

Abstract
Endocytoscopy is an optical biopsy technique which uses a miniaturized camera to capture white light microscopy images through an endoscope. We have developed an alternative design that instead relays images to an external camera via a coherent fiber bundle. In this paper we characterize the device and demonstrate microscopy of porcine tissue ex vivo. One advantage of our approach is the ease with which other bundle-compatible imaging modalities can be deployed simultaneously. We show this by acquiring quasi-simultaneous endocytoscopy and fluorescence confocal endomicroscopy images through a single fiber bundle. This opens up possibilities for multi-modal endomicroscopy, combining white light and fluorescence imaging.
OCIS codes: (170.2150) Endoscopic imaging, (110.0180) Microscopy
1. Introduction
Modern pathology relies on our ability to image human tissue on a cellular scale. This usually requires histology, where one or more tissue samples are excised from the patient and then fixed, sliced and stained before being examined under a microscope. Histology is the ‘gold standard’ for the diagnosis of many diseases, but it is also a somewhat protracted process, typically taking a day or more. The cryosection technique can be used to accelerate the process, but results may still take up to 30 minutes, adding significantly to the operating time [1]. Freezing artefacts may also be introduced, making this technique generally less reliable. There are therefore significant difficulties associated with the use of histology either for rapid diagnosis or for intraoperative surgical guidance.
These limitations have led to extensive interest in ‘optical biopsy’, in which microscopy is performed directly on the patient [2–4]. The availability of live video images not only permits an immediate diagnosis, but also allows for the study of much larger areas of tissue than could be sampled by physical biopsy. For applications where conventional histology is still required to confirm a diagnosis, optical biopsy can have a role in guiding the selection of physical biopsy sites, allowing these to be more effectively targeted.
Optical biopsy requires a microscope which is sufficiently small and flexible so as to be usable in vivo, ideally through the instrument channel of an endoscope. Several microscopy techniques have been successfully miniaturized, including confocal endomicroscopy [5–8], structured illumination endomicroscopy [9, 10], widefield endomicroscopy [11], multi-photon endomicroscopy [12, 13], and optical coherence tomography (OCT) [14, 15]. Commercial in vivo microscopy systems are available, including the Cellvizio probe-based confocal laser endomicroscope (Mauna Kea Technologies) [16] and several endoscopic and intravascular OCT systems (for example C7 XR by Lightlab/St Jude Medical [17]). However, all of these techniques generate images which are substantially different from the white-light transmission micrographs familiar to histopathologists.
Endocytoscopy has recently emerged as a further viable optical biopsy technique, producing images with a resolution and field-of-view comparable to fluorescence endomicroscopy. However, rather than using fluorescence, it instead derives contrast from absorption and scattering of white light by colored stains in a way similar to conventional histology [18]. We note here that the term endocytoscopy might arguably be applied to any endoscopic cellular imaging technique, but we will follow the convention established in the literature and restrict its definition to a particular form of white-light contact microscopy developed by Olympus Medical Systems Corporation [19]. It is distinct from other forms of endoscopic microscopy, including confocal and widefield fluorescence microscopy, which we will refer to as endomicroscopy. It is also distinguished from magnifying endoscopy or chromoendoscopy by its higher, cellular-level resolution and corresponding smaller field-of-view [20].
Contact endoscopy has been used to obtain high resolution, white light images since the early 1980s using a rigid Hopkins lens probe (Karz Storz) [21]. Cervical [22] and head and neck applications [21] were investigated in particular. An early attempt was also made to use fiber bundles for this purpose [23] (as cited by [20]), but the technique was not adopted clinically. More recently, several potential clinical applications of flexible endocytoscopy have been reported, including in the gastro-intestinal tract [24, 25], lung [26], and bladder [27], as well as for intraoperative surgical guidance [28]; a review is available in [18]. These studies were performed using a series of prototype endocytoscopes developed by Olympus. Topical application of a staining agent was required, typically either toluidine blue or methylene blue [29], although a double staining technique using cresyl violet to highlight nuclei and toluidine blue to stain cytoplasm has also been suggested [30].
While numerous reports have shown potential clinical applications, we have been unable to find a detailed technical discussion of flexible endocytoscopy in the academic literature. Nevertheless, one principle of operation has been described in a patent from 2004 [19]. The microscope objective is designed to be placed in direct contact with the tissue, maximizing light collection and providing mechanical stability. The white light illumination field is offset to one side of the objective, meaning that it cannot directly illuminate the tissue beneath the objective. Instead, superficial tissues within the field-of-view are illuminated from behind by light scattered from deeper tissue layers. The scattered light provides a virtual bright-field illumination source, resulting in images with some similarities to those that would be generated from a transmission microscope.
For the existing prototypes developed by Olympus, this image is formed directly onto a miniaturized CCD [31]. In one prototype (GIF-Y0002), this CCD is the same as used for conventional endoscopy, whereas in a second prototype (GIF-Y0001) a second CCD is employed [32]. Our implementation replaces the miniature camera with a coherent fiber imaging bundle. The bundle relays the microscopy image to an external camera; this can be a standard off-the-shelf scientific or commercial grade CCD. While fiber bundles have several inherent drawbacks, including low light throughput and poor resolution for a given field-of-view [7], their use would also have several advantages for in vivo work, with the most obvious being the potential to build much smaller devices. For example, fiber-bundle based fluorescence endomicroscopy probes are available with diameters suitable for use in needles for deep tissue imaging [33].
A second advantage concerns the possibility of sequentially or simultaneously acquiring other types of microscopy images through the same fiber bundle. Below, we illustrate this potential by incorporating an endocytoscope into a standard design for a confocal endomicroscope. This allowed us to obtain simultaneous endocytoscopy and confocal endomicroscopy images, something that was not possible with previous systems.
2. Materials and methods
We describe two devices: (i) a stand-alone fiber bundle endocytoscope, and (ii) a combined endocytoscope and fluorescence confocal endomicroscope, which we will refer to as the ‘Hybrid System’.
