

Abstract
Objectives
This study seeks to correlate the interrelated properties of conversion, shrinkage, modulus and stress as dimethacrylate networks transition from rubbery to glassy states during photopolymerization.
Methods
An unfilled BisGMA/TEGDMA resin was photocured for various irradiation intervals (7–600 s) to provide controlled levels of immediate conversion, which was monitored continuously for 10 min. Fiber optic near-infrared spectroscopy permitted coupling of real-time conversion measurement with dynamic polymerization shrinkage (linometer), modulus (dynamic mechanical analyzer) and stress (tensometer) development profiles.
Results
The varied irradiation conditions produced final conversion ranging from 6 % to more than 60 %. Post-irradiation conversion (dark cure) was quite limited when photopolymerization was interrupted either at very low or very high levels of conversion while significant dark cure contributions were possible for photocuring reactions suspended within the post-gel, rubbery regime. Analysis of conversion-based property evolution during and subsequent to photocuring demonstrated that the shrinkage rate increased significantly at about 40 % conversion followed by late-stage suppression in the conversion-dependent shrinkage rate that begins at about 45–50 % conversion. The gradual vitrification process over this conversion range is evident based on the broad but well-defined inflection in the modulus versus conversion data. As limiting conversion is approached, modulus and, to a somewhat lesser extent, stress rise precipitously as a result of vitrification with the stress profile showing little if any late-stage suppression as seen with shrinkage.
Significance
Near the limiting conversion for this model resin, the volumetric polymerization shrinkage rate slows while an exponential rise in modulus promotes the vitrification process that appears to largely dictate stress development.
Keywords: dental materials, dimethacrylate, polymers, shrinkage, stress, modulus, vitrification, dark cure
Introduction
Polymer-based composites have become the most common dental restorative material with a current use rate more than twice that of amalgam filling materials [1]. These resin composites fulfill many of the requirements for clinical restorative applications, including excellent esthetics, practical clinical manipulation steps for chair-side applications, high mechanical properties, low coefficient of thermal expansion and high resistance to softening and wear. However, a major limitation of the resin phase used to construct the dental composite is its volumetric polymerization shrinkage and even more critically, the accompanying stress evolution that occurs during polymerization of bonded restorations for which free shrinkage is constrained [2–4].
The reduction in free volume based on polymerization shrinkage in dental composites is a direct function of the proportion of the resin phase of the composite, and more specifically, depends on the initial reactive group concentration and the degree of conversion attained within the resin phase during polymerization. This shrinkage, when coupled with the clinical requirement for relatively high modulus restorative materials, creates the potential for high polymerization stresses within the composite and at the interface between the composite and tooth substrate, which adds complexity to the bonding protocol. These acute and chronic stresses severely strain the interfacial bond between the composite and the tooth, leading to small gaps that can allow marginal leakage of saliva and microorganisms that potentially lead to the development of marginal staining and recurrent decay [5]. In addition, the stress can exceed the tensile strength of enamel that might be compromised by the cavity preparation procedures with the result of stress cracking and enamel fracture along the interface [5].
Dental resins are typically composed of mixtures of two or more monomers that combine a relatively viscous dimethacrylate base monomer, such as bisphenol A glycidyl methacrylate (BisGMA) or urethane dimethacrylate (UDMA), with a lower-viscosity diluent dimethacrylate comonomer, such as triethylene glycol dimethacrylate (TEGDMA) [6]. During resin photopolymerization, viscosity, modulus and glass transition temperature (Tg), all increase as the proportion of free monomer and partially reacted pendant monomer is consumed as the polymer network evolves [7–9].
With advancing polymerization, several interrelated physical and kinetic landmarks, are passed, including the gel point, auto-acceleration leading to a rate maximum and vitrification that leaves a substantial degree of residual unsaturation in the final glassy polymer. Therefore, there are several distinct stages to the polymerization process as the reaction progresses from a liquid pregel regime to a rubbery gelled phase and finally reaches a glassy state [8]. This final stage of the polymer network development extends over significant time scales due to vitrification and the associated persistence of active free radicals [10], which allows for small degrees of additional chemical-based conversion, but also due to slow network densification that has been referred to as physical aging [11].
Gel point is defined as the appearance of an insoluble polymer fraction and it involves a continuous network structure, regardless of its density, that spans macroscopic specimen dimensions [12]. Several studies have pointed out that shrinkage strain that occurs prior to gelation does not contribute to stress development since this involves viscous but unrestricted flow [13]. There are limited options to delay the gel point conversion [14], which otherwise occurs at a very early stage in typical dimethacrylate bulk polymerizations. As the reaction progresses through the rubbery post-gel regime, modulus continues to increase along with conversion, until the Tg of the developing polymer becomes limited by the effective cure temperature [15]. Unlike the physically well delineated gel point, vitrification, which involves the transition from a rubbery to a glassy polymeric state, is a gradual process that is extended by the breadth of the evolving tan δ peak (the maximum of which represents an averaged Tg) due to the structural heterogeneity that is characteristic of dimethacrylate network formation. Stress development is known to be concentrated in this late stage of conversion due to a significant increase in modulus that is expected to accompany the rubbery to glassy transition [9].
