

Abstract
In this review, we summarize the recent literature on dapE-encoded N-succinyl-L,L-diaminopimelic acid desuccinylase (DapE) enzymes, with an emphasis on structure–function studies that provide insight into the catalytic mechanism. Crystallographic data have also provided insight into residues that might be involved in substrate and hence inhibitor recognition and binding. These data have led to the design and synthesis of several new DapE inhibitors, which are described along with what is known about how inhibitors interact with the active site of DapE enzymes, including the efficacy of a moderately strong DapE inhibitor.
Keywords: DapE, Metallohydrolase, Antibiotics, Zinc, X-ray crystallography, Inhibitor design, Catalytic mechanism
Introduction
The emergence of antibiotic-resistant bacterial infections has created a significant and growing medical problem throughout the world [1–5]. Antibiotic resistance has been recognized since the introduction of penicillin more than 50 years ago, when penicillin-resistant infections caused by Staphylococcus aureus rapidly appeared [3, 6]. Because bacteria have been exposed to many of the currently available antibiotics such as β-lactams, fluoroquinolones, macrolides, tetracyclines, aminoglycosides, glycopeptides, and trimethoprim combinations for years, they have evolved resistance to these drugs owing to mutation of genes or the acquisition of genes that impart resistance from other organisms [3, 7–10]. In fact, several pathogenic bacteria, some of which were thought to have been eradicated, have made a significant resurgence owing to resistance to antibiotics [3, 6]. For example, tuberculosis is currently the leading cause of death in adults by an infectious disease worldwide, which is significant given that death rates due to tuberculosis had declined to near imperceptible levels in industrial nations [11–13]. According to the Centers for Disease Control and Prevention, several bacterial strains currently exhibit multi-drug resistance, with more than 60 % of hospital-acquired infections in the USA caused by the so-called ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species).
Antibiotics work by interfering with a vital bacterial cell function at a specific cellular target by either killing the bacteria or arresting their multiplication [5]. This allows the patient’s immune system to clear the bacteria from the body. Inhibitors of cell wall biosynthesis (vancomycin and β-lactams, for example) have proven to be very potent antibiotics, providing evidence that interfering with cell wall synthesis has deleterious effects on bacterial cell survival. Enzymes that are targeted by these antibiotics tend to be present in all bacteria and are highly similar in structure and function, such that certain antibiotics kill or inhibit the growth of a broad range of bacterial species (i.e., broad-spectrum antibiotics) [3, 7–10]. Unfortunately, only two new classes of antibacterial drugs have emerged since 1962. According to the Infectious Diseases Society of America, at least ten new systemic antibacterial drugs should enter the market by 2020, but most of these are derivatives of existing classes of antibiotics. Since every antibiotic has a finite lifetime, as resistance will ultimately occur, particularly if the same enzymes are repeatedly targeted, development of new classes of inhibitors that target previously untargeted cellular enzymes is essential to retain control of infectious disease [14, 15].
Lysine biosynthetic pathway
From bacterial genetic information, the meso-diaminopimelate (m-DAP)/lysine biosynthetic pathway offers several potential antibacterial enzyme targets that have yet to be explored (Fig. 1) [16–21]. One of the products of this pathway, lysine, is required in protein synthesis and is also used in the peptidoglycan layer of Gram-positive bacterial cell walls. A second product of this pathway, m-DAP, is an essential component of the peptidoglycan cell wall for Gram-negative bacteria, providing a link between poly-saccharide strands. Since lysine is an essential amino acid and is not synthesized by humans, it must be ingested. However, most bacteria, plants, and algae synthesize lysine and m-DAP from aspartic acid through three related pathways that diverge after the production of L-tetrahydrodipicolinate [16, 17, 22]. The presence of multiple biosynthetic pathways in bacteria for the synthesis of m-DAP/lysine highlights the importance of m-DAP/lysine for bacterial cell survival.
Fig. 1.

Biosynthetic pathways of meso-diaminopimelic acid and lysine in bacteria
The succinylase pathway is the primary biosynthetic pathway for m-DAP/lysine and is used by all Gram-negative and most Gram-positive bacteria [16]. The dehydrogenase pathway forms m-DAP directly from L-tetrahydrodipicolinate, but this is a high-energy transformation and is limited to only a few Bacillus species [16]. The acetylase pathway is also a minor biosynthetic pathway for m-DAP production and is also limited to only a few Bacillus species [17]. One of the enzymes in the succinylase pathway, the dapE-encoded N-succinyl-L,L-diaminopimelic acid desuccinylase (DapE), is a Zn(II)-containing metallohydrolase. It has been shown that deletion of the gene encoding DapE is lethal to Helicobacter pylori and Mycobacterium smegmatis [23, 24]. Even in the presence of lysine-supplemented media, H. pylori was unable to grow, suggesting that lysine cannot be synthesized by other pathways or imported. Therefore, DapE enzymes appear to be essential for cell growth and proliferation and are part of a biosynthetic pathway that is the only source of lysine in most bacteria. Since there are no similar biosynthetic pathways in mammals, DapE enzymes appear to be potential targets for inhibitors that may possess antimicrobial activity [16].