2.1 Stand alone endocytoscope
For the stand alone system, shown in Fig. 1 , we evaluated two probe designs, a low resolution probe in which the fiber bundle was placed in direct contact with the tissue, and a high resolution probe which included a micro-objective at the distal tip. Both probes used a 2 m long Fujikura fiber bundle (FIGH-30-800G), with approximately 30,000 cores, a useful imaging diameter of 750 µm, and an outer diameter (including coating) of 950 µm. The core-core spacing in the bundles was approximately 5 µm, a factor which influenced the lateral resolution of the system.
Fig. 1.

Stand-alone fiber-bundle endomicroscopy system. (a) Schematic of optics; (b) photograph of system and close-up view of high resolution probe. Abbreviations: MO: 4X microscope objective, LED: light emitting diode (warm white), MM: multimode.
For the high resolution probe we incorporated a micro-objective (GRINTech GT-MO-080-0415-488) to project a 1.93x demagnified image of the fiber bundle onto the tissue at a depth of 80 µm. The micro-objective consisted of GRIN lens coupled to a plano-convex lens, giving an object NA of 0.8 and an image (fiber bundle) NA of 0.42. This provided an approximate halving of the lateral resolution in comparison with the low resolution probe, at the cost of reducing the maximum field-of-view from 750 µm to 390 µm. The micro-objective also introduced pincushion distortion and chromatic aberrations which we discuss in more detail below.
To both probes, we added a multimode fiber which was used to deliver illumination light to the tissue. The end portion was stripped to bare fiber, giving an outer diameter of 425 µm. For the high resolution probe this ran on the outside of the stainless steel tube enclosing the micro-objective, with the distal face level with the end of the lens assembly. The fiber was held in place using heart-shrink tubing, giving a final (long axis) outer diameter of 3.2 mm. While this arrangement was acceptable for a prototype system, a more robust and compact assembly would require the illumination fiber to be run inside the stainless steel tube and to be protected by a glass window. For the low resolution probe the multimode fiber was separated from the fiber bundle by approximately 1.3 mm (center to center) using heat shrink tubing, and attached by a second piece of tubing. The final outer diameter of this probe was 3.4 mm. The diameter could have been reduced by shifting the illumination fiber closer to the fiber bundle but, as discussed in Section 3.1, this would result in a greater inhomogeneity of illumination intensity across the image.
The proximal end of the multimode illumination fiber was coupled to a 7 mW warm-white light LED (Thorlabs MWWHF1). The proximal end of the fiber bundle was imaged onto an 8 bit color CCD camera (Thorlabs DCU223C) by a 4X finite-conjugate plan microscope objective (Edmund Optics #67-706). With the CCD camera plane sited at the nominal tube length of the microscope objective, the 750 µm diameter active area of the fiber bundle was magnified to a size of approximately 3 mm on the CCD. The CCD had a pixel size of 4.65 µm, giving approximately 3 pixels per core spacing. However, so as to provide color images the CCD is overlayed with a Bayer color filter array. As a result, there are approximately 2 green pixels and 1.5 red and blue pixels available per core spacing. This was sufficient for the identification of individual cores in the combined image, as is demonstrated in Section 3.1
Raw images from the camera were handled by a custom software application developed in Labview (National Instruments); copies of the code are available on request. Raw images from the camera contain a honeycomb-like structure arising from the fiber bundle cores. To remove this pattern, we convolved the raw images with a 2D Gaussian kernel with a standard deviation (sigma) of 1.4 pixels (equivalent to 1.2 µm on the face of the fiber bundle). The brightness and contrast of the image was then adjusted automatically prior to live display at 15 frames per second. For images shown here, the electronic gains on the camera were set at 40%, 0% and 30% of maximum for the red, green and blue channels respectively. It is worth noting that a setting of 0% gain on the camera means that there is no additional gain, so this is effectively a true gain of 1. The master gain and the image integration time were manually adjusted during imaging to optimize use of the available bit depth. Data recording was via streaming of the processed image to an uncompressed AVI and, for the data presented in this paper, raw data was also streamed to disk for archiving purposes. No post-processing was applied to the images shown below except for brightness and contrast adjustments, cropping and resizing by cubic interpolation.
2.2 Hybrid system
To demonstrate one of the advantages of a fiber bundle approach to endocytoscopy, we combined our system with a fluorescence confocal endomicroscope. The principle of operation for the confocal endomicroscope is broadly similar to other systems reported in the literature [5, 7]. The illumination source was a 50 mW, 488 nm laser (Vortran Stradus). This was reflected by a dichroic beamsplitter (Semrock LPD01-488RS-25) onto a two axis scanning system composed of a conventional galvanometric scanning mirror (Thorlabs GVS011) and an 8 kHz resonant scanning mirror (Cambridge Technology CRS). A raster pattern was generated on the proximal face of the fiber bundle using a telescope and a X10 microscope objective (Edmund Optics #43-907), as shown in Fig. 2(a) . The scanning pattern was transferred to the tissue by the fiber bundle and, in the case of the high resolution probe, the distal micro-objective.
Fig. 2.
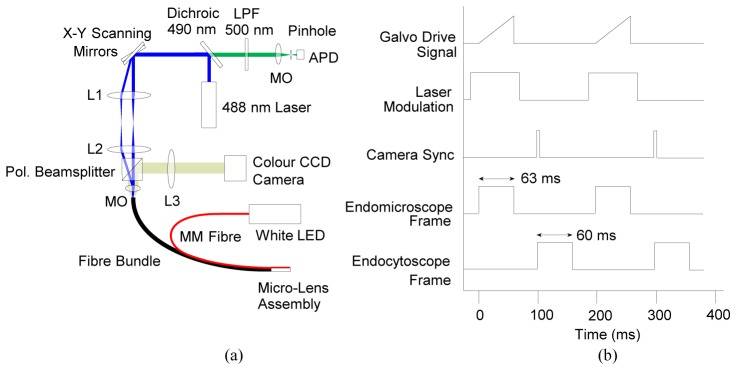
Hybrid endocytoscopy and endomicroscopy system. (a) Schematic of optical system; (b) timing diagram showing synchronization of galvanometer voltage (slow axis scan), laser modulation, camera trigger TTL and acquisition of endomicroscope and endocytoscope image frames. Note that that pulse lengths are illustrative and not precisely to scale, and that the pulse heights are arbitrary. Abbreviations: MM: multimode, Pol.: polarizing, LED: light emitting diode, MO: microscope objective, APD: avalanche photodiode, LPF: long pass wavelength filter, L1 & L3: achromatic doublet, f = 50 mm, L2: achromatic doublet, f = 75 mm.