Several prior studies have focused on the conversion-dependent evolution of polymerization shrinkage stress and modulus development during polymerization [16–18], but conversion measurements typically involved separate specimens and even different specimen geometries or curing protocols compared with those used to analyze property development. With the current investigation, efforts are directed to obtain true conversion-indexed property development with an emphasis on the critical vitrification stage of the polymerization process where significant changes in modulus and stress evolution are anticipated. This information may provide improved insights into new methods and materials approaches that can ultimately lower polymerization stress without jeopardizing the degree of conversion or important polymeric performance properties.
Therefore, the aim of the present study is to correlate the interrelated properties of conversion, shrinkage, modulus and stress as a model dimethacrylate network transitions from the rubbery to the glassy state during and to a limited extent, following photopolymerization.
Materials and Methods
A resin composed of bisphenol A glycidyl methacrylate (BisGMA; Esstech, Essington, PA, USA) and triethylene glycol dimethacrylate (TEGDMA; Esstech) in a 70:30 mass ratio was used. A photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (DMPA; Aldrich, Milwaukee, WI, USA), was added at 0.1 wt% to render the resin photo-curable with UV irradiation. The UV source is acknowledged to introduce differences in light transmission and initiator efficiency compared with visible light irradiation, but it does not alter the structure of the polymer formed at a given initiation rate. The rationale for the use of the UV curing was to allow uniform irradiation of the entire specimen in one exposure cycle. In addition, significantly lower irradiance was used compared with conventional dental curing units to allow adequate sampling of the various evolving properties. This difference would be expected to affect the exothermic response during polymerization and the final conversion limits but does not otherwise affect the fundamental property development processes described here.
Degree of conversion and photopolymerization reaction kinetics
Real-time monitoring of the polymerization kinetics based on the methacrylate =CH2 absorption at 6165 cm−1 [19] was carried out using near–infrared (NIR) spectroscopy at 2 scans per spectrum with 4 wavenumber resolution, which provides a greater than 2 Hz data acquisition rate. Regardless of the irradiation interval, kinetic data was collected continuously for 10 min. Samples (n=3) were irradiated for the following exposure times: 10, 15, 20, 30, 60 and 600 s with the filtered 365 ± 10 nm output of a mercury arc lamp (Acticure 4000, EFOS, Mississaguga, Canada) at an incident irradiance of 10 mW/cm2 with a 6 cm distance between the liquid light guide (6 mm output diameter) and the specimen to assure uniform irradiance over at least a 25 mm spot size. Specimens were 10 mm in diameter and 0.8 mm thick laminated between two glass slides. In selected specimens, an embedded mini-thermocouple (120 μm diameter) with a 1 Hz sampling rate (Tecpel DTM-322, Taipei, Taiwan) was used to track temperature change during and following the irradiation. The average conversion (n=2) at which the temperature maximum was observed rise is reported.
Volumetric polymerization shrinkage
Volumetric shrinkage was measured using a linometer (ACTA; Amsterdam, The Netherlands). A standardized volume of the resin was placed onto an aluminum disc and covered with a glass slide through which the UV curing light irradiation at 10 mW/cm2 was applied under the previously described conditions. The space between the aluminum disc and glass slide provided a fixed specimen thickness of 1.25 mm with a diameter of approximately 6 mm. An alignment guide was attached to the linometer to orient two 100 μm optical fibers such that a real-time NIR signal was transmitted along the transverse axis of the sample. The specimens were irradiated for 10, 15, 20, 30, 60 and 600 s. This approach allows simultaneous monitoring of the degree of conversion with the dynamic volumetric shrinkage [12]. Use of a thin layer of grease to minimize substrate adhesion with the polymer permits the real-time measured displacement caused by linear shrinkage to be converted to the corresponding volumetric shrinkage. The dynamic shrinkage data and degree of conversion were recorded during and extended beyond the specified irradiation intervals for a total period of 10 min. Three measurements were carried out for each exposure time.