DapE enzymes contain a dinuclear Zn(II) active site
DapE enzymes catalyze the hydrolysis of N-succinyl-L,L-diaminopimelic acid (L,L-SDAP), forming L,L-diaminopimelic acid and succinate (Fig. 2) [22]. DapE enzymes have been purified from multiple sources and genes have been identified in a large number of pathogenic Gram-positive and Gram-negative bacteria, including all of the ESKAPE pathogens [23–28]. Alignment of several of the DapE gene sequences shows a minimum of 49 % identity [29]. Significantly, all of the residues that are metal binding ligands in the M28 family of dinuclear Zn(II)-dependent metalloproteases [30], including the aminopeptidase from Vibrio proteolyticus (Aeromonas proteolytica, AAP) and carboxypeptidase G2 from Pseudomonas sp. strain-RS-16 (CPG2) [31, 32], are strictly conserved in all DapE enzymes. Since the catalytic activity of DapE enzymes requires Zn(II), and both CPG2 and AAP possess (μ-aquo)(μ-carboxylato)dizinc(II) active sites with one terminal carboxylate and one histidine residue at each metal site, a similar active site was proposed for DapE enzymes [22, 33–35]. Evidence for a dinuclear Zn(II) active site in DapE enzymes was obtained via zinc K-edge extended X-ray absorption fine structure (EXAFS) spectra of the DapE from Haemophilus influenzae in the presence of 1 and 2 equiv of Zn(II) (i.e., [Zn_(DapE)] and [ZnZn(DapE)]) [36]. Fourier transforms of the zinc EXAFS spectrum are dominated by a peak at approximately 2.0 Å, which was best fit assuming approximately five (N,O) scatterers at 1.96 and 1.98 Å for [Zn_(DapE)] and [ZnZn(DapE)], respectively. Inclusion of a sulfur atom provided poorer fits based on Debye–Waller factors. A second-shell feature at approximately 3.34 Å appears in the [ZnZn(DapE)] EXAFS spectrum but is significantly diminished in the [Zn_(DapE)] EXAFS spectrum. These data confirmed that DapE enzymes can bind two Zn(II) ions that form a dinuclear site.
Fig. 2.
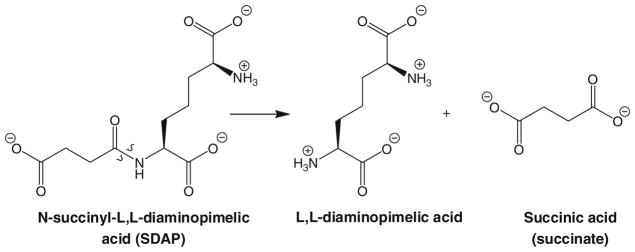
Reaction catalyzed by dapE-encoded N-succinyl-L,L-diaminopimelic acid desuccinylase (DapE)
Both AAP and CPG2 contain active-site histidine residues that function as ligands to the Zn1 and Zn2 centers. On the basis of sequence alignment of the DapE from H. influenzae with AAP and CPG2, both H67 and H349 were predicted to be Zn(II) ligands [29]. In an effort to clearly define the active-site residues and provide insight into the structural properties of each divalent metal ion in DapE enzymes, the H67A and H349A DapE mutant enzymes were prepared. The H67A DapE enzyme exhibited a decrease in catalytic efficiency (about 180-fold) compared with wild-type DapE toward the substrate L,L-SDAP. No catalytic activity was observed for H349A under the experimental conditions used. Electron paramagnetic resonance (EPR) and UV–vis data indicated that the Co(II) ion bound to H349A DapE is analogous to that of wild-type DapE after the addition of a single Co(II) ion. The addition of 1 equiv of Co(II) to H67A DapE provided spectra that are very different from the spectrum for the first Co(II) binding site of the wild-type enzyme, but are similar to the spectrum for the second binding site. The UV–vis and EPR data, in conjunction with the kinetic data, are consistent with the assignment of H67 and H349 as active-site metal ligands for DapE. Furthermore, these data suggest that H67 is a ligand in the first metal binding site, whereas H349 resides in the second metal binding site.
Sequence alignment with AAP and CPG2 also suggested that the active-site residue in the DapE from H. influenzae, E134, likely functions as the general acid/base during the hydrolysis reaction catalyzed by DapE [22]. To elucidate the catalytic role of E134, the E134A and E134D mutant DapE enzymes were prepared [37]. The Michaelis constant (Km) was found not to change upon substitution with aspartate but the kcat values changed drastically in the following order: glutamate (140 s−1), aspartate (0.13 s−1), and alanine (none detected). Examination of the pH dependence of the kinetic constants kcat and Km for the E134D enzyme revealed ionizations at pH 6.4, 7.4, and approximately 9.7. Isothermal titration calorimetry studies on wild-type DapE provided Kd values for the first and second Zn(II) binding sites of 4.4 and 13.6 μM. Isothermal titration calorimetry experiments on E134D DapE and E134A DapE revealed a statistically significant in metal Kd values of 2.9 and 1.4 times, respectively, for the first metal binding event. Interestingly, UV–vis and EPR spectra obtained on Co(II)-substituted E134D and E134A DapE did not reveal any significant changes, suggesting that both Co(II) ions reside in distorted trigonal bipyramidal coordination geometries [38]. Combination of these data indicates that E134 is intrinsically involved in the hydrolysis reaction catalyzed by DapE and likely plays the role of a general acid/base.
