

Abstract
The absence of thyroid hormone (TH) during late gestation and early infancy can cause irreparable deafness in both humans and rodents. A variety of rodent models have been used in an effort to identify the underlying molecular mechanism. Here, we characterize a mouse model of secondary hypothyroidism, pituitary transcription factor 1 (Pit1dw), which has profound, congenital deafness that is rescued by oral TH replacement. These mutants have tectorial membrane abnormalities, including a prominent Hensen's stripe, elevated β-tectorin composition, and disrupted striated-sheet matrix. They lack distortion product otoacoustic emissions and cochlear microphonic responses, and exhibit reduced endocochlear potentials, suggesting defects in outer hair cell function and potassium recycling. Auditory system and hair cell physiology, histology, and anatomy studies reveal novel defects of hormone deficiency related to deafness: (1) permanently impaired expression of KCNJ10 in the stria vascularis of Pit1dw mice, which likely contributes to the reduced endocochlear potential, (2) significant outer hair cell loss in the mutants, which may result from cellular stress induced by the lower KCNQ4 expression and current levels in Pit1dw mutant outer hair cells, and (3) sensory and strial cell deterioration, which may have implications for thyroid hormone dysregulation in age-related hearing impairment. In summary, we suggest that these defects in outer hair cell and strial cell function are important contributors to the hearing impairment in Pit1dw mice.
Keywords: secondary hypothyroidism, tectorial membrane, KCNQ1, KCNQ4, KCNJ10, prestin, POU1F1
Introduction
The organ of Corti is a neuroepithelium with sensory cells known as inner (IHC) and outer hair cells (OHCs). Its maturation in the early postnatal period in mice and during the late prenatal period in humans is highly sensitive to thyroid hormone (TH), as TH deficiency during these periods can cause irreparable hearing deficits (Deol, 1973; Van't Hoff and Stuart, 1979; Uziel et al., 1983, 1986; Vanderschueren-Lodeweyckx et al., 1983). TH activates and represses expression of many genes by interacting with TH receptors (THR). Some TH regulated genes encode functionally important proteins that are involved in structural development and/or physiological processes of the inner ear. Although several candidate genes have been proposed based on studies with rodent models of hypothyroidism, the molecular basis for the permanent nature of hypothyroidism-induced hearing loss is still unclear (Uziel, 1986; Knipper et al., 1998, 2000; Rusch et al., 1998; Zheng et al., 2000; Abe et al., 2003; Cantos et al., 2003; Rueda et al., 2003; Winter et al., 2006; Brandt et al., 2007; Sendin et al., 2007).
To explore the mechanism whereby TH affects the development and function of the inner ear, we characterized Snell dwarf (Pit1dw, officially Pou1f1dw), a mouse model of secondary hypothyroidism. These mice have an inactivating missense mutation in the Pit1 gene, which encodes a POU-homeodomain transcription factor that is highly expressed in the pituitary gland (Camper et al., 1990; Li et al., 1990). The gene is necessary for development of the cells that produce growth hormone (GH), prolactin (PRL), and thyroid-stimulating hormone (TSH) (Gage et al., 1996). If untreated, homozygous mutants exhibit growth insufficiency, infertility, hypothyroidism, and deafness. Auditory brainstem response (ABR) testing revealed profound deafness in Pit1dw mutants at both 3 and 6 weeks of age, but continuous oral thyroid hormone supplementation initiated late in gestation was effective in preventing hearing deficits (Karolyi et al., 2007). Because TH replacement does not rescue the deficiency in GH or PRL, the deafness is attributable to hypothyroidism. In addition, there are no reports of hearing deficits caused by GH or PRL deficiency, and GH deficient little mutants (Ghrhrlit) have normal ABR thresholds (OMIM, K. R. Johnson, personal communication) (Godfrey et al., 1993; Lin et al., 1993). The Pit1dw mutant mice are viable and healthy, living ∼40% longer than their normal littermates (Brown-Borg et al., 1996). We selected these mutants for further characterization because of their viability, profound deafness, simple, autosomal recessive inheritance of hypothyroidism, and responsiveness to TH supplementation (Karolyi et al., 2007).
We report physiological, morphological, and gene expression analyses over the course of cochlear development in normal and Pit1dw mutant mice. Tectorial membrane (TM) composition, morphology, and ultrastructure are altered. OHC function is permanently compromised, as demonstrated by the absence of distortion product oto-acoustic emissions (DPOAE) and cochlear microphonics (CM). In addition to defects in OHC function, Pit1dw mice have reduced endocochlear potential (EP). We examined expression of prestin and KCNQ4 in OHCs, as well as KCNQ1 and KCNJ10 in the stria vascularis, as candidates for underlying mechanisms. We identified some processes that are developmentally delayed but eventually mature in hormone-deficient mice and others that suffer lasting deficits, likely contributing to the permanent hearing problems caused by hypothyroidism. This study advances our understanding of the role that hormones play in development of normal hearing, including permanent effects on the tectorial membrane, expression of the potassium channel genes KCNQ4 and KCNJ10, the survival of OHCs, and deterioration of the intermediate cells in the stria vascularis.
Materials and Methods
Animal care and genotyping.
All experiments were approved by the University Committee on the Use and Care of Animals and conducted in accord with the principles and procedures outlined in the National Institutes of Health Guidelines for the Care and Use of Experimental Animals. Mice were obtained from The Jackson Laboratory in 1990 and maintained at the University of Michigan. Previously described procedures for animal care and genotyping were used, including feeding mice a higher fat chow designed for breeding (PMI5020), delaying weaning of mutants until ∼35 d, and housing mutants with normal littermates to provide warmth (Karolyi et al., 2007). In all experiments, at least 3 animals of each genotype were analyzed for each age group studied unless stated otherwise. Postnatal day zero (P0) is designated as the day of birth.
Histology and gene expression analysis.
Western blot analysis was performed with organ of Corti protein extracts collected from postnatal animals. Protein samples were homogenized in T-PER buffer (Pierce, Thermo Fisher Scientific), denatured in Laemmli sample buffer, and loaded onto SDS-polyacrylamids gels (12%). Gels were blotted onto nitrocellulose filters, and filters were probed with rabbit anti-β tectorin antibody (gift from Dr. Richardson, University of Sussex, UK) at 1:1000 dilution. As a loading control, blots were subsequently probed with anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Santa Cruz Biotechnology s.c.-25778) at the dilution of 1:2000 and developed with an HRP-conjugated goat anti-rabbit (Pierce Biotechnology cat# 31462). Blots were developed with ECL substrate (Santa Cruz Biotechnology s.c.-2048).
Animals aged P8 and older were killed by CO2 inhalation or decapitation. The temporal bones were avulsed and cochleae were quickly transferred into 4% paraformaldehyde in 0.1 m PBS, pH 7.4. The footplate of the stapes was removed and each cochlea was perfused with the fixative introduced through the oval and round windows and extruded through a small opening made at the apex. The tissues were exposed to the fixative for 2 h, followed by overnight incubation (with shaking) at 4°C in 8% EDTA for decalcification. No decalcification was applied to ears from animals younger than P10.
Fixed tissues were prepared for sectioning by embedding in paraffin or OCT. Soft bone specimens were dehydrated through a graded ethanol series and embedded in paraffin wax, sectioned at 6 μm thickness, mounted, dried for 2 h, and stored at room temperature. Alternatively, soft bone specimens were incubated in 30% sucrose overnight at 4°C, embedded in OCT, cryosectioned at 6 μm thickness, mounted, dried for 15 min, and stored at −20°C.
Cochlear cryosections were blocked with 5% goat or donkey serum and 0.1% Triton X-100 in 0.1 m PBS for 1 h at room temperature, and incubated overnight at 4°C with a rabbit antiserum against KCNJ10 (1:300, Alomone), or a rabbit polyclonal antibody against KCNQ4 (1:300) (Kharkovets et al., 2006), which was kindly provided by T. Jentsch (Hamburg University, Germany). Cochlear paraffin sections were dewaxed, rehydrated and boiled in 10 mm citric acid, pH 6, for 10 min to unmask epitopes followed by 1:1 H2O2:methanol (v/v) for 20 min to quench the endogenous peroxidase activity. The sections were incubated overnight at 4°C with a goat polyclonal antibody against prestin (N-20) (1:200, Santa Cruz) and mouse monoclonal antibody against synaptophysin (1:400, Sigma). Immunolabeling was visualized with TRITC-labeled secondary antibody (1:200, Jackson Immunoresearch) or Alexa Fluor 488 conjugated secondary antibodies (1:200, Invitrogen) and counter-stained with DAPI (Vector Laboratories). Sections from wild-type and mutant animals were processed in parallel for histology and immunostaining. Tissues were analyzed and photographed on a Leica DMRB epifluorescence microscope using identical light and exposures for mutant and wild-type littermates.
For transmission electron microscopy analysis, animals were anesthetized and fixed by intracardiac perfusion with 2.5% glutaraldehyde in 0.15 m cacodylate buffer, pH 7.2, containing 1% tannic acid. The inner ear was removed and immersed into the same fixative for 2 h. The tissues were decalcified for 1 week in 3% EDTA with 0.25% glutaraldehyde at 4°C. The inner ears were postfixed with 1% osmium tetroxide in phosphate buffer for 1 h. The specimens were dehydrated in increasing ethanol concentrations and embedded in Embed 812 epoxy resin. Sections were taken on a Leica Ultracut R using a diamond knife, stained with uranyl acetate and lead citrate, and examined on a Philips CM-100 TEM (Beyer et al., 2000; Russell et al., 2007).
