

Abstract
Autophagy related 16-like 1 (ATG16L1) as a genetic risk factor has exposed the critical role of autophagy in Crohn’s disease (CD)1. Homozygosity for the highly prevalent ATG16L1 risk allele, or murine hypomorphic (HM) activity causes Paneth cell dysfunction2,3. As Atg16l1HM mice do not develop spontaneous intestinal inflammation, the mechanism(s) by which ATG16L1 contributes to disease remains obscure. Deletion of the unfolded protein response (UPR) transcription factor X-box binding protein-1 (Xbp1) in intestinal epithelial cells (IECs), whose human orthologue harbors rare inflammatory bowel disease (IBD) risk variants, results in endoplasmic reticulum (ER) stress, Paneth cell impairment and spontaneous enteritis4. Unresolved ER stress is a common feature of IBD epithelium4,5, and several genetic risk factors of CD affect Paneth cells2,4,6-9. Here we show that impairment in either UPR (Xbp1ΔIEC) or autophagy function (Atg16l1ΔIEC or Atg7ΔIEC) in IECs results in each other’s compensatory engagement, and severe spontaneous CD-like transmural ileitis if both mechanisms are compromised. Xbp1ΔIEC mice exhibit autophagosome formation in hypomorphic Paneth cells, which is linked to ER stress via protein kinase RNA-like endoplasmic reticulum kinase (PERK), elongation initiation factor 2α (eIF2α) and activating transcription factor 4 (ATF4). Ileitis is dependent on commensal microbiota and derives from increased IEC death, inositol requiring enzyme 1α (IRE1α)-regulated NFκB activation and tumor necrosis factor signaling which are synergistically increased when autophagy is deficient. ATG16L1 restrains IRE1α activity and augmentation of autophagy in IECs ameliorates ER stress-induced intestinal inflammation and eases NFκB overactivation and IEC death. ER stress, autophagy induction and spontaneous ileitis emerge from Paneth cell-specific deletion of Xbp1. Genetically and environmentally controlled UPR function within Paneth cells may therefore set the threshold for the development of intestinal inflammation upon hypomorphic ATG16L1 function and implicate ileal CD as a specific disorder of Paneth cells.
The UPR and autophagy are integrally linked pathways10. To investigate their relationship in the intestinal epithelium, we stably transduced the small IEC line MODE-K with a short hairpin Xbp1 (shXbp1) lentiviral vector (Extended Data Fig. 1a)4. We observed increased levels of ATG7, Beclin1 (Extended Data Fig. 1b) and phosphatidylethanolamine (PE)-conjugated microtubule-associated protein 1 light chain 3 β (LC3-II) relative to LC3-I compared to control-silenced (shCtrl) cells (Extended Data Fig. 1c). Increased autophagic flux accounted for this given increased levels of LC3-II relative to LC3-I observed after inhibition of autophagosome-lysosome fusion by bafilomycin11 (Extended Data Fig. 1d). Increased GFP-LC3 punctae were seen in shXbp1 compared to shCtrl MODE-K cells transfected with a green fluorescent protein (GFP)-LC3-expressing vector12 (Extended Data Fig. 1e, f), and increased numbers of autophagic vacuoles in shXbp1 compared to shCtrl cells (Extended Data Fig.1g, h). Accordingly, isolated primary IECs from the small intestine of Villin (V)-Cre+;Xbp1fl/fl (‘Xbp1ΔIEC’)4 mice backcrossed onto C57BL/6 (B6) exhibited nearly complete consumption of LC3-I and a relative increase in LC3-II (Fig. 1a and Extended Data Fig.1i), stable amounts of ATG7 presumably reflecting a combination of increased production and consumption, elevated levels of ATG16L1 and Beclin1 (Fig. 1b and Extended Data Fig. 1j) and autophagosomes and degradative autophagic vacuoles consistent with autophagy induction in hypomorphic4 Paneth cells, and to a lesser extent goblet cells (data not shown), compared to V-Cre−;Xbp1fl/fl (Wt) mice (Fig. 1c and Extended Data Fig. 1k). To gain temporal control of Xbp1 deletion and ability to monitor autophagy in situ and exclude the role of chronic inflammation4 in this induction, we generated V-CreERT2;Xbp1fl/fl (‘Xbp1T-ΔIEC’) mice on a B6 background crossed to GFP-LC3 transgenic mice12. Three days after tamoxifen-induced Xbp1 deletion (Extended Data Fig.2a), although mature Paneth cells remained present with little detectable inflammation (data not shown), punctate GFP signal accumulation was greatest at the bottom of the crypts of Lieberkühn (Fig. 1d, e), and co-localized with lysozyme-positive Paneth cells (Extended Data Fig. 2b). Purified crypts of Xbp1T-ΔIEC mice revealed increased LC3-I/II conversion and reduced p62 compared to Wt mice (Extended Data Fig.2c). Thus, Xbp1 loss in IECs induced autophagy most notably in Paneth cells.