Structural characterization of DapE enzymes
A major limitation in developing a previously undescribed class of antimicrobials that target DapE enzymes was the lack of knowledge about their active-site structure, including potential residues involved in substrate binding. The X-ray crystal structure of the DapE from Neisseria meningitidis was reported at 1.9-Å resolution, but the DapE was in the apo form, providing little information about the Zn(II) active site [39]. Recently, the X-ray crystal structure of the DapE from H. influenzae was reported for both the mono and dinuclear Zn(II) forms at 2.0- and 2.3-Å resolution, respectively [40]. Similar to the DapE from N. meningitidis, the DapE from H. influenzae forms a homodimer where the catalytic domain consists of an α/β globular domain with a twisted β-sheet hydrophobic core sandwiched between α-helices (Fig. 3). The active-site cleft is located in the center of the catalytic domain above the centrally located parallel strands of the β-sheet and is covered by loops. The location and the architecture of the active site are strikingly similar to those of the active sites of CPG2 and AAP [28, 29].
Fig. 3.

Ribbon diagram of the X-ray crystal structure of the DapE from Haemophilus influenzae showing the dimer and catalytic structural domains. Zinc ions are shown in black (Zn1, the catalytic zinc) and gray (Zn2)
The active site of the mononuclear Zn(II) form of DapE ([Zn_(DapE)]) reveals that Zn1 resides in a distorted tetrahedral geometry coordinated by the carboxylate oxygens of D100 and E163 as well as one nitrogen atom of H67 (Fig. 4). The remaining coordination site is filled by an oxygen atom provided from a water molecule. In the dinuclear Zn(II) form of DapE ([ZnZn(DapE)]), the active site contains two Zn(II) ions at an interatomic distance of 3.36 Å, compared with 3.45 Å for AAP and 3.25 Å for CPG2 [31, 32]. Each of the Zn(II) ions adopts a distorted tetrahedral geometry and is coordinated by one imidazole group (H67 for Zn1 and H349 for Zn2) and one carboxylate group (E163 for Zn1 and E135 for Zn2). Both Zn(II) ions are bridged by an additional carboxylate group (D100) on one side and water/hydroxide on the opposite side, forming a (μ-aquo)(μ-carboxylato)dizinc(II) core with one terminal carboxylate and one histidine residue at each metal site. These structures confirm the assignment of H67 and H349 as active-site ligands, with H67 residing in the first metal binding site, as well as the role of Glu134.
Fig. 4.

Active sites of a the mononuclear Zn(II) form of DapE and b the dinuclear Zn(II) form of DapE from H. influenzae
Inspection of the X-ray crystal structures of [Zn_(DapE)] and [ZnZn(DapE)] combined with surface analysis revealed a crescent-shaped cavity that extends along the catalytic domain and surrounds the active site (Fig. 5). This well-defined, negatively charged cavity is shaped from the top by strand β12 and α8 and in the middle by a loop connecting these two elements. The bottom of the cavity is formed by a loop connecting strands β6 and β7, and a loop connecting β5 and α4. Taking into account the linear character of the substrate, we think it is likely that the substrate binds in an extended conformation [41], lining up along the grove with the peptide bond positioned over the active-site metals. It is conceivable that substrate binding is further stabilized by interaction of the substrate carboxylate groups with positively charged amino acid side chains. Potential candidates include K175, R258, and R329 (Fig. 6). In the [ZnZn(DapE)] structure, R258 and R329 form a charged dipole interaction with a sulfate ion, a possible mimic of the carboxylic group of the substrate. R329 is centrally positioned in a positively charged pocket that it forms together with R258. Interestingly, the active-site pocket of CPG2 contains an arginine that is conserved in DapE (R329) and has been proposed to bind the carboxylate group of the side chain. These data, in combination with the findings of reported inhibitor binding studies [35, 36, 42, 43], indicate that DapE is an excellent target for a highly specific drug that should have high efficacy and low toxicity.
Fig. 5.
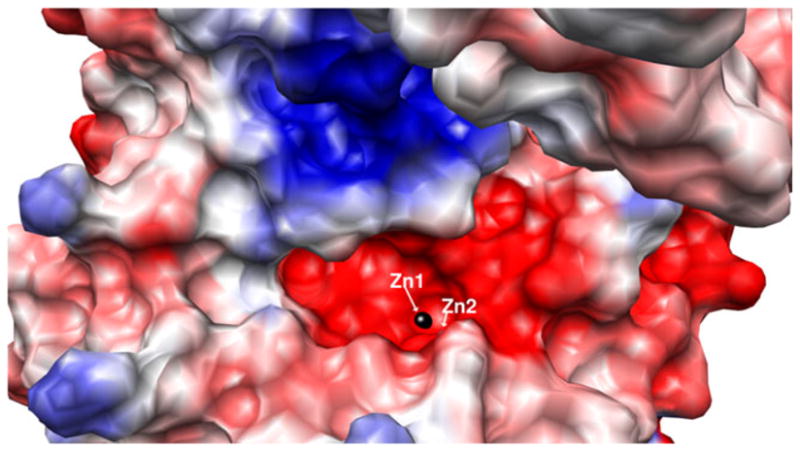
Surface rendering of the dinuclear Zn(II) form of DapE from H. influenzae showing the charge distribution and depicting the crescent-shaped active-site cavity
Fig. 6.