Scanning electron microscopy, whole mount and phalloidin epifluorescence analyses were performed as previously described (Beyer et al., 2000; Mustapha et al., 2007).
Standard tests of auditory physiology.
DPOAEs and EP were conducted as previously described (Karolyi et al., 2007).
CM measurements were performed on mice that were anesthetized (ketamine 65 mg/kg, xylazine 3.5 mg/kg, and acepromazine 2 mg/kg) and given a dose of glycopyrrolate (0.2 mg/kg) to reduce secretions during surgery. Body temperature was maintained through the use of water circulating heating pads and heat lamps. Additional anesthetic (ketamine and xylazine) was administered if needed to maintain anesthesia at a depth sufficient to insure immobilization and relaxation. Mice were placed into a head holder. The external pinna was removed and soft tissue dissected away from the bulla. The bulla wall was opened, and a single ball electrode, ∼100 micrometer in diameter, was placed in the round window niche. The electrode was made from 2T 90% platinum/10% iridium wire and Teflon coated, except for the ball. A Teflon-coated silver return electrode was placed subcutaneously on the jawline. For CM recordings, the output frequency from the SRS 830 lock-in amplifier was stepped from 4 to 30 kHz, with a time constant of one second and a dwell time of 300 microseconds. The signal was passed through a programmable attenuator (TDT PA5) to produce a constant-amplitude signal. The signal from the round window electrode (Teflon-coated silver wire, with reference in the contralateral jawline) was sent through a Grass P15 amplifier (filter 0.1–50 kHz) to the input of the SRS830 lock-in amplifier. Signals were generated and data recorded with a MATLAB script written in-house.
Patch-clamp electrophysiology.
Preparations of semi-intact mouse organ of Corti were obtained from mice 2 to 6 weeks old. Mice were anesthetized by intraperitoneal injection of ketamine (65 mg/kg) and xylazine (7 mg/kg) and killed by rapid decapitation. The bony labyrinth was extracted into cold perilymph-like saline (in mm, 142.0 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 10 HEPES, 0.7 Na2HPO4, 2 Na-pyruvate, 5.8 glucose; pH 7.4; 305 mOsm), supplemented with amino acids and vitamins (1X MEM, Invitrogen). Approximately three-quarters of the apical cochlear coil was separated from the modiolus and spiral ligament. All recordings were from OHC located approximately one-half turn from the cochlear apex. Viable hair cells were identified by their appearance, including features of birefringent membrane, columnar shape and absence of observable Brownian motion in cell organelles. Whole-cell voltage-clamp recordings were made with a MultiClamp 700B and Digidata 1440A data acquisition system using the pClamp 10.0 software suite (Molecular Devices). Data were sampled at 20 kHz and low-pass filtered at 4 kHz. For measurements of nonlinear capacitance, electronic compensation of membrane capacitance was necessarily omitted. Data were obtained from all three rows of OHC. Currents were recorded several minutes after break-in to the whole-cell configuration, to allow the cytoplasm and pipette solution to equilibrate. Electrodes were pulled from borosilicate glass capillaries (World Precision Instruments) to a resistance of 3–6 MΩ. Drug perfusions, when necessary, were delivered locally through a multichannel micromanifold (ALA Scientific). All recordings were made at room temperature (21−26°C). Membrane voltages have not been compensated for junction potentials or residual series resistance errors. Unless otherwise noted, all measurements are reported as means ± 1 SEM.
Voltage-dependent capacitance was estimated from current transients elicited by small, incremental steps in the command voltage. This method, described more fully elsewhere (Huang and Santos-Sacchi, 1993; Oliver and Fakler, 1999), relies on a simplified electrical model of the cell, under conditions where ionic currents have been blocked. The voltage protocol consisted of a stair-step series of voltage commands from −125 to 70 mV, using brief 5 mV steps 10 ms in duration. Extracellular and intracellular solutions were designed to block dominant potassium and calcium currents, resulting in a nearly linear steady-state current-voltage curve. The extracellular solution consisted of the following (in mm): 100 NaCl, 20 CsCl, 20 TEA-Cl, 1.52 MgCl2, 2 CoCl2, 10 HEPES, 5 glucose, with pH adjusted to 7.2 using NaOH and osmolarity set to ∼300 mOsm. The intracellular solution contained the following (in mm): 140 CsCl, 2 MgCl2, 10 HEPES, 10 EGTA, with pH adjusted to 7.2 using CsOH and osmolarity set to ∼300 mOsm.
Membrane capacitance was calculated from estimates of input resistance and monoexponential fits to capacitance transients. These calculations were plotted against the voltage command eliciting the transient and fit to the derivative of the Boltzman equation describing charge movement through the membrane electric field:
 |
where Vm is membrane voltage, Clin is the linear, voltage-independent membrane capacitance, Qmax is the maximum charge moved through the membrane electric field, V1/2 is the voltage corresponding to half-maximum charge movement, and α is the Boltzman slope factor describing voltage-sensitivity. The maximum voltage-dependent capacitance (Cnonlin) is equal to Cm(V1/2) − Clin.
For potassium current measurements, the bath solution was the perilymph-like saline used for dissection (see above), and the electrode solution contained (in mm): 135 NaCl, 0.1 CaCl2, 3.5 MgCl2, 5 HEPES, 5 EGTA, and 2.5 Na2ATP, with pH adjusted to 7.2 with KOH and osmolarity set to 292 mOsm. Voltage commands were referenced to a holding potential of −80 mV in all cases. Tail currents were corrected off-line for a linear leak conductance and fit with a first-order Boltzman function:
 |
where Imax is the maximum tail current, Vm is the membrane voltage, V1/2 is the half-maximal activation voltage, and Vs is the Boltzman slope factor describing voltage sensitivity. In many cases, the sum of two first-order Boltzman functions was required to describe the tail currents. The F-statistic from curve fits in pClamp was used to determine whether a single or double Boltzman equation best fit the data. In some cases, the M-current blocker linopirdine (200 μm) was locally applied to selectively block KCNQ4 currents (Marcotti and Kros, 1999).
Results
Altered composition of the tectorial membrane in Pit1dw mutant mice
The morphological maturation of the cochlea is subject to developmental delay in mice with disrupted TH receptors, Thra, Thrb, which are unable to respond to TH. Similar features of developmental delay are noted in many types of hypothyroid mice, including pituitary TSH deficient Cga mutants and pharmacologically induced hypothyroidism (Deol, 1973; Uziel et al., 1983, 1986; Li et al., 1999; Rusch et al., 2001; Karolyi et al., 2007). We compared mid-modiolar cochlear sections of Pit1dw mutants and wild-type littermates using light microscopy. We observed a delay in the opening of the tunnel of Corti in Pit1dw mutants at P12, but by P21 the opening was indistinguishable from wild type (Fig. 1A). These features are similar to those described in other hypothyroid mutants. At P21 the tectorial membrane (TM) in Pit1dw mutants clearly contains an abnormal protrusion (Fig. 1A). This abnormality appears to be a more prominent Hensen's stripe, and it persists through P42 (data not shown). This prominent stripe is evident in another model of secondary hypothyroidism, the Cga mutant that lacks pituitary TSH, but neither the underlying mechanism nor the significance has been explored (Karolyi et al. 2007).
Figure 1.
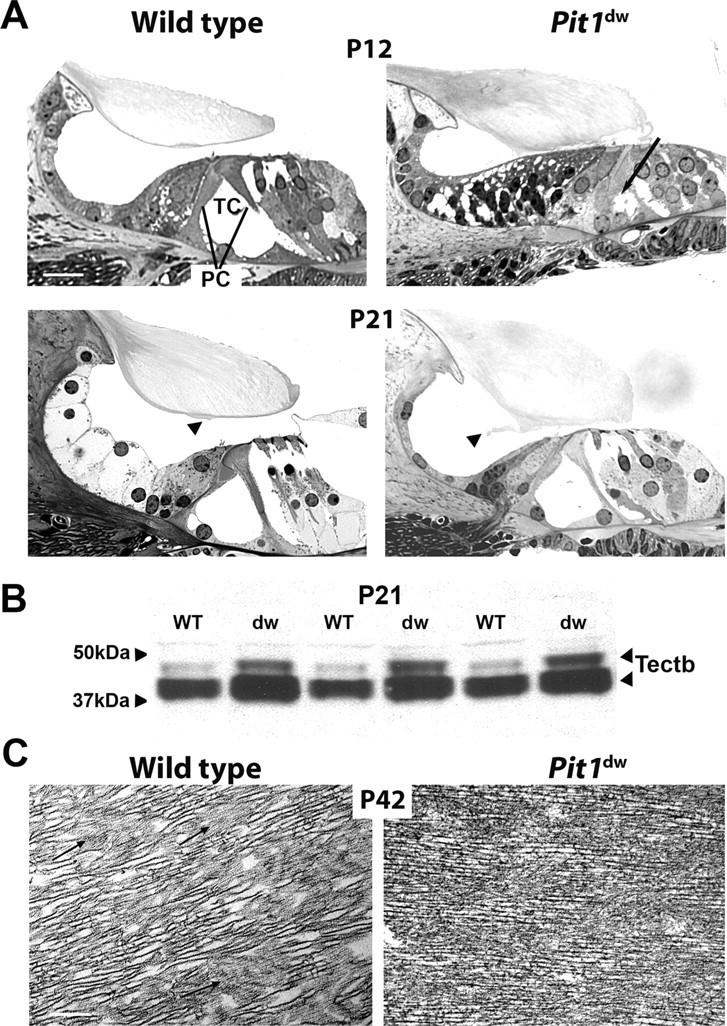
Tectorial membrane abnormalities in Pit1dw mice. A, Plastic sections of the organ of Corti taken from wild-type and Pit1dw mutant mice at P12 and P21 were visualized by light microscopy. The arrow indicates the unopened tunnel of Corti (TC) in the P12 old mutant. OHCs, IHCs, and pillar cells (PC) are indicated. Scale bars: 10 μm. B, Analysis of TECTB content in the tectorial membrane of P21 animals using Western blotting. Two polypeptide bands of TECTB are present at ∼43 and 47 kDa. The blot was reprobed with a GAPDH antibody, and equivalent amounts of immunoreactive ∼36 kDa protein were detected, indicating equivalent amounts of proteins were loaded (data not shown). C, Transmission electron micrographs illustrate the ultrastructure of the tectorial membrane in P42 old animals. Regions shown are from the central core of the tectorial membrane overlying the organ of Corti. Fine diameter filaments (arrows) forming the striated sheet matrix are different in the wild-type mice and the mutants. Scale bars: 2 μm.