Dynamic Mechanical Analysis
For the dynamic studies to track modulus development during the later stages of polymerization, a technique is introduced here that allows simultaneous real-time monitoring of conversion and modulus achieved by integration of fiber optic transmission NIR spectroscopy with a photo-accessible dynamic mechanical analyzer (DMA; PerkinElmer 8000, Waltham, MA, USA; Figure 1). Bar-shaped specimens (2×2×25 mm) were prepared by partial photopolymerization to a post-gel stage (approximately 30–40 % conversion) using a bifurcated light guide to achieve simultaneous, uniform irradiance of both the upper and lower surfaces. The specimens (n=3) were subjected to ambient temperature, single cantilever DMA testing (2.5 % strain at 1 Hz) along with the introduction of a secondary irradiation interval (20, 60, 120, 600 s) at an incident UV irradiance of 10 mW/cm2 (again supplied via bifurcated light guides at a 6 cm distance). An additional group of pre-cured specimens (n=3) were subjected to 600 s of 100 mW/cm2 incident irradiance in the same manner. Simultaneous scanning of the storage modulus development and degree of conversion were recorded for 20 min from the start of the second curing cycle. To accurately assign conversion before and during the secondary reaction in the DMA, the NIR aromatic absorbance associated with BisGMA (4620 cm−1) was used as an internal reference [18].
Figure 1.
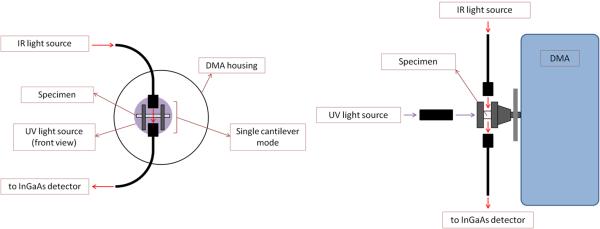
Schematic front (a) and side (b) views of a dynamic mechanical analyzer instrument coupled with fiber optic near-IR spectroscopy and UV light guides for monitoring dynamic modulus development and photopolymerization kinetics simultaneously. Partially prepolymerized bar-shaped specimens are attached to a single cantilever fixture that allowed uniform UV irradiation during testing.
Polymerization shrinkage stress
The shrinkage stress was determined using a tensometer (ADAF-PRC, Gaithersburg, MD). Specimens with dimensions of 1 mm thick × 6 mm diameter were prepared for each exposure time (n=3) and photopolymerized through the quartz rod attached to the bottom of the specimen for the previously mentioned exposure times with appropriate intensity to obtain the same standardized 10 mW/cm2 incident irradiance condition. Along with the dynamic stress development profile, simultaneous real-time conversion data during polymerization were collected via NIR fiber optic cables [20].
Statistical Analysis
Normally distributed data were subjected to one-way ANOVA and Student-Newman-Keuls post-hoc test, and any data that failed the normality test were then subjected to Kruskal-Wallis one-way analysis of variance on rank and Dunn's post-hoc test. All of the dynamic data presented represent averaged results rather than individual specimen profiles.
Results and Discussion
Reaction kinetics
Statistical analysis of the discrete kinetics data revealed that statistically higher final conversion values were achieved as the exposure times were increased (p< 0.001) (Table 1), except for the 20 s and 30 s irradiation times (p=0.083) (Figure 2a). However, there was no statistically significant difference regarding maximum rate of conversion associated with the different irradiation times (p=0.778) (Figure 2b) since all the exposure intervals matched or exceeded that necessary to reach the rate maximum under these curing conditions. The kinetic data demonstrated that the rate maximum consistently occurs at approximately 18 % conversion (Table 1). These results are in agreement with other studies where the rate maxima in photopolymerizations of a variety dental resins and composites were encountered over the conversion range of approximately 10–20 % [6, 21]. The kinetic plot (Figure 2b) for the 60 s irradiation interval shows an inflection at approximately 45 % conversion. This kinetic transition point has been suggested as an indication of the vitrification stage of the polymerization [22] where the reaction continues at a very low rate over an extended time span. While not examined here, macrogelation of this dimethacrylate resin would be anticipated to occur at or below approximately 5 % conversion [14]. A single sample was photocured for only 7 s at the same 10 mW/cm2 irradiance level. In that case, conversion at the end of the irradiation was approximately 2 %, which is well ahead of the expected rate maximum and also presumably close to the gel point. While the reaction rate quickly dropped to near zero when the light was shuttered under this circumstance, the conversion plateau displayed a slight but clearly positive slope ending at a conversion of approximately 6 % within the 10 min observation interval. This indicates that even in this highly mobile early stage of polymerization, persistent radicals are present in the incipient heterogeneous polymer.
Table 1.
Conversion and photopolymenzation reaction kinetics associated with different irradiation intervals
| Irradiation time, s | 10 | 15 | 20 | 30 | 60 | 600 |
| Conversion at light-off, % (SD) | 14.4 (1.0) | 32.1 (3.1) | 40.0 (2.0) | 46.4 (1.3) | 51.3 (1.0) | 62.0 (1.0) |
| Final conversion, % (SD) | 44.2a (0.6) | 48.5b (1.6) | 51.8c (1.5) | 53.6c (1.2) | 56.3d (0.8) | 62.0e (1.0) |
| Dark cure, % of final conversion | 67.3 | 34.0 | 23.0 | 13.4 | 9.0 | - |
| Maximum rate, %/s (SD) | 4.0 (0.1) | 4.4 (0.9) | 4.3 (0.4) | 4.2 (0.2) | 4.0 (0.3) | 4.2 (0.4) |
| Conversion at max rate, % (SD) | 17.0 (0.3) | 18.5 (1.6) | 18.7 (1.4) | 18.6 (0.7) | 18.0 (0.2) | 18.8 (0.8) |
Lower case letters indicate statistical differences within a row (Student-Newman-Keuls test, α=0.5).