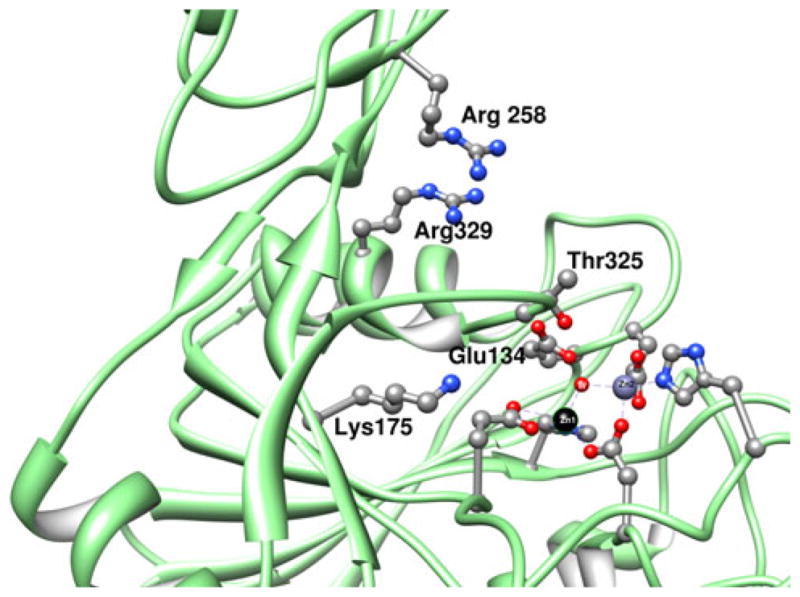
Charged residues near the dinuclear active site of DapE that may play a role in substrate recognition and binding
Proposed catalytic mechanism of DapE
The X-ray structures of [Zn_DapE] and [ZnZn_DapE] provide a structural foundation for a proposed reaction mechanism of DapE [22, 35, 37]. Analysis of the available structures along with the previously reported kinetic and spectroscopic data for DapE enzymes allowed a detailed mechanism of catalysis for DapE enzymes to be proposed (Fig. 7) [40]. On the basis of the proposed catalytic mechanism for AAP [44, 45], the first step in catalysis for DapE enzymes is likely recognition of the L,L-SDAP side chain by the crescent-shaped cavity adjacent to the Zn1 site. Next, the peptide carbonyl oxygen of L,L-SDAP coordinates to Zn1 and expands its coordination number from four to five, activating the carbonyl for nucleophilic attack. Deprotonation of the metal-bound water molecule by E134 to form a nucleophilic hydroxide moiety is consistent with the postulated pKa of the zinc-bound water molecule [22]. Once the zinc-bound hydroxide has formed, it can attack the activated carbonyl carbon of the substrate, forming an γ-1-μ-transition-state complex [36]. Solvent kinetic isotope effect studies yielded an inverse isotope effect that was explained by the attack of a zinc-bound hydroxide on the amide carbonyl [22]. E134 may provide a proton to the penultimate amino nitrogen, similar to what is observed for AAP, returning it to its ionized state and thus facilitating product release. Once the products have been released, a water molecule bridging the two metal ions is replaced. In the absence of the second metal ion, the catalytic mechanism does not likely change markedly as H349 is in a position to assist in orienting the substrate properly in the active site through the formation of a hydrogen bond with a carboxylate side chain of the substrate, thereby stabilizing the transition-state intermediate, reminiscent of proposals for the monometalated forms of AAP and the methionine aminopeptidase from Escherichia coli [46–48]. In the presence of a dinuclear site, the second metal ion likely coordinates either the peptide carbonyl oxygen in a bridging fashion or a carboxylate side chain of the substrate.
Fig. 7.

Proposed catalytic mechanism of a monozinc, and b dizinc DapE
Design and synthesis of DapE inhibitors
The design and synthesis of novel DapE inhibitors requires information regarding substrate specificity. Therefore, the four isomers of N-succinyldiaminopimelic acid (SDAP) as well as a number of acetylated amino acids have been examined as potential substrates. DapE was not able to hydrolyze the D,D, L,D, or D,L isoforms of SDAP, confirming that L,L-SDAP is the only known biological substrate for DapE enzymes. Moreover, no hydrolytic degradation was observed for any acetylated amino acids tested [28, 35]; thus, the DapE active site has strict substrate specificity with regard to both functional groups and stereochemistry. These data also suggest that the carboxylate of the succinyl moiety forms an important interaction within the active site of DapE since the acetylated amino acids could not be hydrolyzed. Alternatively, the acetylated versions of these amino acids may introduce a repulsive steric interaction because of bulky alkyl groups, thus preventing hydrolysis. Given the lack of hydrolytic activity toward SDAP isoforms, a series of potential DapE inhibitors based on SDAP bearing different N-linked acyl side chains terminated with (1) a carboxyl group or (2) a lipophilic moiety were synthesized. Unfortunately, none of these compounds functioned as potent inhibitors of DapE [35, 36, 42, 43]. Finally, D,L-succinyl aminopimelate was examined; this differs from the natural substrate, L,L-SDAP, only by the absence of the amine group on the amino acid side chain. Interestingly, DapE could not hydrolyze this compound, implying that the amine provides an important interaction for substrate binding.