The tectorial membrane is comprised of collagens (Col11a2, Col9a3, and Col9a1) and noncollagen proteins including α-tectorin (Tecta) and β-tectorin (Tectb) and otogelin (Otog), all of which are critical for normal hearing (Richardson et al., 2008). We compared the concentration of TECTB in cochlear protein extracts from normal and Pit1dw mutant mice at P21 using Western blotting (Fig. 1B). Two expected TECTB polypeptides are detected in all mutant and wild-type samples, consistent with previous reports (Knipper et al., 2001). Both isoforms of TECTB are consistently elevated in the Pit1dw mutant samples compared with wild-type littermates. Similar results were obtained at P35 (data not shown). The elevation of TECTB in Pit1dw mutants contrasts with the significant reduction of TECTB in drug-induced hypothyroid rats (Knipper et al., 2001).
Structural changes in the TM have been reported in a variety of models with altered TH (Knipper et al., 2001; Rusch et al., 2001; Richardson et al., 2008). Transmission electron microscopy (TEM) revealed abnormalities in the structure of the striated-sheet matrix in the TM in Pit1dw mutants relative to wild type (Fig. 1C).
OHC loss in Pit1dw mutant mice
To examine maturation of cochlear sensory cells and supporting cells we prepared whole mounts of the organ of Corti from normal and mutant mice and stained them with FITC-phalloidin, which reveals actin-containing regions of the cytoskeleton and cell-cell contacts (Fig. 2A–D). At P14 and P25 the stereocilia of IHCs and OHCs and the junctions between pillar cells are clearly visible in wild-type mice (Fig. 2A,C). In contrast, the pillar cells do not stain with phalloidin in P14 mutants (Fig. 2B). At P25 the mutant pillar cells stain with phalloidin, but the organization of the cytoskeleton is abnormal relative to normal mice, and these abnormalities persist through P42 (data not shown). Disruptions are evident in the repeated pattern of OHCs and the orientation of the OHC hair bundles in both P14 and P25 Pit1dw mutants compared with wild type, suggesting the possibility of OHC death, followed by scar formation in the mutants (Fig. 2B,D).
Figure 2.

Pit1dw mutants undergo significant OHC loss compared with wild-type mice. A–D, Whole mounts of the organ of Corti of mice ages P14 and P25 were stained with FITC-phalloidin to reveal actin-rich structures and visualized by light microscopy. E–G, Scanning electron microscopy (E, F) and TEM (G) were used to examine the organ of Corti from P42 mice. The TEM reveals that the width of supporting cells (SC) that neighbor degenerating OHCs (arrow) are larger than SC neighboring healthy OHC. Pillar cells (PC). Arrows indicate OHC degeneration and scar formation. Scale bars: 10 μm. H, Outer hair cell loss was quantified by region (apex, lower apex, mid and upper base turns) in five different animals of both Pit1dw mutant and wild-type mice. The length of each region was 100 μm, encompassing 166–181 OHCs per region. Mean percentage outer hair cell loss was significantly higher in the Pit1dw mutants compared with the wild-type mice (*) in the lower apex (p < 0.01), mid turn (p < 0.01), and upper base (p < 0.05). No OHC loss was observed in wild-type cochlea at the upper base (#). All statistical tests were conducted in SPSS 15.0.1.1. Error bars represent one SEM.
We performed two additional types of analyses on P42 mutant and wild-type mice to examine OHC death more closely. Scanning electron microscopy of the organ of Corti clearly confirmed the presence of scars in mutant mice in the cochlear mid turn, indicating the expansion of supporting cells into spaces created by missing OHCs (Fig. 2E,F). TEM analyses revealed OHCs from the mid turn of P42 Pit1dw mutant in an advanced stage of degeneration (Fig. 2G). To determine the location and extent of OHC death along the cochlea, we analyzed whole mounts of P42 organ of Corti by staining OHCs with FITC-phalloidin (data not shown) and antibody to the motor protein prestin (supplemental Figure S. 1A,B, available at www.jneurosci.org as supplemental material). Four regions of the organ of Corti were examined and quantified for OHC loss (apex, lower apex, mid turn and upper base) in 5 different animals of both Pit1dw mutant and wild-type mice. Two-sided Student's t tests for independent samples were conducted for differences in the mean between mutants and wild-type mice. Gaps and deviations from the normal pattern of three, well organized rows of OHCs were observed throughout the mutant organ of Corti. The most prominent hair cell loss was in the lower apical region of Pit1dw mutants with a mean percentage loss of 15 ± 2% (mean ± 1 SEM) compared with wild-type mice at 3 ± 2% (Fig. 2H). Mean percentage OHC loss was significantly higher in the Pit1dw mutants compared with wild type in the lower apex (p < 0.01), mid turn (p < 0.01), and upper base (p < 0.05), but not significantly higher in the apex. A similar gradient of hair cell loss was described for Barhl1 mutants (Li et al., 2002). OHC degeneration occurs in the Pax8−/− model of hypothyroidism, but Pax8 expression in the otocyst could be a confounding factor (Patuzzi et al., 1989; Christ et al., 2004). Because most Pit1dw OHC survive and have a healthy appearance, the OHC death we observe in Pit1dw mutants is unlikely to account for the profound hearing impairment. The remaining OHCs might be dysfunctional, however.
Adult Pit1dw mice lack DPOAE and CM
OHCs contribute to sound processing by serving as a nonlinear cochlear amplifier. We used standard audiometric techniques to test OHC function by measuring DPOAEs and CMs because normal morphological appearance does not imply normal OHC function. Wild-type 6-week-old (P42) animals had DPOAEs at low and moderate sound levels (≤80 dB SPL) at all frequencies measured (12, 24, and 48 kHz) (shown for 24 kHz) (Fig. 3A). Postmortem DPOAE measurements on wild-type mice define the noise floor. Live, age-matched Pit1dw mutant littermates had DPOAEs that were indistinguishable from the noise floor in mutant or wild-type mice. We also recorded the magnitude and phase of the cochlear microphonic, a measure of the combined sound-driven receptor currents of both IHCs and OHCs. The CM response is dominated by the more numerous OHCs, particularly those OHCs located at the base of the cochlea close to the recording electrode (Patuzzi et al., 1989). CM amplitude was detected at all sound stimulation frequencies (4 through 30 kHz) in 6-week old (P42) wild-type animals (Fig. 3B). In Pit1dw age-matched offspring CM amplitudes were indistinguishable from those recorded in postmortem normal animals, indicating compromised OHC function in the Pit1dw mutants. The lack of a CM response was confirmed by the absence of a frequency-dependent phase shift in the mutants (Fig. 3C). These data suggest that the OHCs in the Pit1dw mutants are dysfunctional, despite their normal morphological appearance. Since both CM and DPOAE responses were absent in mutant mice, an OHC specific dysfunction could arise from deficits in the electrical motor response in these cells as well as from aberrant mechanical performance, which could be associated with abnormal properties of the TM.
Figure 3.

Pit1dw mutant mice lack DPOAE and CM responses. A, DPOAEs were measured in alive wild-type and mutant mice at P42 (black circles and black triangles, respectively) and compared with DPOAEs of postmortem animals (white circles and triangles). Data are shown for the 24 kHz frequency only. Error bars indicate ±1 SEM. B, C, CM potentials were tested at various sound stimulation frequencies (4 through 30 kHz) in P42 wild-type animals (green line), in Pit1dw mutant mice (red line) and in postmortem animals (green dashed line) of age-matched offspring (B). CM amplitudes were confirmed by the phase shift data (C).
Maturation of prestin expression and distribution in Pit1dw mice
Prestin is a member of a distinct family of anion transporters, SLC26, and it is expressed in OHCs but not in the nonmotile IHCs. Prestin, officially known as SLC26A5, is presumably responsible for OHC electromotility and cochlear amplification (Zheng et al., 2000; Liberman et al., 2002). Prestin mutants exhibit reduced DPOAE, CM and nonlinear capacitance compared with wild type (Liberman et al., 2002; Cheatham et al., 2004; Gao et al., 2007; Dallos et al., 2008). It is not clear, however, whether abnormalities in prestin protein levels, subcellular localization, or function contribute to reduced DPOAEs and CM in Pit1dw mice. To explore these possibilities we performed a developmental time course of immunohistochemical staining for prestin in tissues also stained with DAPI to reveal nuclei and synaptophysin, a presynaptic marker of the efferent synapse at the base of the OHCs. In normal mice, prestin immunoreactivity is evenly distributed throughout the entire OHC membrane during the first few days after birth (shown for P8) (Fig. 4A). Between P8 and P13, the subcellular distribution of prestin protein becomes restricted to the lateral regions of the OHC and is depleted from the basal pole of the OHC membrane (shown for P13) (Fig. 4A). This redistribution in prestin localization begins in the basal turn and propagates toward the apical turn of the cochlea (data not shown).