SD = standard deviation.
Final values taken at 600 s from the onset of photopolymerization at 10 mW/cm2.
Figure 2.

Polymerization kinetic plots based on near-IR spectroscopic monitoring of a BisGMA/TEGDMA resin photocured at 10mW/cm2 for different irradiation times showing conversion with respect to time (a) and polymerization rate as a function of conversion for selected irradiation times.
The temperature maximum associated with the reaction exotherm for the 10 s irradiation occurred at approximately 26 % conversion whereas the peak temperature rise for the 15 s through 60 s photopolymerizations all occurred at 40.2 ± 2.7 % conversion (individual data not shown) and resulted in a gradual return to near ambient temperature prior to the end of the 10 min observation interval. The similarity of the positioning of the exothermic maximum, which was observed well after the actual reaction rate maximum with respect to conversion, can be attributed to the fact that the partial cure exposures ended at or beyond the polymerization rate maximum.
The amount of post-irradiation conversion (dark cure) after a 10 s irradiation increased from 14.4 ± 0.8 % to 44.2 ± 0.6 %, which accounts for 67 % of the overall conversion compared to the more limited increase from 51.3 ± 1.0 % to 56.3 ± 0.8 % conversion for the dark cure interval that contributed only 9 % of the final conversion following the 60 s irradiation (Table 1). When irradiation is halted, remaining free radicals continue to propagate and terminate although no new initiating radicals are generated. A similar progressive reduction in the dark cure contribution towards overall conversion with increasing irradiation times was also observed in a prior study [20]. Continuous illumination throughout the 10 min observation interval produced the highest conversion and accounted for an approximately three-fold higher reaction rate in the kinetic plateau region compared with the 60 s irradiation exposure. This relatively small difference between an active irradiation and continued dark-cure conversion during the late stages of polymerization demonstrates the effect that vitrification has on limiting the ultimate level of conversion achievable based on given curing conditions. Caution should be used in interpretations drawn from the shortest irradiation intervals used here since due to the relatively rapid changes (even with the low irradiance) in conversion over the initial stage, slight discrepancies in the timing of the light exposure with the kinetic data could easily account for significant artifacts in the degree of post-cure. As the irradiation times are extended and the rate of change in conversion moderates beyond the rate maximum and especially into the vitrification stage, the effects from any potential indexing errors between the conversion data and the light exposure cycles (or the subsequent property evolution results that will be reported) effectively become negligible.
Beyond the reaction rate maximum, autodeceleration, where the reaction rate decreases faster than expected based on monomer consumption, occurs with increasing degrees of mobility restriction encountered along with the diminishing concentrations of residual reactive groups as the reaction proceeds. These factors combine to explain why the increasing immediate conversion is coupled to an inverse progression in the extent of dark cure even though there is a corresponding increase in the free radical population present but largely trapped within the developing network [23]. The 10 s exposure in which the extinction of the light coincides with the rate maximum is notable for the very high percentage of dark cure that occurs.
Volumetric polymerization shrinkage
The final volumetric polymerization shrinkage ranged from 2.5 ± 1.1 % achieved at a conversion of 47.2 ± 0.3 % after 10 s irradiation time to a value of 5.4 ± 0.5 % obtained at a conversion of 56.1 ± 0.5 % after the 60 s irradiation time (p˂0.001). There was no significant difference between final shrinkage values that resulted from irradiation times of at least 20 s although a clear monotonic correlation is evident between conversion and shrinkage (Table 2). Analysis of the dark cure-based polymerization shrinkage evolution compared with the shrinkage values during irradiation shows that as the irradiation time increases, the contribution of the dark cure-based polymerization shrinkage to the overall final shrinkage result drops dramatically from 52.8 % for the 10 s irradiation time to only 8.3 % for the 60 s exposure due to the anticipated mobility restrictions at higher conversion values. This suppression in the extent of dark cure-based volumetric polymerization shrinkage directly reflects the suppression in the amount of dark cure conversion. Excluding the early irradiation intervals where both conversion and shrinkage rates are most rapid, the proportion of dark cure shrinkage is only modestly greater than the proportion of dark cure conversion with the overall tendency that the two values show a first order association.
Table 2.