The next step in inhibitor design for DapE enzymes involved understanding how inhibitors bind to the active site. To date, no X-ray structural data for a DapE enzyme in the presence of an inhibitor have been reported; however, zinc K-edge EXAFS spectra of [ZnZn(DapE)] in the presence of the competitive inhibitors 2-carboxyethylphosphonic acid (CEPA; Ki = 800 μM) and 5-mercaptopentanoic acid (MSPA; Ki = 6 μM) have been reported [36]. Phosphonic acid containing compounds have been shown to be potent inhibitors of metallohydrolases, including AAP and the bovine lens leucine aminopeptidase, and have been used as probes of the transition state of hydrolysis reactions. The EXAFS data for [ZnZn(DapE)]–CEPA indicate that the average coordination number of each Zn(II) ion is five, and the Zn–Zn distance remains 3.34 Å. An increase in the M–M distance of [ZnZn(AAP)], from 3.5 to 3.9 Å, is observed upon the addition of the transition-state analog inhibitor L-leucine phosphonic acid (LPA), which contains a ligating group similar to that in CEPA [49]. The X-ray crystal structure and EXAFS data for [ZnZn(AAP)]–LPA reveal that the bridging water molecule is displaced by LPA, resulting in an γ-1,2-μ-phosphonate bridge and an increase in the Zn–Zn distance of 0.4 Å. The fact that the Zn–Zn distance is not altered upon CEPA binding to [ZnZn(DapE)] suggests an γ-1-μ-phosphonate bridge exists, similar to the binding mode of LPA with regard to bovine lens leucine aminopeptidase [49, 50]. On the other hand, MSPA binding to [ZnZn(DapE)] has a marked effect on the zinc K-edge, suggesting that the average electronic environment of the dinuclear Zn(II) site has changed significantly. The observed shift in the position of the absorption edge to lower energy is indicative of a net increase in electron density at the dinuclear Zn(II) site, consistent with a sulfur ligand. In addition, the EXAFS data for [ZnZn(DapE)]–MSPA reveals a new feature at 2.3 Å that is highly characteristic of a direct zinc–sulfur interaction. Moreover, the M–M distance is lengthened from 3.34 to 3.64 Å. These data indicate that the thiol group of MSPA binds to one or both of the Zn(II) ions in the active site of DapE.
The fact that thiol-based molecules were shown to be moderately strong inhibitors of DapE provided insight for examination of bifunctional molecules that contained, in addition to a thiol zinc binding group, a carboxylate moiety that could interact with the positively charged lysine and arginine side chains that purportedly reside near the active site. These studies led to the identification of several low micromolar inhibitors of DapE, all of which, like MSPA, contain a thiol functional group [42]. Thiol-containing compounds are typically potent monodentate inhibitors of Zn(II) metalloproteins [51]. One of the better inhibitors was L-penicillamine (Fig. 8), which exhibited an IC50 of 13.7 μM, and a measured Ki of 4.6 μM (competitive). DapE is stereoselective with respect to recognition of inhibitors, as D-penicillamine gave an IC50 of 50 μM. Given the success with these carboxylic acid containing thiols, L-captopril (Fig. 8), an angiotensin-converting enzyme inhibitor, was examined; it contains the requisite zinc binding group and carboxylate functionalities. L-Captopril exhibited an IC50 of 3.3 μM and a measured Ki of 1.8 μM (competitive). Again, the binding is stereoselective, as D-captopril was an order of magnitude less potent, with an IC50 of 42.0 μM. Given the good potency of L-captopril, another angiotensin-converting enzyme inhibitor, enalapril, was screened but did not show any potency toward DapE. 4-Mercaptobutyric acid is also an inhibitor of DapE and exhibits an IC50 of 43 μM, whereas 2-mercaptobenzoic acid has a measured IC50 of 34 μM. For these low micromolar inhibitors, it was hypothesized that a negatively charged carboxylate in the molecule participates in an ionic interaction with a positively charged lysine or arginine near the active site.
Fig. 8.
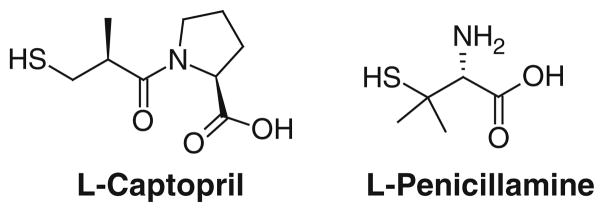
Structures of L-captopril and L-penicillamine
Given that L-captopril functions as a moderately strong inhibitor of DapE, its efficacy was examined using a standard plate assay [42]. Application of L-captopril or L-penicillamine directly to plates cultured with E. coli showed a dose-responsive antibiotic activity for L-captopril. Very little inhibition was observed for 1 mg of L-captopril, but amounts of 5 and 20 mg demonstrated a clear positive antibiotic result. These data suggest that L-captopril can cross the bacterial cell membrane and function to inhibit bacterial cell growth. Whether DapE is the cellular target of L-captopril was not addressed; however, a recent study suggested that DapE is not the main target of L-captopril antimicrobial activity since L-captopril inhibited Salmonella enterica and E. coli in a DapE-independent manner [52]. Clearly, further studies are needed to verify DapE as an antimicrobial target, and such studies will necessitate the discovery of strong binding inhibitors that are specific for DapE enzymes.