Figure 4.

Prestin protein expression and function mature in adult Pit1dw mutant mice. A, Prestin expression and localization was analyzed by staining paraffin-embedded sections from wild-type and Pit1dw mutants at P8, P14, P21, and P42 with prestin-specific antibodies (red). Sections were visualized with light microscopy. Synaptophysin (Syn) staining (green) identifies the basal poles of OHC. Nuclei were labeled using DAPI (blue). Asterisks (*) mark prestin labeling detected at the basal pole of the hair cell. Arrows identify rows of outer hair cells. Scale bars: 10 μm. B, Average capacitance-voltage curves were generated for OHCs from wild-type mice at 2 and 2.5 weeks and for mutants at 2, 4, and 6 weeks of age (respectively, N = 5, 6, 5, 6, 4). Curves were fit to Eq. 1 to estimate linear and nonlinear capacitance components. Fit parameters: WT: 2 weeks, Clin = 7.7 pF, Qmax = 0.82 pC, α = 28.2 mV, V1/2 = −56.3 mV; 2.5 weeks, Clin = 7.2 pF, Qmax = 1.07 pC, α = 26.9 mV, V1/2 = −65.0 mV; Pit1dw: 2 weeks, Clin = 5.9 pF, Qmax = 0.36 pC, α = 28.6 mV, V1/2 = −70.2 mV; 4 weeks, Clin = 6.2 pF, Qmax = 0.61 pC, α = 31.6 mV, V1/2 = −57.5 mV; 6 weeks, Clin = 6.7 pF, Qmax = 0.69 pC, α = 26.8 mV, V1/2 = −56.8 mV. C, Average values for linear and maximum nonlinear capacitance are shown for each experimental group. Error bars represent one SEM. *p < 0.01 with respect to data from 2.5-week-old wild-type OHCs.
Pit1dw mutants exhibit a developmental delay in prestin expression and subcellular localization. Prestin immunoreactivity is qualitatively reduced in Pit1dw mutants at P8 compared with normal littermates (Fig. 4A). The level and localization of prestin expression in Pit1dw mutants are improved at P13, resembling that of wild-type mice at P8. Mutants show continued improvement at P21. By P42 the pattern of prestin expression is indistinguishable from that of 2-week-old wild-type mice. This indicates a developmental delay of 3 to 4 weeks for maturation of prestin expression and localization in Pit1dw mice.
Maturation of prestin function in Pit1dw mice
If the level and subcellular localization of prestin expression is sufficient for normal OHC motor function, then the nonlinear capacitance generated by mature mutant OHCs should be similar to that of hearing mice. Membrane capacitance was estimated from current transients elicited by a stair-step voltage protocol. These data were fit to Eq. 1 to determine the voltage-independent (linear, Clin) and voltage-dependent (nonlinear, Cnonlin) components. Average capacitance curves are shown in Figure 4B for wild-type OHCs at 2 and 2.5 weeks of age (P13–14 and P18–19, respectively) and for Pit1dw OHCs at 2, 4, and 6 weeks of age (P16–17, P30–32, and P43, respectively). Peak total membrane capacitance increased with age for both wild-type and mutant mice. Measures of voltage dependence (V1/2 and α in Eq. 1) were relatively unchanged over the age-ranges studied and across genotype (data not shown; p > 0.01).
For wild-type mice, the increase in membrane capacitance reflected continued improvement in the nonlinear component from 2 to 2.5 weeks of age. Linear capacitance was constant over this age range (Fig. 4C), suggesting that the cells had reached a mature size even while the efficacy of prestin function or density of prestin molecules continued to increase. A previous report has suggested that nonlinear capacitance reaches a plateau ∼2 weeks of age in normal mice (Abe et al., 2007). This subtle difference could represent differences in mouse strain background or husbandry.
For mutant mice, the developmental increase in total membrane capacitance reflected gains in both linear and nonlinear components (Fig. 4C). The gradual growth in linear capacitance from 2 to 6 weeks of age suggested a delay in the maturation of cell size in these animals compared with wild type. To address this possibility, OHCs were mechanically isolated from apical cochlear turns of P15–17 mice, and cell size was measured from captured digital images. Cell length and width for wild-type OHCs were 27.9 ± 0.8 and 6.8 ± 0.3 μm (N = 13), respectively, whereas these measurements in Pit1dw OHCs were 21.3 ± 1.7 and 7.5 ± 0.5 μm (N = 5), respectively. The 24% difference in cell length is statistically significant (p < 0.01). Mean cell surface area was estimated from average length and width measurements, considering the OHC as a simple cylinder. The mean surface area of the wild-type OHCs was 1272 μm2, whereas that for Pit1dw OHCs was 1085 μm2, a reduction of ∼15%. This size difference is similar to the difference in linear capacitance between mutant and wild-type cells at a similar age (18%) (Fig. 4C). Nonlinear capacitance also increased with age in the mutant OHCs. The developmental change in nonlinear capacitance outpaced increases in linear capacitance, suggesting that increases in the nonlinear component must be attributed, at least in part, to a greater density and/or efficacy of prestin in the OHC membrane. These physiological tests reveal delayed maturation of prestin function in adult Pit1dw mutants, consistent with the evidence that adult mutants eventually develop appropriate levels and subcellular localization of prestin protein. Thus, the lack of DPOAEs and CM in Pit1dw mice is not explained by alterations in either nonlinear capacitance or prestin expression and localization. It remains possible, however, that electromotility is compromised in Pit1dw mice through alterations in cytoskeletal structures essential to motor function or in the intrinsic electrical properties of the cell.
Reduced KCNQ4 protein expression specifically in OHC of Pit1dw mutants
The voltage-dependent K+ channel KCNQ4 is responsible for the dominant K+ conductance, IK,n, of mature OHCs (Marcotti and Kros, 1999). KCNQ4 sets the membrane resting potential in cochlear hair cells, and loss of KCNQ4 may cause chronic stress for the cells and lead to their degeneration (Kharkovets et al., 2006). We examined expression of KCNQ4 in the cochlear and vestibular systems using immunohistochemical staining. In normal mice the KCNQ4 immunoreactivity is distributed across the entire OHC membrane during the first few days after birth, eventually shifting from the basolateral surface to the basal pole of the cell by P12-P13 (data not shown). The level of KCNQ4 immunoreactivity in Pit1dw mutants was reduced relative to wild type from P12 through P42 (Fig. 5A). The low level of KCNQ4 immunoreactivity appeared in a normal subcellular pattern. This differs from some rodent models of hypothyroidism that completely lack detectable KCNQ4 expression in OHCs at P12-P13 (Winter et al., 2006). We observed a developmental delay in KCNQ4 expression that followed a basal-apical gradient with expression levels in apical OHCs lagging behind those from the base (data not shown). In striking contrast to the OHC, the level of KCNQ4 expression within the spiral ganglion and vestibular organs of Pit1dw mutant inner ears was indistinguishable from that of wild-type mice at all ages tested (P13, P21, P42 and P60) (supplemental Figure S. 3A, available at www.jneurosci.org as supplemental material). This suggests that the permanent reduction in KCNQ4 expression is specific to the OHC in Pit1dw mutants.
Figure 5.

KCNQ4 protein expression and function are permanently reduced in the cochlear OHC of Pit1dw mutant mice. A, KCNQ4 immunoreactivity is reduced in OHCs (arrows) of mutant mice relative to wild type. Frozen sections obtained from P13, P21, and P42 wild-type and mutant mice were stained for KCNQ4 (green). Nuclei were stained with DAPI (blue). Outer hair cells from Pit1dw mice exhibit a nonmonotonic developmental increase in KCNQ4 currents. B, Normalized tail-currents are shown for 2-week wild-type mice and 2-, 4-, and 6-week-old mutants. Wild-type OHCs uniformly exhibited a dominant low-voltage activated potassium conductance attributed to KCNQ4 function. Mutant OHCs were heterogeneous, regardless of age, in that some cells had KCNQ4 while others did not. C, Average maximum tail-current for low-voltage activated components of the Boltzman fits is shown for wild-type and mutant OHCs. Average currents were lower in mutants than wild type (p < 0.05). Also, there was a statistically reliable effect of age on average KCNQ4 current using a one-way ANOVA (p < 0.01).