Fluorescence light from the tissue returned along the same path and was de-scanned by the scanning system. It was then transmitted by the dichroic mirror, through a 500 nm long-pass filter (Thorlabs FELH0550) and focused by a X20 microscope objective (Edmund Optics #36-132) through a 25 µm pinhole onto an avalanche photodiode (APD) (Thorlabs APD110). The signal from the APD was digitized by an 8 bit high speed digitizer (NI PCI-5114), and image frames were assembled on the board using reference triggers from the scanning mirrors. The frame rate of the confocal-only system was 10 Hz for 500 lines per image.
To combine the system with the endocytoscope, we added a 50/50 polarizing beamsplitter (Thorlabs PBS201) between the final lens of the telescope assembly and the microscope objective. An achromatic doublet (f = 50 mm, Thorlabs) formed an imaging system with an effective magnification of approximately 3X, resulting in a 2.2 mm diameter image of the bundle on a color CCD camera. The beamsplitter was oriented so that a large fraction of the illumination light would be transmitted to the microscope objective. Light returning from the bundle can be expected to have random polarization, and so approximately 50% of both the endomicroscope and endocytoscope images would be sent both to the camera and to the endomicroscope de-scanning system. While this approach is somewhat light inefficient, it provides a simple means of separating the two signals. An alternative would have been to use a dichroic mirror to separate the fluorescence (i.e., the confocal signal) from the white light (i.e., the endocytoscope signal). However, this would mean that some of endocytoscope signal in the green part of the spectrum would not have been collected by the CCD camera, thus affecting the color balance of the images and potentially impairing interpretation.
Laser light reflected from the bundle would interfere with the endocytoscopy images, so the 488 nm illumination was pulsed to allow imaging to alternate between the two modes. A TTL signal line was used to synchronize frame acquisitions. The white LED for the endocytoscope illumination did not need to be modulated as the vast majority of any white light reaching the confocal detection arm was rejected by the pinhole. The timing arrangement for the entire system is summarized in Fig. 2(b).
A second custom Labview software application was developed to handle acquisition of the two imaging channels. Software overheads and jitter in timings limited the net frame rate (for acquisition of one image in each channel) to 5 Hz. This could be improved by a more judicious choice of hardware and better optimized software. On-line processing of the confocal image was similar to that for the endocytoscope images, except for the additional step of digital subtraction of background autofluorescence from the fiber bundle. The two channels were manually rotated and aligned to ensure correspondence.
3. Results
3.1 Endocytoscopy system characterization
In order to evaluate the lateral resolution of the probes we imaged a negative USAF resolution target (Thorlabs R3L3S1N) while illuminated from behind using the white LED. Figure 3 shows the average of 100 image frames for the low and high resolution probes (top and bottom row respectively). As well as the color image, the outputs from three individual color channels of the camera are also shown. The test pattern was visible in single image frames, but the blue channel was slightly noisier than the other two. Averaging over 100 frames ensured that the limiting spatial resolution could be easily assessed in all three channels. For the low resolution probe the insets show a zoom on the last four elements of the 6th group, corresponding to line pair spacings of 12.40, 11.04, 9.80 and 8.78 µm. The images indicate a limiting spatial resolution of approximately 10 µm which is broadly consistent with requiring two fiber cores per resolution element. For the high resolution probe the insets show a zoom on the last four elements of the 7th group, corresponding to line pair spacings of 6.22, 5.52, 4.92 and 4.38 µm. The 4.92 µm line pairs are resolved, suggesting a resolution of better than 5 µm. This is consistent with the approximately X2 magnification provided by the micro-objective.
Fig. 3.

Lateral resolution and field-of-view of (a) high resolution and (b) low resolution probes, evaluated by imaging a USAF resolution target. The three color channels (red, green and blue) of the camera are shown individually and combined (white).
As expected, the high resolution probe performed best in the green channel as this corresponds to its design wavelength. Some lateral chromatic aberrations are visible in the white light image which we did not attempt to correct for. Pincushion distortion introduced by the micro-objective can also be seen; again we did not apply any correction to the images.
For the low resolution probe we observed a non-uniformity of intensity across the images. This arose because the illumination fiber lies to one side of the imaging fiber, and so the tissue is not uniformly illuminated over the field-of-view. Figure 4 shows the relative mean signal detected from porcine bowel tissue, averaged over 400 image frames with the probe in motion, and with a (sigma = 5 pixel) Gaussian filter applied. It can be seen that there is approximately a 50% drop-off in signal across the image. We did not attempt to apply a general correction for this effect as the non-uniformity will depend on the scattering properties of the particular tissue being imaged. When we performed the same operation for the high resolution probe, shown in Fig. 4(b), the effect isn’t as obvious, although there is a slightly lower intensity in the bottom left of the image. In this case the effect may be masked by a large reduction in intensity around the perimeter of the image which is attributable to off-axis aberrations of the micro-objective.
Fig. 4.