Concluding remarks
Bacterial infections, some of which were thought to be eradicated, have made a significant resurgence owing to bacterial resistance to all known antibiotics [2–4]. Consequently, new compounds that combat these pathogens and target enzymes involved in bacterial cell wall synthesis or pathways involved in cell replication are in high demand [53–60]. The m-DAP/lysine biosynthetic pathway offers several potential antibacterial targets that have yet to be explored. Although our understanding of the catalytic mechanism of DapE has improved markedly over the past few years, the mechanism has not yet been entirely delineated. For example, several substrate binding steps have been proposed and the roles of each metal ion during catalysis are still in question. Therefore, more experimental evidence is needed to distinguish between the competing mechanistic proposals for DapE. Even so, the results obtained from the studies described in this review have provided new insight into the structure and function of DapE enzymes and have led to new medicinal chemistry leads. Additional insight into the catalytic mechanism of DapE and the determinants of substrate binding will be critical for the rational design of selective DapE inhibitors that may function as a new class of antimicrobial agents.
Acknowledgments
This work was supported by the National Institutes of Health (R15 AI085559-01A1, R.C.H.).
Molecular graphics were created and analyses were performed with the UCSF Chimera package (http://www.cgl.ucsf.edu/chimera). Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health (National Center for Research Resources grant 2P41RR001081, National Institute of General Medical Sciences grant 9P41GM103311).
Abbreviations
- AAP
Aminopeptidase from Vibrio proteolyticus (Aeromonas proteolytica)
- CEPA
2-Carboxyethylphosphonic acid
- CPG2
Carboxypeptidase G2 from Pseudomonas sp. strain RS-16
- DapE
dapE-encoded N-succinyl-L,L-diaminopimelic acid desuccinylase
- EPR
Electron paramagnetic resonance
- EXAFS
Extended X-ray absorption fine structure
- L,L-SDAP
N-Succinyl-L,L-diaminopimelic acid, LPA, L-Leucine phosphonic acid
- m-DAP
meso-Diaminopimelate
- MSPA
5-Mercaptopentanoic acid
- SDAP
N-Succinyldiaminopimelic acid
Contributor Information
Danuta M. Gillner, Email: danuta.gilner@polsl.pl, Department of Chemistry and Biochemistry, Loyola University-Chicago, 1068 W. Sheridan Rd., Chicago, IL 60626, USA. Department of Chemistry, Silesian University of Technology, ul. Krzywoustego 4, 44-100 Gliwice, Poland
Daniel P. Becker, Department of Chemistry and Biochemistry, Loyola University-Chicago, 1068 W. Sheridan Rd., Chicago, IL 60626, USA
Richard C. Holz, Email: rholz1@luc.edu, Department of Chemistry and Biochemistry, Loyola University-Chicago, 1068 W. Sheridan Rd., Chicago, IL 60626, USA
References
- 1.CfDCa Prevention. MMWR Morb Mortal Wkly Rep. 1995;44:1–13. [Google Scholar]
- 2.Howe RA, Bowker KE, Walsh TR, Feest TG, MacGowan AP. Lancet. 1998;351:601–602. doi: 10.1016/S0140-6736(05)78597-4. [DOI] [PubMed] [Google Scholar]
- 3.Levy SB. Sci Am. 1998;278:46–53. doi: 10.1038/scientificamerican0398-46. [DOI] [PubMed] [Google Scholar]
- 4.Chin J. New Sci. 1996;152:32–35. [Google Scholar]
- 5.Henery CM. C&E News. 2000;78:41–58. [Google Scholar]
- 6.Nemecek S. Sci Am. 1997;276:38–39. [PubMed] [Google Scholar]
- 7.Miller JB. The pharmaceutical century: ten decades of drug discovery. American Chemical Society; Washington: 2000. pp. 52–71. [Google Scholar]
- 8.Lesney MS, Frey R. The pharmaceutical century: ten decades of drug discovery. American Chemical Society; Washington: 2000. pp. 110–129. [Google Scholar]
- 9.Frey R, Lesney MS. The pharmaceutical century: ten decades of drug discovery. American Chemical Society; Washington: 2000. pp. 92–109. [Google Scholar]
- 10.Tweedy BD, Lesney MS. The pharmaceutical century: ten decades of drug discovery. American Chemical Society; Washington: 2000. pp. 72–91. [Google Scholar]