Reduced KCNQ4 currents in Pit1dw mutants
To assess the functional consequences of the permanent reduction of KCNQ4 expression in mutants relative to normal OHC, we measured KCNQ4 currents during development. Voltage activation curves were constructed from tail current analysis and fit with Boltzman functions (Eq. 2). Whole-cell currents in wild-type OHCs were dominated by a low-voltage activated conductance attributed to KCNQ4 channels (Marcotti and Kros, 1999). In the presence of linopirdine, a KCNQ4 blocker, total outward current was reduced due to the elimination of IK,n, revealing a residual high-voltage activated potassium current (supplemental Figure S. 2, available at www.jneurosci.org as supplemental material). The shapes of activation curves from Pit1dw mutant mice were heterogeneous suggesting large variations in the amount of KCNQ4-related conductance. While all cells from 2-week-old wild-type mice exhibited a dominant low-voltage activated KCNQ4 current, mutant OHCs, regardless of age, possessed varying mixtures of low- and high-voltage activated conductances (Fig. 5B). Occasionally, Pit1dw mutant OHCs were best fit by a single Boltzman with a high V1/2 (above −20 mV), indicating the complete absence of measurable KCNQ4 current in those cells (4 of 17 cells in P15–17, 0 of 10 in P28–32, 1 of 18 in P40–45). To compare the amount of KCNQ4 between experimental groups, we determined the maximum tail current from Boltzman fits to the low-voltage activated components in Figure 5B. This is an approximate measure of KCNQ4 current density since the sizes of all cells in this study were comparable (Fig. 4C). The data are plotted for each cell and as averages in Figure 5C. Maximum KCNQ4 current was reduced in all mutant age groups compared with wild type (p < 0.05). In contrast to results for nonlinear capacitance, the KCNQ4 current in mutants did not steadily increase toward mature wild-type levels. Instead, KCNQ4 current in the mutant animals peaked at 4 weeks of age (P28–35). Since KCNQ4 channels are active at negative membrane potentials, loss of KCNQ4 conductance would result in depolarization of the resting membrane potential. Current-clamp was used to measure resting potential in several OHCs from wild-type and mutant mice. The OHCs from mutant mice at 6 weeks of age were depolarized by 10 to 15 mV compared with OHCs from 2-week-old wild-type mice (data not shown).
Substantially reduced endocochlear potential in Pit1dw mice
The EP is the main driving force for the sensory transduction that leads to perception of sound. The potential is generated across the basal cell barrier of the stria vascularis by the K+ channel KCNJ10, which is located in the intermediate cells (Marcus et al., 2002; Rozengurt et al., 2003; Wangemann et al., 2004). We found that the EP in 6-week-old wild-type mice ranged from 105 to 113 mV (N = 2), while the EP in Pit1dw mutant mice was reduced by 45%, ranging from 48 to 50 mV (N = 3) (Fig. 6A). Similarly, the EP of 3-week-old mutants is 50% of wild type (data not shown). The abnormal EP contributes to the lack of DPOAE and CM and indicates that a cochlear defect of peripheral origin is involved in the deafness characteristic of the Pit1dw mutants.
Figure 6.

Endocochlear potential and KCNJ10 expression are lower in Pit1dw mutants than wild type. A, EP were measured in P42 wild-type and age-matched mutant animals. Representative plots of endocochlear potential (in mV) for littermate control and Pit1dw mutant animals are shown. The time depicted includes electrode insertion into and withdrawal from the endolymph. B, Frozen sections of the organ of Corti of wild-type and mutant mice collected at P21 and P42 were stained for KCNJ10 (red) (data shown for P42 only). Nuclei (blue). KCNJ10 immunoreactivity is detected in the intermediate cells of the stria vascularis of these mice.
Reduced KCNJ10 protein expression in Pit1dw mutants is specific to the stria vascularis
The stria vascularis contains marginal cells, intermediate cells and basal cells. Defects in any of these cells can result in hearing deficits (Marcus et al., 2002; Jabba et al., 2006; Knipper et al., 2006; Jin et al., 2008). To investigate the cause of the reduced EP in Pit1dw mutant mice, we examined the expression of the K+ channels KCNQ1 and KCNJ10 in marginal and intermediate cells, respectively, using immunohistochemical staining. The Pit1dw mutant mice have reduced KCNJ10 immunoreactivity in both apical and basal cochlear turns compared with wild-type mice at P21 (data not shown) and P42 (Fig. 6B). The stria of the mutants appears smaller in width relative to wild type at both ages, and the smaller size seemed attributable to diminished contribution of intermediate cells. KCNJ10 expression is normal in Pit1dw mutant spiral ganglion and vestibular system at P21 and P42, revealing that the KCNJ10 deficiency is specific to the stria (supplemental Figure S. 3B, available at www.jneurosci.org as supplemental material). KCNQ1 is important for marginal cell function and maintenance of stria (Rivas and Francis, 2005). No obvious differences in KCNQ1 expression are noted at P21 or P42 (data not shown). Thus, reduced KCNJ10 expression is likely to be a major contributor to the lowered EP observed in Pit1dw mutants, as observed in the KCNJ10 knock-out (Marcus et al., 2002; Rozengurt et al., 2003; Wangemann et al., 2004).
Stria vascularis pathology in adult Pit1dw mutants
We examined the stria vascularis at various ages in wild-type and mutant mice using light microscopy of hematoxylin and eosin stained paraffin sections (data not shown) and TEM (Fig. 7). Ultrastructure analysis reveals abnormalities in the stria that are more obvious in mutants at P42 (N = 3 per genotype and age) (Fig. 7A), than at P12 or P21 (data not shown). The width of the stria is consistently smaller in the mutant than the wild type. Marginal, intermediate, and basal cells all appear to be present, but there is less interdigitation of the intermediate cells with the basal aspect of the marginal cells and less infolding of the basolateral membrane. The smaller size of the mutant stria appears to come from reduced contribution of intermediate cells, consistent with reduced KCNJ10 immunostaining (Fig. 6). In addition, the TEM analysis reveals prominent accumulation of dark deposits in the stria vascularis of P42 old Pit1dw mutants with little or no deposits in wild-type littermates (Fig. 7B). The lipofuscin-like deposits (aging pigment) are found in all cell types of the stria, but are clearly most abundant in the central region of the stria, consistent with intermediate cells. The presence of this pathology suggests that cells in the stria vascularis of Pit1dw mutants are undergoing deterioration.
Figure 7.
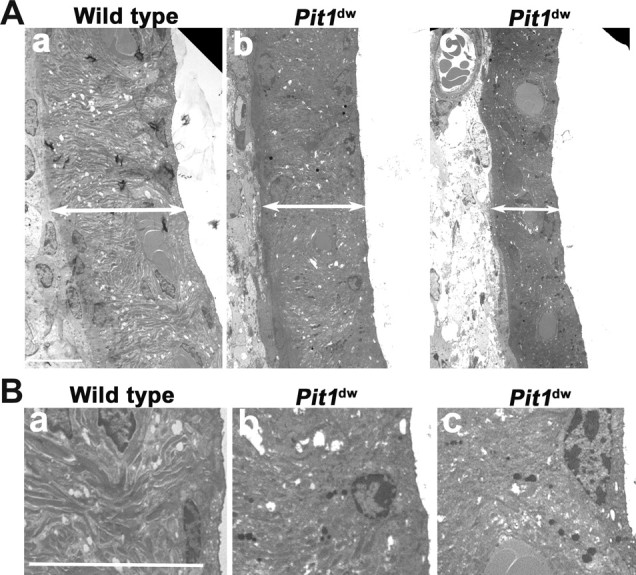
Stria vascularis pathology in adult Pit1 mutants. A, Stria vascularis prepared from P42 wild-type mice (a) and mutant mice (b, c) were analyzed by TEM. Double-headed arrow bars define the width of the stria vascularis. B, Lipofuscin granules are more abundant in the intermediate cells of Pit1dw mutant mice, suggesting that the mutant cells are exhibiting signs of aging. The ultrastructure of wild-type (a) stria vascularis animals was compared with Pit1dw mutants (b, c). Lipofuscin-like granules are more abundant in the mutants. Sections shown are from the basal turns of the cochlea, which were more prominently affected than the apex. Scale bars: 10 μm.
Discussion
Congenital hypothyroidism impairs multiple developmental processes within the inner ear, offering a mechanistic explanation for hypothyroidism-induced deafness. We used Pit1dw/dw as a model of secondary hypothyroidism. These mice have profound congenital deafness but normal balance. We found no evidence of Pit1 expression in the cochlea using multiple approaches indicating that the hearing impairment in Pit1 mutants is caused by the lack of Pit1 activity in the pituitary gland (data not shown).
Pit1dw/dw have abnormalities in TM composition and ultrastructure, endolymph ionic balance, OHC survival, and strial cell health. These features contribute to the lack of DPOAE, CM, and the reduced EP, which can be attributed to long-term defects in OHC KCNQ4 expression and strial KCNJ10 expression. The mutant OHCs eventually exhibit normal prestin function and expression, but the defects in KCNQ4 are permanent. The strial intermediate cells exhibit persistently low KCNJ10 expression, resulting in ionic balance defects that likely contribute to the reduced EP, OHC dysfunction, and sporadic OHC death. The profound hearing impairment characteristic of Pit1dw/dw may result from compounding effects of several hormone-dependent processes.
Pit1dw/dw have tectorial membrane defects
Pit1dw/dw TM abnormalities include increased size of Hensen's stripe and elevation of TECTB composition. Tectb−/− lack Hensen's stripe, suggesting that TECTB is required to develop this structure (Russell et al., 2007). Thus, the increased TECTB in Pit1dw/dw could contribute to their prominent Hensen's stripe. While TSH-deficient Cgatm1/1tm1 also have a prominent Hensen's stripe (Karolyi et al., 2007), TECTB is decreased in pharmacologically induced hypothyroidism (Knipper et al., 2001). TM composition and the prominence of Hensen's stripe have not been assessed quantitatively in other models with impaired TH action, but ultrastructural abnormalities of the TM are common (Rusch et al., 2001; Richardson et al., 2008). The significance of altered TM composition and ultrastructure for OHC function is not clear (Knipper et al., 2001; Rusch et al., 2001; Richardson et al., 2008; Song et al., 2008). Mutations in individual TM components, Tecta, Tectb, and Otog, do not cause profound, congenital deafness characteristic of Pit1dw/dw, although multiple TM defects could have a compounding effect (Legan et al., 2000, 2005; Simmler et al., 2000; Russell et al., 2007).