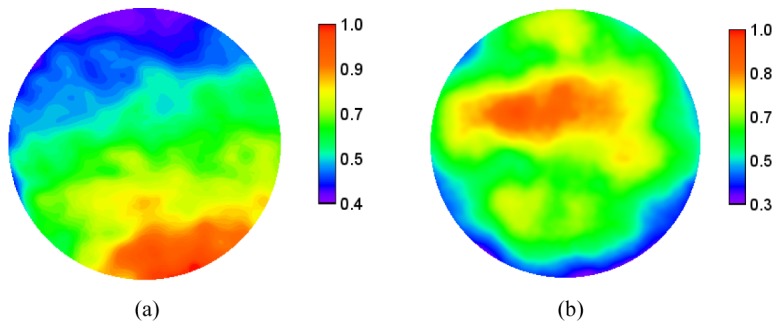
Non uniformity of (a) low resolution and (b) high resolution probes. The scale shows the relative image intensity averaged across 400 frames when imaging porcine large bowel mucosa. The non-uniformity for the low resolution probe arises because of the location of the illumination fiber.
Figure 5 shows the effect of image processing used to remove the honeycomb pattern arising from the discrete fiber bundle cores. Figure 5(a) is a raw image, with a zoom to show the core pattern in Fig. 5(b). This is decomposed into red, green and blue channels in Fig. 5(c), 5(d) and 5(e). Figure 5(f) shows the image after application of the Gaussian filter, with a zoom shown in Fig. 5(g).
Fig. 5.

Removal of fiber bundle core pattern. (a) Raw image of sponge stained with toluidine blue 0.25%, using the high resolution probe; (b) zoom on raw image showing honeycomb pattern of cores; (c,d,e) red, green and blue channels of image in (b), showing that cores can be resolved in each; (f) processed image following application of Gaussian filter; (g) zoom on processed image showing core pattern is no longer visible. Note that the brightness of the color channel images was adjusted for easier viewing. For presentation, the zoomed images were interpolated from 100x100 to 300x300 pixels by cubic interpolation.
3.2 Endocytoscopy tissue validation
We tested the ability of the system to generate usable images by studying several samples of porcine gastrointestinal tract tissue samples ex vivo. All were stained with toluidine blue 0.25%, a nuclear stain [34] which has been shown to be particularly effective for endocytoscopy of the stomach and colon [35]. Several drops of the stain were applied to the tissue surface from a pipette and left in place for approximately two minutes. Excess stain was then washed away using several drops of water from a second pipette.
Figure 6 shows example images using both the high and low resolution probes from mucosa of the large intestine and stomach. Media 1 (3.7MB, AVI) shows short clips of live videos at 15 frames per second (fps). Although this imaging was conducted ex vivo, the sites are accessible by endoscope, and numerous examples are available in the literature of in situ imaging both using camera-based endocytoscopy and fiber-bundle confocal fluorescence endomicroscopy [18]. The mucosal crypts are visible in both Fig. 6(a) and 6(c). They are lined with epithelial cells on the crypt edges, although these are not always clearly defined. In Fig. 6(b) and 6(d) gastric tubular structures are clearly visible, although individual nuclei cannot be identified.
Fig. 6.
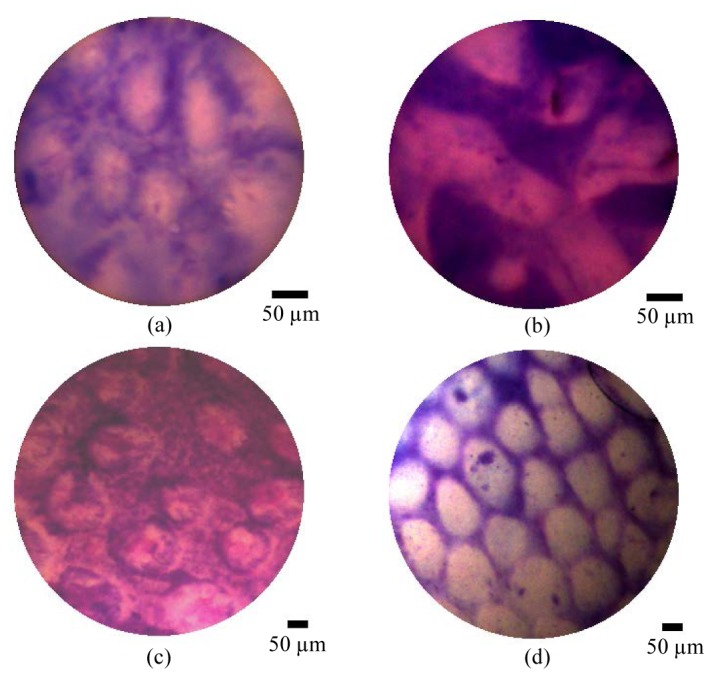
Ex vivo porcine tissue imaging results showing using high resolution probe (a,b) and low resolution probe (c,d). showing (a,c) large bowel mucosa, and (b,d) stomach mucosa. All samples were stained with toluidine blue 0.25% for approximately 2 minutes and washed before imaging. A selection of videos from the two tissue types are shown in Media 1 (3.7MB, AVI) . Scale bars are 50 µm.
3.3 Hybrid system
The hybrid system enables confocal fluorescence and white light endocytoscopy images to be acquired through a single fiber bundle, either sequentially (imaging first with one channel, and then the other) or quasi-simultaneously (acquisition of alternate frames). One of the principal differences between the two channels is the optical sectioning capability of confocal endomicroscopy. We confirmed this rejection of out-of-focus background by measuring the axial sensitivity of the confocal channel using both probes. We removed the emission filter so as to detect reflected light and placed the probes in contact with a mirror. A motorized stage was then used to drive the probes away from the mirror. An image frame was acquired for every 1 µm of axial motion. We averaged over a 35 x 35 µm square area (on the proximal end of the bundle) to calculate the sensitivity at each axial position. While this measures the sensitivity profile in reflection, rather than fluorescence mode, and hence can only be considered an approximation to the true profile, this method removes the need for a single surface fluorescent target.