Figure 1. PERK/eIF2α signaling induces autophagy in Xbp1-deficient intestinal epithelial cells.
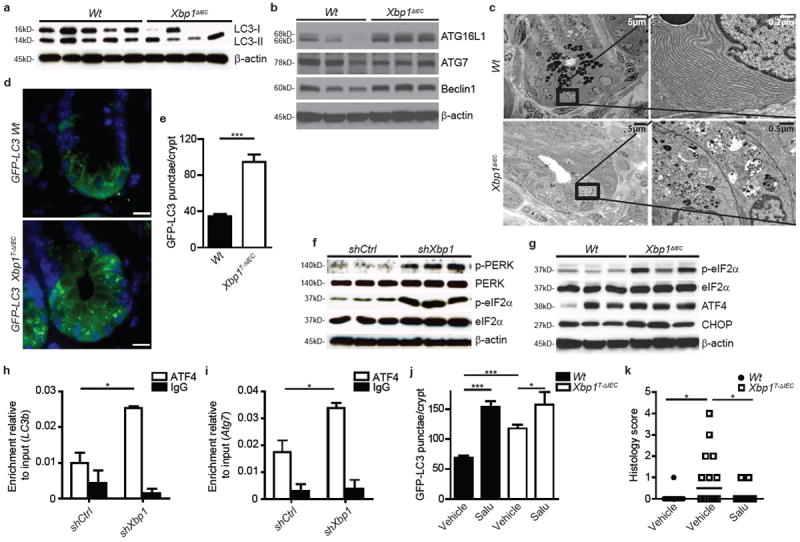
a, b, Immunoblot for LC3 conversion in isolated primary IECs (a) (n=5/4) and for autophagy proteins in primary IEC scrapings (b) (n=3). c, Transmission electron microscopy (TEM) of crypts. Note autophagic vacuoles in various stages of evolution in Xbp1ΔIEC hypomorphic Paneth cells. d, e, Crypt showing GFP-LC3 punctae (d), quantified in (e) (n=10; unpaired Student’s t-test). Bar, 5 μm. f, g, Immunoblot of silenced MODE-K cells (f) and primary IEC scrapings (g) for the PERK/eIF2α branch (n=3). h, i, Promoter sequence qPCR for Map1lc3b (LC3b) (h) and Atg7 (i) after anti-ATF4 ChIP (unpaired Student’s t-test). j, GFP-LC3 punctae per crypt after treatment with tamoxifen for 3 days and vehicle or salubrinal (n=10; one-way ANOVA with post-hoc Bonferroni). (k) Enteritis histology score after salubrinal and tamoxifen co-treatment (n=12/14/13; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). Results represent three (a, f, g) or two (c, e, h, i) independent experiments. *P < 0.05, ***P < 0.001.