TH is not required for prestin expression or function
Prestin is a motor protein that underlies OHC motility. TH regulates its transcription, and hypothyroidism causes abnormalities in prestin expression and subcellular distribution (Rusch et al., 2001; Weber et al., 2002; Winter et al., 2006). These abnormalities are transient in Pit1dw/dw, representing delayed development that eventually matures. While data on prestin distribution and function in older drug treated and TR mutant animals are unavailable, we predict that prestin activity will mature functionally in pharmacologically treated animals as it does in Pit1dw/dw. OHC motility may be compromised by observed defects in cytoskeletal specializations that support motor movement, but it seems unlikely that deficits in OHC motor function account completely for the absence of Pit1dw/dw DPOAEs and CM.
Maintenance of healthy cochlear cells is hormone dependent
KCNQ4 expression and current is detectable in Pit1dw/dw OHCs throughout adulthood, and the developmental profile of KCNQ4 currents is qualitatively similar to normal mice (Beisel et al., 2005). The persistently reduced KCNQ4 expression and function in Pit1dw/dw contrasts with the eventual maturation of prestin expression and function. Abnormal KCNQ4 expression results in chronic depolarization of Pit1dw/dw OHCs, which may lead to the untimely demise of OHCs that we observed in 6-week-old Pit1dw/dw. The level of depolarization we observed in Pit1dw/dw OHCs was similar to that of Kcnq4−/− (Kharkovets et al., 2006). Mice with altered KCNQ4 conductance, through pharmacological block or genetic disruption, exhibit progressive OHC loss (Nouvian et al., 2003). This supports the idea that sporadic OHC loss occurs concomitant with reduced KCNQ4 conductance and a depolarized resting potential. We considered whether reduced Igf1 levels might cause OHC loss because complete lack of IGF1 causes OHC loss and severe hearing impairment (Camarero et al., 2001; Cediel et al., 2006). We concluded that the OHC loss in deaf Pit1dw/dw is not likely to be attributable to the reduced IGF1 levels because both Pit1dw/dw and normal-hearing Igf1 +/− animals have a ∼50% reduction in circulating IGF1 (Woods and Savage, 1996; Le Roith et al., 2001).
The OHC loss in Pit1dw/dw is unlikely to account for their profound congenital deafness because the timing is late and the fraction of hair cell death is small (Bohne et al., 1990). Stria vascularis dysfunction is likely to be an important contributor to the deafness. Normal EP is required for OHC function, including the generation of DPOAE and CM. Pit1dw/dw have approximately half the normal EP. Persistent reduction in KCNJ10 expression in the stria vascularis contributes to their low EP. This conclusion is supported by the observations that Kcnj10−/− mice lack EP and have compromised hearing (Marcus et al., 2002) and Slc26a4−/−, a euthyroid model of Pendred syndrome, lack both EP and KCNJ10 (Rusch et al., 2001; Wangemann et al., 2004). We predict that KCNJ10 expression is reduced in the stria vascularis of hypothyroid and TR mutant animals. This would be expected to reduce, but not eliminate, DPOAE and ABR responses (Gow et al., 2004). Thus, the absence of DPOAE in Pit1dw/dw arises from a combination of several deficits.
Pit1dw/dw stria are small with abnormalities in ultrastructure including accumulation of lipofuscin-like deposits. The decreased stria size in Pit1dw/dw appears independent of KCNJ10 because complete absence of KCNJ10 in Slc26a4 and Kcnj10 mutants does not cause this morphological effect (Marcus et al., 2002; Wangemann et al., 2004). Lipofuscin accumulation accelerates in aging cochleae (Nadol, 1979; Kazee and West, 1999), brains of Alzheimer's and Parkinson's patients (Eichhoff et al., 2008), and eyes of patients with atrophic age-related macular degeneration (Holz et al., 1999; Schmitz-Valckenberg et al., 2004). Thus, the stria vascularis pathology suggests that Pit1dw/dw cells are undergoing changes associated with physiological stress or premature aging. This is surprising because Pit1dw/dw and animals with GH defects have reduced cellular stress and enhanced longevity (Leiser et al., 2006). The etiology of the stria pathology in Pit1dw/dw is not known, but stria development and maintenance are hormone-dependent.
Many genetic defects disrupt development and function of both the vestibular apparatus and the cochlea, yet hypothyroidism only affects the cochlea. The basis for this specificity is unknown. Intriguingly, the alteration in KCNQ4 and KCNJ10 expression in Pit1dw/dw is confined to OHCs and stria, respectively. This cell type specific dysregulation suggests that the same genes have different requirements for hormonal regulation in various cell types within the same organ. This could be achieved by many mechanisms, such as utilization of different cell-type specific transcription components and/or deiodinases. Alternatively, variation in the critical timing for hormone exposure could underlie differences in hormone sensitivity, where cells differentiating early benefit from maternal hormones during gestation, while cell types differentiating later depend on neonatal hormone production.
Why are Pit1dw/dw profoundly deaf?
Thyroid hormone regulates many processes in the developing cochlea. Individual defects in TM, DPOAE, CM, EP, prestin, Kcnq4, and Kcnj10 are consistent with progressive and/or moderate to severe hearing impairment. The profound hearing impairment observed in Pit1dw/dw could be explained by a compounding effect of genes that individually have moderate effects or there could be additional processes involved. Given the broad effects of TH on gene expression, other processes, such as IHC function, neurotransmission, and innervation, are likely compromised (Brandt et al., 2007; Sendin et al., 2007).
Advantages of the Pit1dw/dw model
The advantages of studying Pit1dw/dw are their viability and responsiveness to TH supplementation, which makes it possible to distinguish the aspects of TH deficiency that lead to permanent hearing defects from those that cause developmental delay. For example, the developmental delay in prestin expression is not permanent, but the alterations in the TM and defective KCNJ10 and KCNQ4 expression and function are. In addition, Pit1dw/dw reveal the importance of TH for stria vascularis and OHC function and for avoiding pathological changes and cell death. The prominence of hypothyroidism and presbycusis in older individuals suggests that our studies may be important for understanding the mechanisms contributing to age related hearing loss (ARHL).
Is hypothyroidism a risk factor presbycusis?
There are several lines of evidence that suggest hormone deficiency may be a risk factor for presbycusis. OHC loss and strial abnormalities of the Pit1dw/dw cochleae are also found in cases of age-related and environmentally induced hearing loss. In addition, the Pit1dw/dw exhibit reduced Kcnq4 expression, a gene mutated in autosomal dominant hearing loss is also associated with presbycusis (Van Eyken et al., 2006).
Hypothyroidism is common in older individuals (Fransen et al., 2003) and GH production decreases with age (Lanfranco et al., 2003; Bartke, 2005). Since hormones are necessary for cochlear cell survival and function in young mice, it is possible that reduced hormone production in adults is a risk factor for ARHL, or that individuals born from mothers with hypothyroidism during pregnancy could be predisposed to ARHL. While this is speculative, the mouse could be an excellent tool to investigate genetic and environmental modifiers of ARHL (Johnson and Zheng, 2002; Nemoto et al., 2004).
Footnotes
This work was supported in part by the Deafness Research Foundation (M.M.), March of Dimes Grant 6-FY08-262 (S.A.C.), National Institutes of Health Grants DC05053 (S.A.C.), P30 DC05188 (D.F.D.), and DC05401 (Y.R.), the Margaret G. Bertsch Research Endowment (Margaret I. Lomax), and the University of Michigan Office of the Vice President for Research, Biomedical Research Council, and a Clinical and Translational Science Award (S.A.C.). The immunohistochemical analyses by whole mount and sectioning, and animal colony maintenance were done by M.M. and Q.F. DPOAE, CM, and EP were done by D.F.D. T.-W.G. contributed to the study of OHC loss. Y.R. analyzed the TEM. Nonlinear capacitance and KCNQ4 current measurements were done by R.K.D. Conceptualization of the experimental design and preparation of this manuscript was done by M.M., S.A.C., and R.K.D. We thank the following individuals for their important contributions to this work: Margaret I. Lomax for intellectual contributions and financial support, Alicia Giordimaina and Albert Chow for statistical analysis, Lisa A. Beyer for TEM and scanning electron microscopy, Karin Halsey and Jennifer Benson for physiological tests (DPOAE, CM and EP), Dr. Hassan Chaib for Western blot analysis, Dr. Guy Richardson for the β-tectorin antibody, and Dr. Jentsch for the KCNQ4 antibody.