Figures 7(a) and 7(b) show 35 µm wide cross section through the axial stack for the low and high resolution probes respectively. The sensitivity profiles, acquired by averaging across (a) and (b), are shown in Fig. 7(c). The sectioning depth range is, using a half-maximum criterion, approximately 21 µm and 16 µm for the low and high resolution probes respectively. Note that for the low resolution probe, maximum sensitivity is obtained when the target is in direct contact with the fiber bundle, making the sensitivity profile single-sided. In contrast, the high resolution probe has a finite working distance and hence a more conventional double-sided sensitivity profile. These values are large compared with a typical confocal microscope, partly due to the choice of a large effective pinhole size. A smaller sectioning range could be obtained by decreasing this pinhole size, although the sectioning range is ultimately limited by the effective size of the fiber bundle cores as projected onto the tissue.
Fig. 7.

Axial sensitivity profile measurements for confocal channel. (a) Cross-sectional image of mirror using high resolution probe; (b) cross-sectional image of mirror using low resolution probe; (c) axial sensitivity profiles for high (blue) and low (red) resolution probes.
Validation of the principle of combined imaging was performed in two stages. First we performed sequential imaging of tissue stained first with acriflavine hydrochloride, and then with toluidine blue using the combined system described above. Figure 8(a) shows an example image frame from porcine large bowel, stained with 0.05% acriflavine hydrochloride for approximately two minutes before washing. The morphology of the mucosal crypts is clearly visible. We would expect the images to appear somewhat different if staining was instead performed using intravenous fluorescein, as is the case for most in vivo endomicroscopy studies. We then stained the same sample with 0.25% toluidine blue and imaged approximately the same area of tissue with the endocytoscopy channel. An example image frame is shown in Fig. 8(b).
Fig. 8.

Ex vivo porcine large bowel mucosa imaged using the combined endocytoscopy and fluorescence confocal endomicroscopy system, using the high resolution probe. The tissue was first stained with acriflavine hydrochloride 0.05% and imaged with the fluorescence channel (a) and subsequently stained with toluidine blue 0.25% and imaged with the endocytoscopy channel (b). Scale bars are 50 µm.
To demonstrate the potential to use the two channels simultaneously, we first imaged lens tissue paper stained with 0.05% acriflavine. Figures 9(a) and 9(b) demonstrate that the tissue structure was clearly visible in both channels. Repeating this experiment with porcine large bowel tissue proved more difficult because toluidine blue absorbs strongly in the wavelength range at which acriflavine hydrochloride fluoresces, meaning that much of the fluorescence was quenched. We lowered the toluidine blue concentration to 0.01% and obtained the image shown in Fig. 9(c) and 9(d). While the crypts are now clearly visible in both channels, the image quality remains markedly reduced. It therefore seems likely that a different staining regime will be required in order to allow effective simultaneous use of the two channels. One stain with potential is cresyl violet which has previously been used both in endocytoscopy [18] and confocal fluorescence endomicroscopy [36]. Alternatively, intravenous staining with fluorescein may provide clearer images.
Fig. 9.

Simultaneous fluorescence confocal endomicroscopy (a,c) and endocytoscopy (b,d). (a,b) Lens tissue paper stained with acriflavine hydrochloride 0.05%, imaged with high resolution probe; (b,d) porcine large bowel mucosa ex vivo stained with acriflavine hydrochloride 0.05% and toluidine blue 0.1%, imaged with high resolution probe; (e) merged fluorescence endomicroscopy and endocytoscopy images of large bowel mucosa with fluorescence shown in green. Scale bar is 50 µm
It may be desirable to merge the two channels to create a single image. An example is shown in Fig. 9(e) where the fluorescence image has been overlayed in green onto the endocytoscope image. Such merged images must be treated with some caution as there is (in the present implementation) an average of 100 ms delay between the acquisition of the two channels. This could potentially lead to motion artifacts in the merged image. In this case we specifically chose a point at which the probe was not in motion, although a small offset can nevertheless be observed.
4. Discussion
The new endocytoscope device reported in this paper was developed mainly as a proof-of-concept prototype and a number of improvements could be made, some of which are discussed below. Nevertheless, the images shown in Fig. 6 demonstrate that fiber bundle endocytoscopy is a viable imaging technique. Direct comparisons with results in the literature from the Olympus prototypes are difficult as the imaging conditions were somewhat different and, in the case of the images shown here, it is likely that the staining procedure was not optimal. However, by way of example we suggest comparison with Figs. 4 and 5 of Ref [35]. which shows images of porcine stomach and large bowel using various concentrations of toluidine blue. Given that we were able to visualize important morphological features such as mucosal crypts in the large bowel mucosa, we anticipate that future studies using fiber bundle endocytoscopy will demonstrate diagnostic potential.
However, replacement of the camera with a fiber bundle is not without its drawbacks. Firstly, the resolution of the microscope is limited by the spacing between individual fiber cores, as well as by cross-coupling between cores along the bundle. This problem can be reduced by incorporating magnifying optics into the distal tip of the probe, such that the fiber bundle is projected to a smaller diameter on the tissue. However, this also reduces the field-of-view by the same factor. The resolution we obtained here was poorer than that reported for the Olympus prototypes, but we also obtained a larger field-of-view. There would be no difficulty in principle in incorporating a higher magnification micro-objective into the probe, although it is important that this is well corrected across the visible range of wavelengths.
The micro-objective used for the high resolution probe was optimized for fluorescence imaging around 500 nm rather than for this particular application. As a result, chromatic aberrations were present, including a manufacturer-specified focal depth shift of approximately 0.5 µm/nm. In principle it should be possible to incorporate a better-corrected micro-objective. For example, designs have been suggested for achromatic objectives for use in endomicroscopy [37] and probes for the Cellvizio Dual Band Endomicroscope (Mauna Kea Technologies) allow simultaneous excitation at 488 and 660 nm [38].
A second, related problem is that the raw images are corrupted by a honeycomb-like pattern corresponding to the individual cores and shared cladding of the bundle. A number of image processing methods have been suggested for removing this kind of pattern, ranging from simple spatial filtering [39] to more complex methods involving segmentation of individual cores [5]. Here we used a simple Gaussian filter to remove the pattern; this was selected for robustness and to avoid the need for calibration. It would certainly be possible to incorporate more complex reconstruction procedures which may yield some improvement in resolution and signal to noise ratio.