- 11.Snider DE, Raviglione M, Kochi A. In: Global burden of tuberculosis. Bloom BR, editor. ASM Press; Washington: 1994. pp. 3–11. [Google Scholar]
- 12.Dolin PJ, Raviglione MC, Kochi A. Bull WHO. 1994;72:213–220. [PMC free article] [PubMed] [Google Scholar]
- 13.Raviglione MC, Snider DE, Kochi A. JAMA. 1995;273:220–226. [PubMed] [Google Scholar]
- 14.Teuber M. Cell Mol Life Sci. 1999;56:755–763. doi: 10.1007/s000180050022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller JR, Dunham S, Mochalkin I, Banotai C, Bowman M, Buist S, Dunkle B, Hanna D, Harwood HJ, Huband MD, Karnovsky A, Kuhn M, Limberakis C, Liu JY, Mehrens S, Mueller WT, Narasimhan L, Ogden A, Ohren J, Prasad JV, Shelly JA, Skerlos L, Sulavik M, Thomas VH, VanderRoest S, Wang L, Wang Z, Whitton A, Zhu T, Stover CK. Proc Natl Acad Sci USA. 2009;106:1737–1742. doi: 10.1073/pnas.0811275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scapin G, Blanchard JS. Adv Enzymol. 1998;72:279–325. doi: 10.1002/9780470123188.ch8. [DOI] [PubMed] [Google Scholar]
- 17.Born TL, Blanchard JS. Cur Opin Chem Biol. 1999;3:607–613. doi: 10.1016/s1367-5931(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 18.Girodeau J-M, Agouridas C, Masson M, Pineau R, Le Goffic F. J Med Chem. 1986;29:1023–1030. doi: 10.1021/jm00156a021. [DOI] [PubMed] [Google Scholar]
- 19.Cox RJ, Sutherland A, Vederas JC. Bioorg Med Chem. 2000;8:843–871. doi: 10.1016/s0968-0896(00)00044-4. [DOI] [PubMed] [Google Scholar]
- 20.Vederas JC. Can J Chem. 2006;84:1197–1207. [Google Scholar]
- 21.Hutton CA, Perugini MA, Gerrard JA. Mol Biosyst. 2007;3:458–465. doi: 10.1039/b705624a. [DOI] [PubMed] [Google Scholar]
- 22.Born TL, Zheng R, Blanchard JS. Biochemistry. 1998;37:10478–10487. doi: 10.1021/bi9806807. [DOI] [PubMed] [Google Scholar]
- 23.Karita M, Etterbeek ML, Forsyth MH, Tummuru MR, Blaser MJ. Infect Immun. 1997;65:4158–4164. doi: 10.1128/iai.65.10.4158-4164.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavelka MS, Jacobs WR. J Bacteriol. 1996;178:6496–6507. doi: 10.1128/jb.178.22.6496-6507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouvier J, Richaud C, Higgins W, Bögler O, Stragier P. J Bacteriol. 1992;174:5265–5271. doi: 10.1128/jb.174.16.5265-5271.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuchs TM, Schneider B, Krumbach K, Eggeling L, Gross R. J Bacteriol. 2000;182:3626–3631. doi: 10.1128/jb.182.13.3626-3631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw-Reid CA, McCormick MM, Sinskey AJ, Stephanopoulos G. Appl Microbiol Biotechnol. 1999;51:325–333. doi: 10.1007/s002530051398. [DOI] [PubMed] [Google Scholar]
- 28.Lin Y, Myhrman R, Schrag ML, Gelb MH. J Biol Chem. 1988;263:1622–1627. [PubMed] [Google Scholar]
- 29.Gillner DM, Bienvenue DL, Nocek BP, Joachimiak A, Zachary V, Bennett B, Holz RC. J Biol Inorg Chem. 2009;14:1–10. doi: 10.1007/s00775-008-0418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of proteolytic enzymes. Academic; London: 1998. [Google Scholar]
- 31.Rowsell S, Pauptit RA, Tucker AD, Melton RG, Blow DM, Brick P. Structure. 1997;5:337–347. doi: 10.1016/s0969-2126(97)00191-3. [DOI] [PubMed] [Google Scholar]
- 32.Desmarais W, Bienvenue DL, Bzymek KP, Petsko GA, Ringe D, Holz RC. J Biol Inorg Chem. 2006;11:398–408. doi: 10.1007/s00775-006-0093-x. [DOI] [PubMed] [Google Scholar]
- 33.Chevrier B, Schalk C, D’Orchymont H, Rondeau J-M, Moras D, Tarnus C. Structure. 1994;2:283–291. doi: 10.1016/s0969-2126(00)00030-7. [DOI] [PubMed] [Google Scholar]
- 34.Greenblatt HM, Almog O, Maras B, Spungin-Bialik A, Barra D, Blumberg S, Shoham G. J Mol Biol. 1997;265:620–636. doi: 10.1006/jmbi.1996.0729. [DOI] [PubMed] [Google Scholar]
- 35.Bienvenue DL, Gilner DM, Davis RS, Bennett B, Holz RC. Biochemistry. 2003;42:10756–10763. doi: 10.1021/bi034845+. [DOI] [PubMed] [Google Scholar]