Polymerization shrinkage at different irradiation times and degrees of conversion
| Irradiation time, s | 10 | 15 | 20 | 30 | 60 | 600 |
| Final conversion, % (SD) | 47.2 (0.3) | 51.5 (0.8) | 53.0 (0.3) | 55.5 (0.8) | 56.1 (0.5) | 59.8 (0.4) |
| Polymerization shrinkage at light-off, % (SD) | 1.2 (1.0) | 2.82 (0.4) | 3.25 (1.0) | 4.1 (0.2) | 5.0 (0.5) | 5.7 (0.7) |
| Final polymerization shrinkage, % (SD) | 2.5a (1.1) | 3.5a,b (0.6) | 4.1b,c (0.8) | 4.7b,c (0.2) | 5.4c (0.5) | 5.7c (0.7) |
| Dark cure, % of final conversion | 15.2 | 10.4 | 9.0 | 8.3 | 6.2 | - |
| Dark cure polymerization shrinkage, % of final shrinkage | 52.8 | 20.3 | 20.1 | 12.6 | 8.3 | - |
Lower case letters indicate statistical differences within a row (Student-Newman-Keuls test, α=0.5).
SD = standard deviation.
Final values taken at 600 s from the onset of photopolymerization at 10 mW/cm2.
However, the conversion-based (rather than time-based, which is shown in Figure 3a) rate of volumetric polymerization shrinkage is not uniform and it increases significantly at about 40 % conversion (Figure 3b), which coincides with the exotherm-based temperature maximum under these polymerization conditions. The increase in shrinkage with respect to conversion at this stage results in the maximum for the conversion-based rate of shrinkage while the time-based shrinkage rate maximum occurs at approximately 20 % conversion, which is achieved during the first 5–10 s of irradiation. In the final stage of the polymerization reaction, which accounts for the majority of the reaction time, conversion advances very slowly due to the progressive vitrification process. Within this regime, consistent late-stage suppression in the conversion-dependent shrinkage rate beginning at about 45–50 % conversion (Figure 3b) indicates that excess free volume is retained during vitrification as the timescale for bulk network contraction exceeds that for continued propagation through residual free and pendant monomer. This suppression of the shrinkage rate at high conversion values in glassy polymers has been discussed previously and indirectly probed [9, 12, 22, 24]. It should be noted that the real-time NIR-based conversion data obtained here were validated by normalization with the aromatic internal reference peak located at 4620 cm−1 [19]. Within experimental error, identical conversion was obtained with or without application of the internal reference normalization, which indicates that any dimensional distortion of the shrinkage specimen during polymerization does not contribute to anomalous conversion results.
Figure 3.

Dynamic volumetric shrinkage as a function of time (a) or conversion (b) obtained at different irradiation times.
Dynamic mechanical analysis
The time-based development of storage modulus for the partially irradiated bars subjected to a second-stage continuation of the photopolymerization process to different levels of conversion is shown in Figure 4a. More informative is the dramatic increases in modulus that are evident over relatively small conversion increments at the advanced stages of the polymerization process (Figure 4b). Once again, the post-irradiation (dark cure) conversion increase was significantly higher for the lowest irradiation interval, which was correlated with relatively minimal modulus evolution over this range of conversion (Table 3). At higher conversion, the post-cure modulus development was amplified dramatically as a result of vitrification. This is apparent from the diverging proportions of conversion and stress development during the dark cure interval as overall conversion increases (Table 3), which is contrary to the convergent relationship between dark cure conversion and shrinkage (Table 2). The gradual nature of the vitrification process is evident based on the broad but well-defined inflection in the dynamic conversion-dependent modulus data.
Figure 4.

Dynamic flexural modulus evolution with respect to time (a) or conversion (b) obtained in single cantilever mode for partially cured bars exposed to varied secondary irradiation times and intensities.
Table 3.
Flexural modulus data obtained from ambient dynamic mechanical analysis of partially pre-cured specimen bars during further exposure with different irradiance and exposure interval conditions
| Irradiation time, s | 20 | 60 | 120 | 600 | 600 sat 100 mW/cm2 |
| Final conversion, % (SD) | 46.9 (0.4) | 49.1 (4.9) | 53.1 (2.3) | 56.4 (1.5) | 65.7 (1.9) |
| Modulus at light-off, MPa (SD) | 30.6 (10.1) | 47.0 (17.5) | 130.1 (1.0) | 337.5 (12.4) | 682.8 (35.3) |
| Final modulus, MPa (SD) | 73.3a (18.2) | 107.0a (6.3) | 224.2a (3.4) | 415.2b (23.1) | 1092.6c (154.4) |
| Dark cure, % of final conversion | 67.1 | 21.4 | 18.3 | 4.1 | 1.4 |
| Dark cure modulus, % of final modulus | 58.3 | 56.1 | 42.0 | 18.7 | 37.5 |
Different lower case letters indicates statistical differences within a row (Dunn's Method, α=0.5).