References
- Abe S, Katagiri T, Saito-Hisaminato A, Usami S, Inoue Y, Tsunoda T, Nakamura Y. Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am J Hum Genet. 2003;72:73–82. doi: 10.1086/345398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Kakehata S, Kitani R, Maruya S, Navaratnam D, Santos-Sacchi J, Shinkawa H. Developmental expression of the outer hair cell motor prestin in the mouse. J Membr Biol. 2007;215:49–56. doi: 10.1007/s00232-007-9004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke A. Minireview: role of the growth hormone/insulin-like growth factor system in mammalian aging. Endocrinology. 2005;146:3718–3723. doi: 10.1210/en.2005-0411. [DOI] [PubMed] [Google Scholar]
- Beisel KW, Rocha-Sanchez SM, Morris KA, Nie L, Feng F, Kachar B, Yamoah EN, Fritzsch B. Differential expression of KCNQ4 in inner hair cells and sensory neurons is the basis of progressive high-frequency hearing loss. J Neurosci. 2005;25:9285–9293. doi: 10.1523/JNEUROSCI.2110-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer LA, Odeh H, Probst FJ, Lambert EH, Dolan DF, Camper SA, Kohrman DC, Raphael Y. Hair cells in the inner ear of the pirouette and shaker 2 mutant mice. J Neurocytol. 2000;29:227–240. doi: 10.1023/a:1026515619443. [DOI] [PubMed] [Google Scholar]
- Bohne BA, Gruner MM, Harding GW. Morphological correlates of aging in the chinchilla cochlea. Hear Res. 1990;48:79–91. doi: 10.1016/0378-5955(90)90200-9. [DOI] [PubMed] [Google Scholar]
- Brandt N, Kuhn S, Münkner S, Braig C, Winter H, Blin N, Vonthein R, Knipper M, Engel J. Thyroid hormone deficiency affects postnatal spiking activity and expression of Ca2+ and K+ channels in rodent inner hair cells. J Neurosci. 2007;27:3174–3186. doi: 10.1523/JNEUROSCI.3965-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Camarero G, Avendano C, Fernandez-Moreno C, Villar A, Contreras J, de Pablo F, Pichel JG, Varela-Nieto I. Delayed inner ear maturation and neuronal loss in postnatal Igf-1-deficient mice. J Neurosci. 2001;21:7630–7641. doi: 10.1523/JNEUROSCI.21-19-07630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camper SA, Saunders TL, Katz RW, Reeves RH. The Pit-1 transcription factor gene is a candidate for the Snell dwarf mutation. Genomics. 1990;8:586–590. doi: 10.1016/0888-7543(90)90050-5. [DOI] [PubMed] [Google Scholar]
- Cantos R, López DE, Merchán JA, Rueda J. Olivocochlear efferent innervation of the organ of corti in hypothyroid rats. J Comp Neurol. 2003;459:454–467. doi: 10.1002/cne.10620. [DOI] [PubMed] [Google Scholar]
- Cediel R, Riquelme R, Contreras J, Díaz A, Varela-Nieto I. Sensorineural hearing loss in insulin-like growth factor I-null mice: a new model of human deafness. Eur J Neurosci. 2006;23:587–590. doi: 10.1111/j.1460-9568.2005.04584.x. [DOI] [PubMed] [Google Scholar]
- Cheatham MA, Huynh KH, Gao J, Zuo J, Dallos P. Cochlear function in Prestin knockout mice. J Physiol. 2004;560:821–830. doi: 10.1113/jphysiol.2004.069559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ S, Biebel UW, Hoidis S, Friedrichsen S, Bauer K, Smolders JW. Hearing loss in athyroid pax8 knockout mice and effects of thyroxine substitution. Audiol Neurootol. 2004;9:88–106. doi: 10.1159/000076000. [DOI] [PubMed] [Google Scholar]
- Dallos P, Wu X, Cheatham MA, Gao J, Zheng J, Anderson CT, Jia S, Wang X, Cheng WH, Sengupta S, He DZ, Zuo J. Prestin-based outer hair cell motility is necessary for mammalian cochlear amplification. Neuron. 2008;58:333–339. doi: 10.1016/j.neuron.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deol MS. Congenital deafness and hypothyroidism. Lancet. 1973;2:105–106. doi: 10.1016/s0140-6736(73)93310-2. [DOI] [PubMed] [Google Scholar]
- Eichhoff G, Busche MA, Garaschuk O. In vivo calcium imaging of the aging and diseased brain. Eur J Nucl Med Mol Imaging. 2008;(Suppl 1):S99–S106. doi: 10.1007/s00259-007-0709-6. [DOI] [PubMed] [Google Scholar]
- Fransen E, Lemkens N, Van Laer L, Van Camp G. Age-related hearing impairment (ARHI): environmental risk factors and genetic prospects. Exp Gerontol. 2003;38:353–359. doi: 10.1016/s0531-5565(03)00032-9. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Brinkmeier ML, Scarlett LM, Knapp LT, Camper SA, Mahon KA. The Ames dwarf gene, df, is required early in pituitary ontogeny for the extinction of Rpx transcription and initiation of lineage specific cell proliferation. Mol Endocrinol. 1996;10:1570–1581. doi: 10.1210/mend.10.12.8961267. [DOI] [PubMed] [Google Scholar]
- Gao J, Wang X, Wu X, Aguinaga S, Huynh K, Jia S, Matsuda K, Patel M, Zheng J, Cheatham M, He DZ, Dallos P, Zuo J. Prestin-based outer hair cell electromotility in knockin mice does not appear to adjust the operating point of a cilia-based amplifier. Proc Natl Acad Sci U S A. 2007;104:12542–12547. doi: 10.1073/pnas.0700356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey P, Rahal JO, Beamer WG, Copeland NG, Jenkins NA, Mayo KE. GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nat Genet. 1993;4:227–232. doi: 10.1038/ng0793-227. [DOI] [PubMed] [Google Scholar]
- Gow A, Davies C, Southwood CM, Frolenkov G, Chrustowski M, Ng L, Yamauchi D, Marcus DC, Kachar B. Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J Neurosci. 2004;24:7051–7062. doi: 10.1523/JNEUROSCI.1640-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz FG, Schütt F, Kopitz J, Völcker HE. [Introduction of the lipofuscin-fluorophor A2E into the lysosomal compartment of human retinal pigment epithelial cells by coupling to LDL particles. An in vitro model of retinal pigment epithelium cell aging] Ophthalmologe. 1999;96:781–785. doi: 10.1007/s003470050496. [DOI] [PubMed] [Google Scholar]
- Huang G, Santos-Sacchi J. Mapping the distribution of the outer hair cell motility voltage sensor by electrical amputation. Biophys J. 1993;65:2228–2236. doi: 10.1016/S0006-3495(93)81248-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabba SV, Oelke A, Singh R, Maganti RJ, Fleming S, Wall SM, Everett LA, Green ED, Wangemann P. Macrophage invasion contributes to degeneration of stria vascularis in Pendred syndrome mouse model. BMC Med. 2006;4:37. doi: 10.1186/1741-7015-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Ulfendahl M, Jarlebark L. Spatiotemporal loss of K+ transport proteins in the developing cochlear lateral wall of guinea pigs with hereditary deafness. Eur J Neurosci. 2008;27:145–154. doi: 10.1111/j.1460-9568.2007.05994.x. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY. Ahl2, a second locus affecting age-related hearing loss in mice. Genomics. 2002;80:461–464. [PMC free article] [PubMed] [Google Scholar]
- Karolyi IJ, Dootz GA, Halsey K, Beyer L, Probst FJ, Johnson KR, Parlow AF, Raphael Y, Dolan DF, Camper SA. Dietary thyroid hormone replacement ameliorates hearing deficits in hypothyroid mice. Mamm Genome. 2007;18:596–608. doi: 10.1007/s00335-007-9038-0. [DOI] [PubMed] [Google Scholar]
- Kazee AM, West NR. Preservation of synapses on principal cells of the central nucleus of the inferior colliculus with aging in the CBA mouse. Hear Res. 1999;133:98–106. doi: 10.1016/s0378-5955(99)00058-1. [DOI] [PubMed] [Google Scholar]
- Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006;25:642–652. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipper M, Bandtlow C, Gestwa L, Köpschall I, Rohbock K, Wiechers B, Zenner HP, Zimmermann U. Thyroid hormone affects Schwann cell and oligodendrocyte gene expression at the glial transition zone of the VIIIth nerve prior to cochlea function. Development. 1998;125:3709–3718. doi: 10.1242/dev.125.18.3709. [DOI] [PubMed] [Google Scholar]
- Knipper M, Zinn C, Maier H, Praetorius M, Rohbock K, Köpschall I, Zimmermann U. Thyroid hormone deficiency before the onset of hearing causes irreversible damage to peripheral and central auditory systems. J Neurophysiol. 2000;83:3101–3112. doi: 10.1152/jn.2000.83.5.3101. [DOI] [PubMed] [Google Scholar]
- Knipper M, Richardson G, Mack A, Müller M, Goodyear R, Limberger A, Rohbock K, Köpschall I, Zenner HP, Zimmermann U. Thyroid hormone-deficient period prior to the onset of hearing is associated with reduced levels of beta-tectorin protein in the tectorial membrane: implication for hearing loss. J Biol Chem. 2001;276:39046–39052. doi: 10.1074/jbc.M103385200. [DOI] [PubMed] [Google Scholar]
- Knipper M, Claussen C, Ruttiger L, Zimmermann U, Lullmann-Rauch R, Eskelinen EL, Schroder J, Schwake M, Saftig P. Deafness in LIMP2-deficient mice due to early loss of the potassium channel KCNQ1/KCNE1 in marginal cells of the stria vascularis. J Physiol. 2006;576:73–86. doi: 10.1113/jphysiol.2006.116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranco F, Gianotti L, Giordano R, Pellegrino M, Maccario M, Arvat E. Ageing, growth hormone and physical performance. J Endocrinol Invest. 2003;26:861–872. doi: 10.1007/BF03345237. [DOI] [PubMed] [Google Scholar]