There are also considerable light collection inefficiencies involved in the use of a fiber bundle, as not all of the light collected by the objective will be successfully coupled into the bundle. However, this limitation is countered somewhat by the possibility of using a scientific grade CCD camera with a higher quantum efficiency and reduced noise. The CCD camera used here was a reasonably low-end (less than $2000 USD) scientific camera, and so we would expect a low noise camera to yield less noisy images. However, inspection of the images suggests that the quality is not limited by shot or electronic noise.
Results shown in Fig. 4 demonstrate that there is a non-uniformity of intensity across the image due to the offset of the illumination fiber. In practice, we do not expect this to significantly impact clinical interpretation of the images since non-uniformity will also arise from a number of other sources, including the probe-tissue contact pressure and local variations in quality of the staining. In principle, the non-uniformity could be reduced by using an array of illumination fibers encircling the imaging bundle, or by using some of the outer cores of the bundle to provide illumination.
High quality imaging requires a careful and consistent approach to staining, and it is certainly possible that these images could have been improved further by a variation in the staining technique. In particular we did not apply a mucolytic agent to break down mucus [40]. A particular disadvantage of endocytoscopy in comparison with fluorescence endomicroscopy is the need to apply a topical stain rather than making an intravenous administration of fluorescein. This topical staining process is time consuming [20], and so may limit the area of surveillance. Endocytoscopy is also limited to very superficial tissue layers [20], whereas confocal endomicroscopy can, in principle, penetrate up to a few hundred microns [8]. However, these are problems which are also faced by camera-based endocytoscopy rather than particular limitations of our approach. Indeed, by demonstrating that fluorescence confocal endomicroscopy can be acquired sequentially or simultaneously we have opened up the possibility of using both techniques to their advantage. Fluorescence endomicroscopy could be used initially to survey a large area of tissue and endocytoscopy to examine particular areas in more detail. Whether this has practical applications will depend on the diagnostic value of endocytoscopy relative to endomicroscopy.
A number of fiber bundle microscopy approaches have been reported, including confocal fluorescence [41], confocal reflectance [42], widefield fluorescence [11], widefield reflectance [43], structured illumination [9, 10] and line-scanning endomicroscopy [44]. The confocal fluorescence modality has so far shown the widest range of applicability and has been well validated in a number of clinical trials, and it was for this reason that we used this technique to demonstrate dual modality imaging, However, widefield endomicroscopy has been reported to be a simple and low cost alternative for applications where topical (rather than intravenous) fluorescent stains can be used [11, 45, 46]. We would expect that widefield techniques would also be compatible with fiber bundle endocytoscopy, and so there are several possibilities to develop low cost, multi-modal endomicroscopes. For example, one possibility would be to simultaneously acquire widefield fluorescence and endocytoscopy images using a single camera (fitted with a filter to reject the fluorescence excitation light), leading to a highly compact and versatile system.
5. Conclusion
We have demonstrated that endocytoscopy may be feasible using a fibre-bundle based probe rather than a miniaturized camera. The low light throughput of imaging bundles does not appear to be a limiting factor, even with a relatively weak illumination source. The bundle does unfortunately limit the lateral resolution, but this could be improved using a high magnification, achromatic micro-objective, something which is well within current technological limits. The most promising benefit arising from the use of a fiber bundle is the possibility of combination with confocal endomicroscopy. We have shown that effective imaging is possible when the two channels are used sequentially, and that simultaneous imaging is also possible in principle. Further work will be required to identify an optimal staining approach for this ‘dual-mode’ imaging, but the additional information it could provide may help to make to the optical biopsy a more effective alternative to histology.
Acknowledgments
We would like to thank Petros Giataganas, Vasiliki Simaiaki and Siyang Zuo for assistance with tissue preparation. This work was supported by EPSRC Grant EP/I027769/1: SMART Endomicroscopy.
References and links
- 1.Riedl O., Fitzal F., Mader N., Dubsky P., Rudas M., Mittlboeck M., Gnant M., Jakesz R., “Intraoperative frozen section analysis for breast-conserving therapy in 1016 patients with breast cancer,” Eur. J. Surg. Oncol. 35(3), 264–270 (2009). 10.1016/j.ejso.2008.05.007 [DOI] [PubMed] [Google Scholar]