SD = standard deviation.
Final values taken at 1200 s from the onset of the secondary curing cycle at 10 mW/cm2 except where noted otherwise.
Towards the latter stages of polymerization, crosslink density, which is related directly to modulus in the rubbery state, increases rapidly with respect to conversion. The proportion of free monomer relative to pendant reactive groups decreases, with the relatively slow continued late-stage conversion leading mainly to crosslink formation. Even when a free monomer molecule is reacted during the final stage of polymerization, which adds to network mass without appreciably affecting network density, it diminishes a local plasticizing contribution that then effectively raises Tg and modulus as well. Cooling associated with both the declining reaction exotherm and any post-irradiative thermal effects, also contribute to an increase in modulus that is largely independent of continued conversion, as demonstrated by the very steep modulus development profile of the extended, higher irradiance condition when the lamp is extinguished. The higher effective curing temperature associated with the longer, higher intensity curing cycle results in temporary reductions in shrinkage and modulus while causing delayed mobility restriction, which raises the limiting conversion and further increases final modulus. The proportion of post-irradiation modulus rise relative to the corresponding increase in dark cure conversion for the extended 100 mW/cm2 exposure condition is several-fold greater than that observed with the lower irradiance level (Table 3), which can be explained by the cooling effect suppressing the rate of additional conversion while simultaneously enhancing bulk polymer density and modulus development. The results indicate that as polymer network formation transitions to the glassy state, the reaction rate slows several orders of magnitude but network density, and thus the polymer properties, such as modulus that are dependent on crosslink density, continue to develop. It should be recognized that the use of a pre-cure stage in the photocuring process alters the exothermic potential of the subsequent second-stage reaction [25]; however, the staged curing is not expected to significantly alter the final polymer network structure since other prior work that used a wide range of curing protocols [26] (although not pulse-cure procedures specifically) found polymer Tg and modulus values that were predictable based only on the final conversion achieved rather than the route taken to achieve a given level of conversion.
An alternate approach to modulus development can be taken that involves the static modulus data reflecting the range of conversion associated with the partial and full cure photopolymerizations conducted here. A plot of modulus as a function of conversion for the range of partial cure specimens before and again after the application of the secondary curing cycles (Figure 5) shows good agreement between the combined static data and individual dynamic modulus data. As with the dynamic modulus evolution, the static data shows a vitrification-based inflection at approximately 45 % conversion for this resin and these reaction conditions. The displays of modulus development in Figures 4 and 5 use a linear scale rather than log scale (more typical of modulus data) so as not to obscure the conversion-dependency.
Figure 5.

Correlation between degree of conversion and the static flexural modulus of pre-cured specimens before the secondary photopolymerization (open circles) and after the additional photocure and post-cure intervals (filled circles).
Polymerization shrinkage stress
The plots showing the rapid rise in stress as a function of time (Figure 6a; summarized in Table 4) are complemented by the corresponding conversion-related plots (Figure 6b), which demonstrate a gradual initial rise in the post-gelation rubbery state followed by a rapid increase again beginning at approximately 45 % conversion, which coincides reasonably well with the observed reduction in the shrinkage rate and a rapid rise in modulus as well as the shoulder present in the simple reaction kinetics data. Given the polymerization conditions used here, this stage of conversion can be considered as the onset of vitrification with the recognition that it represents the macroscopic vitrification process since nano- and micro-vitrified regimes are present at much lower conversion levels as evidenced by the extended dark cure processes noted here at the early stages of these network-forming photopolymerizations. As glassy network development extends into the spaces between the initial microgel regions to create a continuous vitrified polymer phase [27], the bulk mechanical properties are dramatically affected. The differential between the proportions of dark cure stress rise and dark cure conversion increase becomes greater as the irradiation times increase (Table 4). The conversion dependent late-stage stress development associated with vitrification shows limited (if any) stress rate suppression (Figure 6b) as was observed with the end-stage polymerization shrinkage.
Figure 6.

Stress evolution profiles with respect to time (a) or as a function of conversion (b) for different irradiation times at a 10mW/cm2 irradiance.
Table 4.
Stress evolution at different irradiation times and degrees of conversion
| Irradiation time, s | 10 | 15 | 20 | 30 | 60 | 600 |
| Final conversion, % (SD) | 45.6 (1.1) | 53.5 (0.7) | 54.4 (0.7) | 55.3 (0.6) | 57.6 (2.4) | 59.5 (4.0) |
| Stress at light-off, MPa (SD) | 0.1 (0.1) | 0.6 (0.1) | 0.5 (0.1) | 0.8 (0.2) | 1.0 (0.1) | 1.6 (0.4) |
| Final stress, MPa (SD) | 0.3a (0.2) | 1.1b (0.2) | 1.0b (0.05) | 1.3b (0.2) | 1.4b (0.2) | 1.6b (0.4) |
| Dark cure, % of final conversion | 21.6 | 9.5 | 10.0 | 11.0 | 6.3 | - |
| Dark cure stress, % of final stress | 73.6 | 50.0 | 49.5 | 43.3 | 26.2 | - |
Lower case letters indicate statistical differences within a row (Student-Newman-Keuls test, α=0.5).