- Legan PK, Lukashkina VA, Goodyear RJ, Kössi M, Russell IJ, Richardson GP. A targeted deletion in alpha-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron. 2000;28:273–285. doi: 10.1016/s0896-6273(00)00102-1. [DOI] [PubMed] [Google Scholar]
- Le Roith D, Scavo L, Butler A. What is the role of circulating IGF-I? Trends Endocrinol Metab. 2001;12:48–52. doi: 10.1016/s1043-2760(00)00349-0. [DOI] [PubMed] [Google Scholar]
- Legan PK, Lukashkina VA, Goodyear RJ, Lukashkin AN, Verhoeven K, Van Camp G, Russell IJ, Richardson GP. A deafness mutation isolates a second role for the tectorial membrane in hearing. Nat Neurosci. 2005;8:1035–1042. doi: 10.1038/nn1496. [DOI] [PubMed] [Google Scholar]
- Leiser SF, Salmon AB, Miller RA. Correlated resistance to glucose deprivation and cytotoxic agents in fibroblast cell lines from long-lived pituitary dwarf mice. Mech Ageing Dev. 2006;127:821–829. doi: 10.1016/j.mad.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Li D, Henley CM, O'Malley BW., Jr Distortion product otoacoustic emissions and outer hair cell defects in the hyt/hyt mutant mouse. Hear Res. 1999;138:65–72. doi: 10.1016/s0378-5955(99)00150-1. [DOI] [PubMed] [Google Scholar]
- Li S, Crenshaw EB, 3rd, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature. 1990;347:528–533. doi: 10.1038/347528a0. [DOI] [PubMed] [Google Scholar]
- Li S, Price SM, Cahill H, Ryugo DK, Shen MM, Xiang M. Hearing loss caused by progressive degeneration of cochlear hair cells in mice deficient for the Barhl1 homeobox gene. Development. 2002;129:3523–3532. doi: 10.1242/dev.129.14.3523. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Gao J, He DZ, Wu X, Jia S, Zuo J. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature. 2002;419:300–304. doi: 10.1038/nature01059. [DOI] [PubMed] [Google Scholar]
- Lin SC, Lin CR, Gukovsky I, Lusis AJ, Sawchenko PE, Rosenfeld MG. Molecular basis of the little mouse phenotype and implications for cell type-specific growth. Nature. 1993;364:208–213. doi: 10.1038/364208a0. [DOI] [PubMed] [Google Scholar]
- Marcotti W, Kros CJ. Developmental expression of the potassium current IK,n contributes to maturation of mouse outer hair cells. J Physiol. 1999;520:653–660. doi: 10.1111/j.1469-7793.1999.00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol. 2002;282:C403–C407. doi: 10.1152/ajpcell.00312.2001. [DOI] [PubMed] [Google Scholar]
- Mustapha M, Beyer LA, Izumikawa M, Swiderski DL, Dolan DF, Raphael Y, Camper SA. Whirler mutant hair cells have less severe pathology than shaker 2 or double mutants. J Assoc Res Otolaryngol. 2007;8:329–337. doi: 10.1007/s10162-007-0083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadol JB., Jr Electron microscopic findings in presbycusic degeneration of the basal turn of the human cochlea. Otolaryngol Head Neck Surg. 1979;87:818–836. doi: 10.1177/019459987908700617. [DOI] [PubMed] [Google Scholar]
- Nemoto M, Morita Y, Mishima Y, Takahashi S, Nomura T, Ushiki T, Shiroishi T, Kikkawa Y, Yonekawa H, Kominami R. Ahl3, a third locus on mouse chromosome 17 affecting age-related hearing loss. Biochem Biophys Res Commun. 2004;324:1283–1288. doi: 10.1016/j.bbrc.2004.09.186. [DOI] [PubMed] [Google Scholar]
- Nouvian R, Ruel J, Wang J, Guitton MJ, Pujol R, Puel JL. Degeneration of sensory outer hair cells following pharmacological blockade of cochlear KCNQ channels in the adult guinea pig. Eur J Neurosci. 2003;17:2553–2562. doi: 10.1046/j.1460-9568.2003.02715.x. [DOI] [PubMed] [Google Scholar]
- Oliver D, Fakler B. Expression density and functional characteristics of the outer hair cell motor protein are regulated during postnatal development in rat. J Physiol. 1999;519:791–800. doi: 10.1111/j.1469-7793.1999.0791n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patuzzi RB, Yates GK, Johnstone BM. Changes in cochlear microphonic and neural sensitivity produced by acoustic trauma. Hear Res. 1989;39:189–202. doi: 10.1016/0378-5955(89)90090-7. [DOI] [PubMed] [Google Scholar]
- Richardson GP, Lukashkin AN, Russell IJ. The tectorial membrane: one slice of a complex cochlear sandwich. Curr Opin Otolaryngol Head Neck Surg. 2008;16:458–464. doi: 10.1097/MOO.0b013e32830e20c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas A, Francis HW. Inner ear abnormalities in a Kcnq1 (Kvlqt1) knockout mouse: a model of Jervell and Lange-Nielsen syndrome. Otol Neurotol. 2005;26:415–424. doi: 10.1097/01.mao.0000169764.00798.84. [DOI] [PubMed] [Google Scholar]
- Rozengurt N, Lopez I, Chiu CS, Kofuji P, Lester HA, Neusch C. Time course of inner ear degeneration and deafness in mice lacking the Kir4.1 potassium channel subunit. Hear Res. 2003;177:71–80. doi: 10.1016/s0378-5955(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Rueda J, Prieto JJ, Cantos R, Sala ML, Merchán JA. Hypothyroidism prevents developmental neuronal loss during auditory organ development. Neurosci Res. 2003;45:401–408. doi: 10.1016/s0168-0102(03)00009-9. [DOI] [PubMed] [Google Scholar]
- Rusch A, Erway LC, Oliver D, Vennstrom B, Forrest D. Thyroid hormone receptor beta-dependent expression of a potassium conductance in inner hair cells at the onset of hearing. Proc Natl Acad Sci U S A. 1998;95:15758–15762. doi: 10.1073/pnas.95.26.15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch A, Ng L, Goodyear R, Oliver D, Lisoukov I, Vennstrom B, Richardson G, Kelley MW, Forrest D. Retardation of cochlear maturation and impaired hair cell function caused by deletion of all known thyroid hormone receptors. J Neurosci. 2001;21:9792–9800. doi: 10.1523/JNEUROSCI.21-24-09792.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell IJ, Legan PK, Lukashkina VA, Lukashkin AN, Goodyear RJ, Richardson GP. Sharpened cochlear tuning in a mouse with a genetically modified tectorial membrane. Nat Neurosci. 2007;10:215–223. doi: 10.1038/nn1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Valckenberg S, Bültmann S, Dreyhaupt J, Bindewald A, Holz FG, Rohrschneider K. Fundus autofluorescence and fundus perimetry in the junctional zone of geographic atrophy in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:4470–4476. doi: 10.1167/iovs.03-1311. [DOI] [PubMed] [Google Scholar]
- Sendin G, Bulankina AV, Riedel D, Moser T. Maturation of ribbon synapses in hair cells is driven by thyroid hormone. J Neurosci. 2007;27:3163–3173. doi: 10.1523/JNEUROSCI.3974-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmler MC, Cohen-Salmon M, El-Amraoui A, Guillaud L, Benichou JC, Petit C, Panthier JJ. Targeted disruption of otog results in deafness and severe imbalance. Nat Genet. 2000;24:139–143. doi: 10.1038/72793. [DOI] [PubMed] [Google Scholar]
- Song L, McGee J, Walsh EJ. The influence of thyroid hormone deficiency on the development of cochlear nonlinearities. J Assoc Res Otolaryngol. 2008;9:464–476. doi: 10.1007/s10162-008-0140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel A. Periods of sensitivity to thyroid hormone during the development of the organ of Corti. Acta Otolaryngol Suppl. 1986;429:23–27. doi: 10.3109/00016488609122726. [DOI] [PubMed] [Google Scholar]
- Uziel A, Legrand C, Ohresser M, Marot M. Maturational and degenerative processes in the organ of Corti after neonatal hypothyroidism. Hear Res. 1983;11:203–218. doi: 10.1016/0378-5955(83)90079-5. [DOI] [PubMed] [Google Scholar]
- Vanderschueren-Lodeweyckx M, Debruyne F, Dooms L, Eggermont E, Eeckels R. Sensorineural hearing loss in sporadic congenital hypothyroidism. Arch Dis Child. 1983;58:419–422. doi: 10.1136/adc.58.6.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyken E, Van Laer L, Fransen E, Topsakal V, Lemkens N, Laureys W, Nelissen N, Vandevelde A, Wienker T, Van De Heyning P, Van Camp G. KCNQ4: a gene for age-related hearing impairment? Hum Mutat. 2006;27:1007–1016. doi: 10.1002/humu.20375. [DOI] [PubMed] [Google Scholar]
- Van't Hoff W, Stuart DW. Deafness in myxoedema. Q J Med. 1979;48:361–367. [PubMed] [Google Scholar]
- Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004;2:30. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zimmermann U, Winter H, Mack A, Köpschall I, Rohbock K, Zenner HP, Knipper M. Thyroid hormone is a critical determinant for the regulation of the cochlear motor protein prestin. Proc Natl Acad Sci U S A. 2002;99:2901–2906. doi: 10.1073/pnas.052609899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter H, Braig C, Zimmermann U, Geisler HS, Fränzer JT, Weber T, Ley M, Engel J, Knirsch M, Bauer K, Christ S, Walsh EJ, McGee J, Köpschall I, Rohbock K, Knipper M. Thyroid hormone receptors TRalpha1 and TRbeta differentially regulate gene expression of Kcnq4 and prestin during final differentiation of outer hair cells. J Cell Sci. 2006;119:2975–2984. doi: 10.1242/jcs.03013. [DOI] [PubMed] [Google Scholar]
- Woods KA, Savage MO. Laron syndrome: typical and atypical forms. Baillieres Clin Endocrinol Metab. 1996;10:371–387. doi: 10.1016/s0950-351x(96)80500-3. [DOI] [PubMed] [Google Scholar]
- Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:149–155. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]