- 2.Shukla R., Abidi W. M., Richards-Kortum R., Anandasabapathy S., “Endoscopic imaging: How far are we from real-time histology?” World J Gastrointest Endosc 3(10), 183–194 (2011). 10.4253/wjge.v3.i10.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J. T. C., Loewke N. O., Mandella M. J., Levenson R. M., Group C., “Review article: Modern trends in imaging ii point-of-care pathology with miniature microscopes,” Pathology 34, 81–98 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton R. C., Kemp S. V., Shah P. L., Elson D., Darzi A., Shibuya K., Mulgrew S., Yang G.-Z., “Progress toward optical biopsy: Bringing the microscope to the patient,” Lung 189(2), 111–119 (2011). 10.1007/s00408-011-9282-7 [DOI] [PubMed] [Google Scholar]
- 5.Goualher G. L., Perchant A., Genet M., Cav C., Viellerobe B., Abrat B., Ayache N., “Towards optical biopsies with an integrated fibered confocal fluorescence microscope,” MICCAI 2004, 761–768 (2004). [Google Scholar]
- 6.Gmitro A. F., Aziz D., “Confocal microscopy through a fiber-optic imaging bundle,” Opt. Lett. 18(8), 565–567 (1993). 10.1364/OL.18.000565 [DOI] [PubMed] [Google Scholar]
- 7.Jabbour J. M., Saldua M. A., Bixler J. N., Maitland K. C., “Confocal endomicroscopy: Instrumentation and medical applications,” Ann. Biomed. Eng. 40(2), 378–397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polglase A. L., McLaren W. J., Skinner S. A., Kiesslich R., Neurath M. F., Delaney P. M., “A fluorescence confocal endomicroscope for in vivo microscopy of the upper- and the lower-gi tract,” Gastrointest. Endosc. 62(5), 686–695 (2005). 10.1016/j.gie.2005.05.021 [DOI] [PubMed] [Google Scholar]
- 9.Bozinovic N., Ventalon C., Ford T., Mertz J., “Fluorescence endomicroscopy with structured illumination,” Opt. Express 16(11), 8016–8025 (2008). 10.1364/OE.16.008016 [DOI] [PubMed] [Google Scholar]
- 10.Ford T. N., Lim D., Mertz J., “Fast optically sectioned fluorescence hilo endomicroscopy,” J. Biomed. Opt. 17(2), 021105 (2012). 10.1117/1.JBO.17.2.021105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muldoon T. J., Anandasabapathy S., Maru D., Richards-Kortum R., “High-resolution imaging in barrett’s esophagus: A novel, low-cost endoscopic microscope,” Gastrointest. Endosc. 68(4), 737–744 (2008). 10.1016/j.gie.2008.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helmchen F., Fee M. S., Tank D. W., Denk W., “A miniature head-mounted two-photon microscope. High-resolution brain imaging in freely moving animals,” Neuron 31(6), 903–912 (2001). 10.1016/S0896-6273(01)00421-4 [DOI] [PubMed] [Google Scholar]
- 13.Göbel W., Kerr J. N. D., Nimmerjahn A., Helmchen F., “Miniaturized two-photon microscope based on a flexible coherent fiber bundle and a gradient-index lens objective,” Opt. Lett. 29(21), 2521–2523 (2004). 10.1364/OL.29.002521 [DOI] [PubMed] [Google Scholar]
- 14.Tearney G. J., Brezinski M. E., Bouma B. E., Boppart S. A., Pitris C., Southern J. F., Fujimoto J. G., “In vivo endoscopic optical biopsy with optical coherence tomography,” Science 276(5321), 2037–2039 (1997). 10.1126/science.276.5321.2037 [DOI] [PubMed] [Google Scholar]
- 15.Zysk A. M., Nguyen F. T., Oldenburg A. L., Marks D. L., Boppart S. A., “Optical coherence tomography: A review of clinical development from bench to bedside,” J. Biomed. Opt. 12(5), 051403 (2007). 10.1117/1.2793736 [DOI] [PubMed] [Google Scholar]
- 16.Osdoit A., Lacombe F., Cavé C., Loiseau S., Peltier E., “To see the unseeable: Confocal miniprobes for routine microscopic imaging during endoscopy,” Proc. SPIE 6432, 64320F (2007). 10.1117/12.701004 [DOI] [Google Scholar]
- 17.Barlis P., Schmitt J. M., “Current and future developments in intracoronary optical coherence tomography imaging,” EuroIntervention: journal of EuroPCR in collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology 4, 529 (2009). [DOI] [PubMed]
- 18.Neumann H., Fuchs F. S., Vieth M., Atreya R., Siebler J., Kiesslich R., Neurath M. F., “Review article: In vivo imaging by endocytoscopy,” Aliment. Pharmacol. Ther. 33(11), 1183–1193 (2011). 10.1111/j.1365-2036.2011.04647.x [DOI] [PubMed] [Google Scholar]
- 19.N. Hasegawa, “Magnifying image pickup unit for an endoscope, an endoscope for in vivo cellular observation that uses it, and endoscopic, in vivo cellular observation methods,” US Patent 7267648 (2007).
- 20.Inoue H., Kudo S. E., Shiokawa A., “Technology insight: Laser-scanning confocal microscopy and endocytoscopy for cellular observation of the gastrointestinal tract,” Nat. Clin. Pract. Gastroenterol. Hepatol. 2(1), 31–37 (2005). 10.1038/ncpgasthep0072 [DOI] [PubMed] [Google Scholar]
- 21.Szeto C., Wehrli B., Whelan F., Franklin J., Nichols A., Yoo J., Fung K., “Contact endoscopy as a novel technique in the detection and diagnosis of mucosal lesions in the head and neck: A brief review,” J. Oncol. 2011, 196302 (2011). 10.1155/2011/196302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamou J., “Microhysteroscopy — A new technique in endoscopy and its applications,” Acta Endoscopica 10(5-6), 415–422 (1980). 10.1007/BF02973531 [DOI] [Google Scholar]
- 23.Tada M., Nishimura S., Watanabe Y., “A new method for the ultra-magnifying observation of the colon mucosa,” Kyoto Pref. Univ. Med. 91, 349–354 (1982). [Google Scholar]
- 24.Dekker E., Kara M., Offerhaus J., Fockens P., “Endocytoscopy in the colon: Early experience with a new real-time contact microscopy system,” Gastrointest. Endosc. 61(5), AB224 (2005). 10.1016/S0016-5107(05)01230-7 [DOI] [Google Scholar]
- 25.Pohl H., Koch M., Khalifa A., Papanikolaou I. S., Wiedenmann B., Roesch T., “Evaluation of an endocytoscopy system in the surveillance of patients with barrett esophagus,” Gastrointest. Endosc. 63(5), AB239 (2006). 10.1016/j.gie.2006.03.618 [DOI] [PubMed] [Google Scholar]