SD = standard deviation.
Final values taken at 600 s from the onset of photopolymerization at 10 mW/cm2.
This means that stress development is sensitive to relatively small variations in the final conversion, which includes differences due to the time and intensity associated with the photocuring process as well as the post-irradiation storage time that allows for additional small incremental increases in conversion. It should be noted that the variation in the stress levels as a function of conversion is narrower than the range observed for the modulus. This may reflect the fact that while modulus changes quite rapidly in the final stage of polymerization, the contribution of shrinkage is minimal over the same range of late-stage conversion. Post-cure storage time would also allow for potential physical network densification related to delayed shrinkage arising from the suppressed shrinkage (residual excess free volume) during vitrification. This work demonstrates the complexity of polymer property development and also provides context for other prior studies focused on conversion-dependent stress build up [12, 28–30].
In conclusion, for a model dimethacrylate glassy network approaching its limiting conversion, the conversion-dependent volumetric polymerization shrinkage rate is suppressed due to the vitrification process while modulus shows a dramatic increase. As a result, stress evolves at an intermediate rate that appears more dependent on modulus change during vitrification. The minimal and practically unavoidable differences in final conversion or density that occur for photocured resin or composite materials can be expected to provide considerable differences in polymerization stress. In a clinical setting, the combined desire for short curing cycles, large potential differences between curing light outputs and the variety of materials as well as thickness of restorative increments placed all promote variations in overall conversion. This especially applies for conversion at the base of a restoration where lower local stress may result but this would coincide with very significantly reduced local modulus. In addition, water uptake with its corresponding plasticization effect, which may also affect the gradual longer-term post-cure, complicates the physical and chemical aging process that likely continues to alter polymerization-based stress over extended timescales well beyond those studied here. The timing and extent of temperature variations that accompany the polymerization process, which has implications for the use of high intensity curing protocols, may have some modest effect on the final modulus and stress levels attained, but the level of conversion achieved appears to primarily govern these critical polymer properties.
Acknowledgements
This study was supported by NIH/NIDCR R01DE014227 and the King Abdulaziz University Vice-Presidency for Graduate Studies and Academic Research. The monomers used were kindly donated by Esstech.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Am Dent Assoc 2005–06 Survey of Dental Services Rendered, Table 35. p. 31.
- 2.Pfeifer CS, Ferracane JL, Sakaguchi PL, Braga RR. Factors affecting photopolymerization stress in dental composites. J Dent Res. 2008;87:1043–1047. doi: 10.1177/154405910808701114. [DOI] [PubMed] [Google Scholar]
- 3.Lu H, Stansbury JW, Dickens SH, Eichmiller FC, Bowman CN. Probing the origins and control of shrinkage stress in dental resin-composites: II. Novel method of simultaneous measurement of polymerization shrinkage stress and conversion. J Biomed Mater Res Part B: Appl Biomater. 2004;71B:206–213. doi: 10.1002/jbm.b.30088. [DOI] [PubMed] [Google Scholar]
- 4.Cramer NB, Stansbury JW, Bowman CN. Recent advances and developments in composite dental restorative materials. J Dent Res. 2011;90:402–416. doi: 10.1177/0022034510381263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennison JB, Sarrett DC. Prediction and diagnosis of clinical outcomes affecting restoration margins. J Oral Rehabil. 2012;39:301–318. doi: 10.1111/j.1365-2842.2011.02267.x. [DOI] [PubMed] [Google Scholar]
- 6.Dickens SH, Stansbury JW, Choi KM, Floyd CJE. Photopolymerization kinetics of methacrylate dental resins. Macromolecules. 2003;36:6043–6053. [Google Scholar]
- 7.Hale A, Macosko CW, Bair HE. Glass transition temperature as a function of conversion in thermosetting polymers. Macromolecules. 1991;24:2610–2621. [Google Scholar]
- 8.Venditti RA, Gillham JK. Relationship between the glass transition temperature (Tg) and fractional conversion for thermosetting systems. J Appl Polym Sci. 1997;64:3–14. [Google Scholar]