Although Xbp1-deficient IECs exhibited broad evidence of ER stress4 (Extended Data Fig. 2d-j), an examination of shXbp1, relative to shCtrl, MODE-K cells demonstrated a particularly significant increase in phosphorylated (p)-PERK and its substrate p-eIF2α (Fig. 1f) with the latter reversed by Perk-silencing identifying it as the factor responsible for p-eIF2α formation (Extended Data Fig. 3a). Increased p-eIF2α was also detected in primary IECs of Xbp1ΔIEC (Fig. 1g and Extended Data Fig. 3b) and Xbp1T-ΔIEC mice (Extended Data Fig. 3c). Consistent with PERK-eIF2α involvement in autophagy induction, ATF4, a transcriptional effector of this pathway, and its transcriptional target, C/EBP-homologous protein (CHOP; encoded by Ddit3), were increased in primary IECs of Xbp1ΔIEC mice (Fig. 1g and Extended Data Fig. 3b), and chromatin-immunoprecipitation (ChIP) with anti-ATF4 demonstrated increased binding to the Map1lc3b (LC3b) (Fig. 1h) and Atg7 (Fig. 1i) promoters, both of which contain ATF4 binding sites13, in shXbp1 relative to shCtrl MODE-K cells. ATG7 is essential for the formation of the ATG12-ATG5 conjugate during autophagy10,14. shXbp1 MODE-K cells exhibited increased LC3b and Atg7 expression compared to shCtrl MODE-K cells (Extended Data Fig. 3d), and Perk co-silencing abrogated ATG7 induction observed in shXbp1 compared to shCtrl MODE-K cells (Extended Data Fig. 3a). Salubrinal, a selective inhibitor of eIF2α dephosphorylation15 (Extended Data Fig.2a), increased the accumulation of GFP-LC3 punctae primarily in Paneth cells, in both Xbp1-sufficient and –deficient IECs (Fig. 1j and Extended Data Fig. 2b and 3e), and provoked increased levels of LC3-II relative to LC3-I (Extended Data Fig. 3f) and CHOP (Extended Data Fig. 3f) in Xbp1T-ΔIEC mice, along with, importantly, an amelioration of the acute enteritis (Fig. 1k and Extended Data Fig. 3g). Similarly, silencing of growth arrest and DNA damage-inducible protein 34 (Gadd34; encoded by Ppp1r15a), part of the protein phosphatase 1 complex that dephosphorylates eIF2α16, led to increased eIF2α phosphorylation and ATG7 expression in shXbp1 compared to shCtrl MODE-K cells (Extended Data Fig. 3h, i). Xbp1ΔIEC;Gadd34+/− mice with hypomorphic GADD34 function exhibited increased p-eIF2α and ATG7 in purified crypt epithelial cells compared to Xbp1ΔIEC;Gadd34+/+ mice (Extended Data Fig. 3j). Thus, PERK-p-eIF2α is a critical mediator of UPR-induced autophagy primarily in Paneth cells consequent to XBP1-deficiency.
These studies let us hypothesize that autophagy may function as a compensatory mechanism in IECs upon sustained ER stress. We therefore generated V-Cre+;Atg7fl/fl;Xbp1fl/fl (‘Atg7/Xbp1ΔIEC’) mice14. IECs from Atg7ΔIEC mice lacked LC3-II formation and the ATG5-ATG12;ATG16L1 complex (Extended Data Fig. 4a). Atg7/Xbp1ΔIEC mice demonstrated a complete absence of UPR-induced autophagy (Fig. 2a and Extended Data Fig. 4a-c), and a remarkable worsening of ileitis compared to Xbp1ΔIEC mice. In notable contrast to Xbp1ΔIEC mice, where inflammation was limited to the mucosa, >70% of Atg7/Xbp1ΔIEC mice developed discontinuous submucosal or transmural inflammation, characterized by acute and chronic inflammation extending in an abrupt knife-like fashion to muscularis propria and serosa, closely resembling the early fissuring ulcerations and fistulous tracts observed in human CD (Fig. 2b, c and Extended Data Fig. 4d). In contrast to Xbp1ΔIEC mice, enteritis in Atg7/Xbp1ΔIEC mice progressed over the 18 week observation period such that at this time point all animals exhibited submucosal or transmural disease (Extended Data Fig. 4e, f).