- 26.Neumann H., Vieth M., Neurath M. F., Fuchs F. S., “In vivo diagnosis of small-cell lung cancer by endocytoscopy,” J. Clin. Oncol. 29(6), e131–e132 (2011). 10.1200/JCO.2010.31.8097 [DOI] [PubMed] [Google Scholar]
- 27.Ohigashi T., Kozakai N., Mizuno R., Miyajima A., Murai M., “Endocytoscopy: Novel endoscopic imaging technology for in-situ observation of bladder cancer cells,” J. Endourol. 20(9), 698–701 (2006). 10.1089/end.2006.20.698 [DOI] [PubMed] [Google Scholar]
- 28.Rao G. V., Pradeep R., Mansard M. J., Ramji C., Banerjee R., Nageshwar Reddy D., “Endocytoscopy assists in the intraoperative diagnosis of carcinoma in a patient with chronic pancreatitis,” Endoscopy 39(S1Suppl 1), E317–E318 (2007). 10.1055/s-2007-966798 [DOI] [PubMed] [Google Scholar]
- 29.Neumann H., Atreya R., Vieth M., Neurath M. F., “Toluidine blue is as effective as methylene blue for endocytoscopy,” Gastrointest. Endosc. 75(4), 479 (2012). 10.1016/j.gie.2012.03.1306 [DOI] [Google Scholar]
- 30.Inoue H., Yokoyama A., Kudo S. E., “Ultrahigh magnifying endoscopy: Development of cm double staining for endocytoscopy and its safety,” Nippon Rinsho 68(7), 1247–1252 (2010). [PubMed] [Google Scholar]
- 31.Ohigashi T., Kozakai N., Mizuno R., Miyajima A., Murai M., “Endocytoscopy: Novel endoscopic imaging technology for in-situ observation of bladder cancer cells,” J. Endourol. 20(9), 698–701 (2006). 10.1089/end.2006.20.698 [DOI] [PubMed] [Google Scholar]
- 32.Minami H., Inoue H., Yokoyama A., Ikeda H., Satodate H., Hamatani S., Haji A., Kudo S., “Recent advancement of observing living cells in the esophagus using cm double staining: Endocytoscopic atypia classification,” Dis. Esophagus 25(3), 235–241 (2012). 10.1111/j.1442-2050.2011.01241.x [DOI] [PubMed] [Google Scholar]
- 33.Chang K., “Eus-guided needle-based confocal laser induced endomicroscopy (ncle): A correlation study of “through the needle” imaging with normal histology in a porcine model,” Endoscopy 43(S 03), A14 (2011). 10.1055/s-0031-1292085 [DOI] [Google Scholar]
- 34.Sridharan G., Shankar A. A., “Toluidine blue: A review of its chemistry and clinical utility,” J Oral Maxillofac Pathol 16(2), 251–255 (2012). 10.4103/0973-029X.99081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kodashima S., Fujishiro M., Takubo K., Kammori M., Nomura S., Kakushima N., Muraki Y., Tateishi A., Kaminishi M., Omata M., “Ex-vivo study of high-magnification chromoendoscopy in the gastrointestinal tract to determine the optimal staining conditions for endocytoscopy,” Endoscopy 38(11), 1115–1121 (2006). 10.1055/s-2006-944915 [DOI] [PubMed] [Google Scholar]
- 36.Goetz M., Toermer T., Vieth M., Dunbar K., Hoffman A., Galle P. R., Neurath M. F., Delaney P., Kiesslich R., “Simultaneous confocal laser endomicroscopy and chromoendoscopy with topical cresyl violet,” Gastrointest. Endosc. 70(5), 959–968 (2009). 10.1016/j.gie.2009.04.016 [DOI] [PubMed] [Google Scholar]
- 37.Kyrish M., Tkaczyk T. S., “Achromatized endomicroscope objective for optical biopsy,” Biomed. Opt. Express 4(2), 287–297 (2013). 10.1364/BOE.4.000287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vercauteren T., Doussoux F., Cazaux M., Schmid G., Linard N., Durin M.-A., Gharbi H., Lacombe F., “Multicolor probe-based confocal laser endomicroscopy: A new world for in vivo and real-time cellular imaging,” Proc. SPIE 8575, 857504 (2013). 10.1117/12.2002490 [DOI] [Google Scholar]
- 39.Han J.-H., Lee J., Kang J. U., “Pixelation effect removal from fiber bundle probe based optical coherence tomography imaging,” Opt. Express 18(7), 7427–7439 (2010). 10.1364/OE.18.007427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh R., Chen Yi Mei S. L., Tam W., Raju D., Ruszkiewicz A., “Real-time histology with the endocytoscope,” World J. Gastroenterol. 16(40), 5016–5019 (2010). 10.3748/wjg.v16.i40.5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.G. L. Goualher, A. Perchant, M. Genet, C. Cav, B. Viellerobe, B. Abrat, and N. Ayache, “Towards optical biopsies with an integrated fibered confocal fluorescence microscope,” in MICCAI (2004), pp. 761–768. [Google Scholar]
- 42.Liang C., Descour M. R., Sung K. B., Richards-Kortum R., “Fiber confocal reflectance microscope (fcrm) for in-vivo imaging,” Opt. Express 9(13), 821–830 (2001). 10.1364/OE.9.000821 [DOI] [PubMed] [Google Scholar]
- 43.Liu X., Huang Y., Kang J. U., “Dark-field illuminated reflectance fiber bundle endoscopic microscope,” J. Biomed. Opt. 16(4), 046003 (2011). 10.1117/1.3560298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makhlouf H., Gmitro A. F., Tanbakuchi A. A., Udovich J. A., Rouse A. R., “Multispectral confocal microendoscope for in vivo and in situ imaging,” J. Biomed. Opt. 13(4), 044016 (2008). 10.1117/1.2950313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pierce M. C., Vila P. M., Polydorides A. D., Richards-Kortum R., Anandasabapathy S., “Low-cost endomicroscopy in the esophagus and colon,” Am. J. Gastroenterol. 106(9), 1722–1724 (2011). 10.1038/ajg.2011.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Regunathan R., Woo J., Pierce M. C., Polydorides A. D., Raoufi M., Roayaie S., Schwartz M., Labow D., Shin D., Suzuki R., Bhutani M. S., Coghlan L. G., Richards-Kortum R., Anandasabapathy S., Kim M. K., “Feasibility and preliminary accuracy of high-resolution imaging of the liver and pancreas using fna compatible microendoscopy (with video),” Gastrointest. Endosc. 76(2), 293–300 (2012). 10.1016/j.gie.2012.04.445 [DOI] [PMC free article] [PubMed] [Google Scholar]