- 9.Stansbury JW. Dimethacrylate network formation and polymer property evolution as determined by the selection of monomers and curing conditions. Dent Mater. 2012;28:13–22. doi: 10.1016/j.dental.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leprince J, Lamblin G, Truffier-Boutry D, Demoustier-Champagne S, Devaux J, Mestdagh M, et al. Kinetic study of free radicals trapped in dental resins stored in different environments. Acta Biomaterialia. 2009;5:2518–2524. doi: 10.1016/j.actbio.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 11.Hutchinson JM. Physical aging of polymers. Prog Polym Sci. 1995;20:703–760. [Google Scholar]
- 12.Stansbury JW, Lemon MT, Lu H, Ding X, Lin Y, Ge J. Conversion-dependent shrinkage stress and strain in dental resins and composites. Dent Mater. 2005;21:56–67. doi: 10.1016/j.dental.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Versluis A, Tantbirojn D, Douglas WH. Do dental composites always shrink toward the light? J Dent Res. 1998;77:1435–1445. doi: 10.1177/00220345980770060801. [DOI] [PubMed] [Google Scholar]
- 14.Pfeifer CS, Wilson ND, Shelton ZR, Stansbury JW. Delayed gelation through chain-transfer reactions: mechanism for stress reduction in methacrylate networks. Polymer. 2011;52:3295–3303. doi: 10.1016/j.polymer.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lange J, Ekelof R, George GA. Indications of micro-vitrification during chainwise cross-linking polymerization. Polymer. 1999;40:3595–3598. [Google Scholar]
- 16.Ferracane JL. Developing a more complete understanding of stress produced in dental composites during polymerization. Dent Mater. 2005;21:36–42. doi: 10.1016/j.dental.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Goncalves F, Pfeifer CS, Ferracane JL, Braga RR. Contraction stress determinants in dimethacrylate composites. J Dent Res. 2008;87:367–371. doi: 10.1177/154405910808700404. [DOI] [PubMed] [Google Scholar]
- 18.Antonucci JM, Giuseppetti AA, O'Donnell JNR, Schumacher GE, Skrtic D. Polymerization stress development in dental composites: Effect of cavity design factor. Materials. 2009;2:169–180. doi: 10.3390/ma2010169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stansbury JW, Dickens SH. Determination of double bond conversion in dental resins by near infrared spectroscopy. Dent Mater. 2001;17:71–79. doi: 10.1016/s0109-5641(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 20.Lu H, Stansbury JW, Bowman CN. Towards the elucidation of shrinkage stress development and relaxation in dental composites. Dent Mater. 2004;20:979–986. doi: 10.1016/j.dental.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Trujillo M, Newman SM, Stansbury JW. Use of near-IR to monitor the influence of external heating on dental composite photopolymerization. Dent Mater. 2004;20:766–777. doi: 10.1016/j.dental.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt LE, Leterrier Y, Schmah D, Manson JAE, James D, Gustavsson E, Svensson LS. Conversion analysis of acrylated hyperbranched polymers UV-cured below their ultimate glass transition temperature. J Appl Polym Sci. 2007;104:2366–2376. [Google Scholar]
- 23.Zhang Y, Kranbuehl DE, Sautereau H, Seytre G, Dupuy J. Study of UV cure kinetics resulting from a changing concentration of mobile and trapped radicals. Macromolecules. 2008;41:708–715. [Google Scholar]
- 24.Anseth KS, Kline LM, Walker TA, Anderson KJ, Bowman CN. Reaction-kinetics and volume relaxation during polymerizations of multiethylene glycol dimethacrylates. Macromolecules. 1995;28:2491–2499. [Google Scholar]
- 25.Lu H, Stansbury JW, Bowman CN. Impact of curing protocol on conversion and shrinkage stress. J Dent Res. 2005;84:833–826. doi: 10.1177/154405910508400908. [DOI] [PubMed] [Google Scholar]
- 26.Lovell LG, Lu H, Elliott JE, Stansbury JW, Bowman CN. The effect of cure rate on the mechanical properties of dental resins. Dent Mater. 2001;17:504–511. doi: 10.1016/s0109-5641(01)00010-0. [DOI] [PubMed] [Google Scholar]
- 27.Krzeminski M, Molinari M, Troyon M, Coqueret X. Characterization by atomic force microscopy of the nanoheterogeneities produced by the radiation-induced cross-linking polymerization of aromatic diacrylates. Macromolecules. 2010;43:8121–8127. [Google Scholar]
- 28.Francis LF, McCormick AV, Vaessen DM, Payne JA. Development and measurement of stress in polymer coatings. J Mater Sci. 2002;37:4897–4911. [Google Scholar]
- 29.Stolov AA, Xie T, Penelle J, Hsu SL, Stidham HD. An analysis of photopolymerization kinetics and stress development in multifunctional acrylate coatings. Polym Eng Sci. 2001;41:314–328. [Google Scholar]
- 30.Vaessen DM, Ngantung FA, Palacio MLB, Francis LF, McCormick AV. Effect of lamp cycling on conversion and stress development in ultraviolet-cured acrylate coatings. J Appl Polym Sci. 2002;84:2784–2793. [Google Scholar]