles JP, Elliott LD, Alder RW, Booker-Milburn KI. Angew. Chem. Int. Ed. 2013;52:1499–1502. doi: 10.1002/anie.201208892. [DOI] [PubMed] [Google Scholar]; (d) Šterk D, Jukič M, Časar Z. Org. Process. Res. Dev. 2013;17:145–151. [Google Scholar]; (e) Junkers T, Conradi M. J. Photochem. Photobiol., A. 2013;259:41–46. [Google Scholar]; (f) Harrowven DC, Mohamed M, Gonçalves TP, Whitby RJ, Bolien D, Sneddon HF. Angew. Chem. Int. Ed. 2012;51:4405–4408. doi: 10.1002/anie.201200281. [DOI] [PubMed] [Google Scholar]; (g) Lévesque F, Seeberger PH. Angew. Chem. Int. Ed. 2012;51:1706–1709. doi: 10.1002/anie.201107446. [DOI] [PubMed] [Google Scholar]; (h) Anderson BG, Bauta WE, Cantrell WR., Jr. Org. Process. Res. Dev. 2012;16:967–975. [Google Scholar]; (i) Terao K, Nishiyama Y, Tanimoto H, Morimoto T, Oelgemöller M, Kakiuchi K. J. Flow Chem. 2012;2:73–76. [Google Scholar]
- 26.Hook BDA, Dohle W, Hirst PR, Pickworth M, Berry MB, Booker-Milburn KI. J. Org. Chem. 2005;70:7558–7564. doi: 10.1021/jo050705p. [DOI] [PubMed] [Google Scholar]
- 27. For additional details regarding the construction of the reactor and its operation see the experimental section.
- 28.Shintani R, Murakami M, Hayashi T. Org. Lett. 2009;11:457–459. doi: 10.1021/ol802569q. For example, see: [DOI] [PubMed] [Google Scholar]
- 29.Park KK, Lee JL, Ryu J. Tetrahedron. 2003;59:7651–7659. For the photo-Fries rearrangement of sulfonamides, see. and references cited therein. [Google Scholar]
- 30.School M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 31.Jiménez MC, Márquez F, Miranda MA, Tormos R. J. Org. Chem. 1994;59:197. For a discussion on the photocyclization of allylphenols and related compounds, see: and references therein. [Google Scholar]
- 32. Improved yields in the benzannulation were obtained by treating the crude product with KOH in methanol. This serves to hydrolyze any esters that were formed by trapping of the hydroxyl group in the product phenol with ketene intermediates during the reaction.
- 33. In this case reaction with TBAF led to cleavage of the phenolic triflate.
- 34.(a) Mehta G, Kotha S. Tetrahedron. 2001;57:625–659. For reviews on cyclobutarene chemistry, see. [Google Scholar]; (b) Sadana AK, Saini RK, Billups WE. Chem. Rev. 2003;103:1539–1602. doi: 10.1021/cr010022j. [DOI] [PubMed] [Google Scholar]
- 35.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 36.(a) Charlton JL, Alauddin MM. Tetrahedron. 1987;43:2873–2889. Reviewed in. [Google Scholar]; (b) Segura JL, Martin N. Chem. Rev. 1999;99:3199–3246. doi: 10.1021/cr990011e. [DOI] [PubMed] [Google Scholar]
- 37.Zhong Y-L, Shing Y-L, Zhong TKM. J. Org. Chem. 1997;62:2622–2624. doi: 10.1021/jo9621581. [DOI] [PubMed] [Google Scholar]
- 38.(a) Watson SC, Eastham JF. J. Organomet. Chem. 1967;9:165–167. For the titration of n-butyllithium see: [Google Scholar]; (b) Ellison RA, Griffin R, Kotsonis FN. J. Organomet. Chem. 1972;36:209–213. [Google Scholar]; (c) Lin H-S, Paquette L. Synth. Commun. 1994;24:2503–2505. For the titration of i-PrMgBr see: [Google Scholar]
- 39.Roulland E, Monneret C, Florent J-C. J. Org. Chem. 2002;67:4399–4406. doi: 10.1021/jo010481k. [DOI] [PubMed] [Google Scholar]
- 40.He Y, Lin M, Li Z, Liang X, Li G, Antilla JC. Org. Lett. 2011;13:4490–4493. doi: 10.1021/ol2018328. [DOI] [PubMed] [Google Scholar]
- 41.Williams JW, Hurd CD, Hanford WE, Sauer JC. J. Org. Chem. Org. React. 1940;1958;53:122, 108–140. See also. [Google Scholar]
- 42.Kohnen AL, Mak XY, Lam TY, Dunetz JR, Danheiser RL. Tetrahedron. 2006;62:3815–3822. doi: 10.1016/j.tet.2005.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu N, Allen AD, Chan W, Kobayashi S, Tidwell TT, Tahmassebi D, Aguilar A, Cabrera EP, Godoy J. Can. J. Chem. 2008;86:333–341. [Google Scholar]
- 44. The reaction tubes were positioned such as to allow efficient stirring with a magnetic stir motor. This required that the bottoms of the reaction tubes were lower than the lamp’s anode, as opposed to having the solution aligned with the center of the arc.
- 45. The bath temperature rose to 29 °C during the irradiation.
- 46. All solvents and solutions used in this reaction were degassed prior to use by bubbling argon through the solution for 10 min.
- 47. Because of the facile air oxidation of the product indole to the corresponding p-quinone, HCl and all of the workup and chromatography solutions were purged with argon prior to use.
- 48. Quick neutralization is essential once the product indole forms to avoid acid-promoted dimerization/decomposition.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.