- 36.Cosper NJ, Bienvenue DL, Shokes JE, Gilner DM, Tsukamoto T, Scott R, Holz RC. J Am Chem Soc. 2003;125:14654–14655. doi: 10.1021/ja036650v. [DOI] [PubMed] [Google Scholar]
- 37.Davis R, Bienvenue D, Swierczek SI, Gilner DM, Rajagopal L, Bennett B, Holz RC. J Biol Inorg Chem. 2006;11:206–216. doi: 10.1007/s00775-005-0071-8. [DOI] [PubMed] [Google Scholar]
- 38.Bennett B. Metals in biology. In: Hanson G, Berliner L, editors. Biological magnetic resonance. Vol. 29. Springer; New York: 2010. pp. 345–370. [Google Scholar]
- 39.Badger J, Sauder JM, Adams JM, Antonysamy S, Bain K, Bergseid MG, Buchanan SG, Buchanan MD, Batiyenko Y, Christopher JA, Emtage S, Eroshkina A, Feil I, Furlong EB, Gajiwala KS, Gao X, He D, Hendle J, Huber A, Hoda K, Kearins P, Kissinger C, Laubert B, Lewis HA, Lin J, Loomis K, Lorimer D, Louie G, Maletic M, Marsh CD, Miller I, Molinari J, Muller-Dieckmann HJ, Newman JM, Noland BW, Pagarigan B, Park F, Peat TS, Post KW, Radojicic S, Ramos A, Romero R, Rutter ME, Sanderson WE, Schwinn KD, Tresser J, Winhoven J, Wright TA, Wu L, Xu J, Harris TJ. Proteins. 2005;60:787–796. doi: 10.1002/prot.20541. [DOI] [PubMed] [Google Scholar]
- 40.Nocek BP, Gillner DM, Fan Y, Holz RC, Joachimiak A. J Mol Biol. 2010;397:617–626. doi: 10.1016/j.jmb.2010.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyndall JDA, Nall T, Fairlie DP. Chem Rev. 2005;105:973–999. doi: 10.1021/cr040669e. [DOI] [PubMed] [Google Scholar]
- 42.Gillner DM, Armoush N, Holz RC, Becker D. Bioorg Med Chem Lett. 2009;19:6350–6352. doi: 10.1016/j.bmcl.2009.09.077. [DOI] [PubMed] [Google Scholar]
- 43.Vaněk V, Pícha J, Buděšínský M, Šanda M, Jiráček J, Holz RC, Hlaváček J. Protein Pept Lett. 2010;17:405–409. doi: 10.2174/092986610790780387. [DOI] [PubMed] [Google Scholar]
- 44.Ustynyuk L, Bennett B, Edwards T, Holz RC. Biochemistry. 1999;38:11433–11439. doi: 10.1021/bi991090r. [DOI] [PubMed] [Google Scholar]
- 45.Stamper CC, Bienvenue DL, Moulin A, Bennett B, Ringe D, Petsko GA, Holz RC. Biochemistry. 2004;43:9620–9628. doi: 10.1021/bi049126p. [DOI] [PubMed] [Google Scholar]
- 46.Copik AJ, Swierczek SI, Lowther WT, D’souza VM, Matthews BW, Holz RC. Biochemistry. 2003;42:6283–6292. doi: 10.1021/bi027327s. [DOI] [PubMed] [Google Scholar]
- 47.Ye QZ, Xie SX, Ma ZQ, Huang M, Hanzlik RP. Proc Natl Acad Sci USA. 2006;103:9470–9475. doi: 10.1073/pnas.0602433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holz RC. Coord Chem Rev. 2002;232:5–26. [Google Scholar]
- 49.Stamper CC, Bennett B, Edwards T, Holz RC, Ringe D, Petsko GA. Biochemistry. 2001;40:7035–7046. doi: 10.1021/bi0100891. [DOI] [PubMed] [Google Scholar]
- 50.Sträter N, Lipscomb WN. Biochemisty. 1995;34:9200–9210. doi: 10.1021/bi00028a033. [DOI] [PubMed] [Google Scholar]
- 51.Jacobsen FE, Lewis JA, Cohen SM. Chem Med Chem. 2007;2:152–171. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 52.Uda NR, Creus M. Bioinorg Chem Appl. 2011;2011:306465. doi: 10.1155/2011/306465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arfin SM, Kendall RL, Hall L, Weaver LH, Stewart AE, Matthews BW, Bradshaw RA. Proc Natl Acad Sci USA. 1995;92:7714–7718. doi: 10.1073/pnas.92.17.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ben-Bassat A, Bauer AK, Chang S-Y, Myambo K, Boosman A, Chang S. J Bacteriol. 1987;169:751–757. doi: 10.1128/jb.169.2.751-757.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ben-Bassat A, Bauer K. Nature. 1987;326:315. [Google Scholar]
- 56.Chang S-Y, McGary EC, Chang S. J Bacteriol. 1989;171:4071–4072. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzales T, Robert-Baudouy J. FEMS Microbiol Rev. 1996;18:319–344. doi: 10.1111/j.1574-6976.1996.tb00247.x. [DOI] [PubMed] [Google Scholar]
- 58.Taylor A. FASEB J. 1993;7:290–298. doi: 10.1096/fasebj.7.2.8440407. [DOI] [PubMed] [Google Scholar]
- 59.Taylor A. Trends Biochem Sci. 1993;18:167–172. [PubMed] [Google Scholar]
- 60.Taylor A, editor. Aminopeptidases. Landes; Austin: 1996. [Google Scholar]