Figure 2. Impairment of ER stress-induced compensatory autophagy results in severe transmural inflammation.
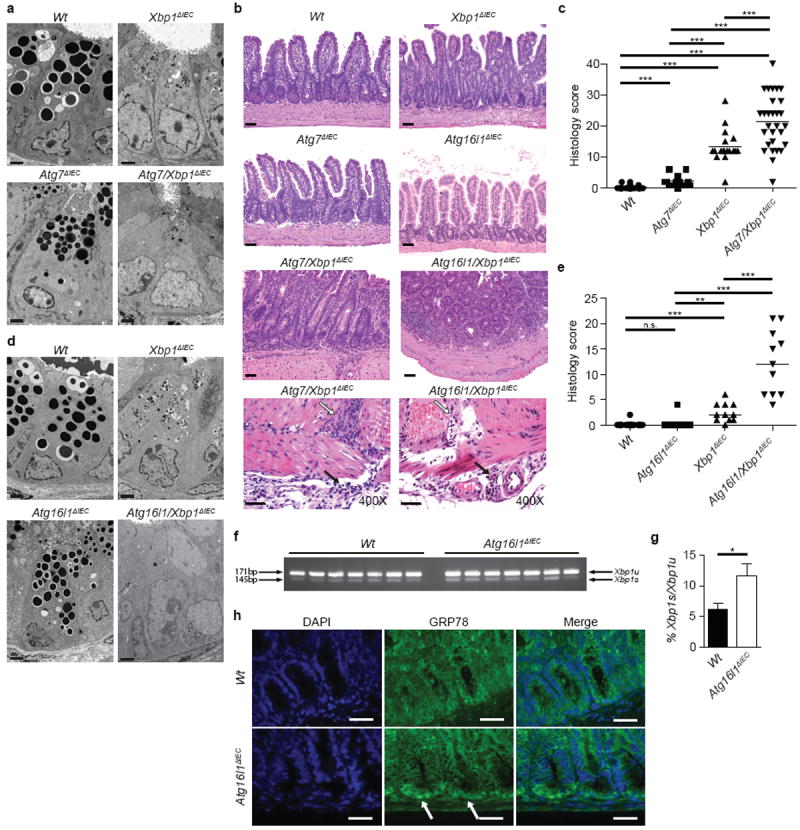
a, TEM of crypts of Lieberkühn (n=2). Bar, 2 μm. b, Representative H&E stainings. Note transmural inflammation extending through muscularis propria (white arrow) into serosa (black arrow) in Atg7/Xbp1ΔIEC and Atg16l1/Xbp1ΔIEC mice scored in (c) and (e). Bar, 50 and 10 μm, respectively. c, Enteritis histology score (n=26/12/18/27; 10-18 weeks; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). d, Crypt TEM in indicated genotypes (n=2). Bar, 2 μm. e, Enteritis histology score (n=11; 18 weeks; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). f, Xbp1 mRNA splicing (Xbp1u, unspliced; Xbp1s, spliced) of crypts, densitometry in (g) (n=7; unpaired Student’s t-test). h, GRP78 (green) immunofluorescence, white arrows indicate GRP78+ crypts (DAPI, blue; n=5). Bar, 10 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
ATG16L1 is a major genetic risk factor for CD1,17, especially ileal CD18. Complex formation of ATG16L1 protein with ATG12-ATG5 defines the site of LC3 PE conjugation during autophagosome formation19,20. ATG16L1 protein expression was markedly increased in Xbp1ΔIEC compared to Wt primary IECs (Fig. 1b and Extended Data Fig. 1j), presumably consequent to PERK/eIF2α/ATF4-dependent transactivation of Atg7 and LC3b promoters and stabilization by the ATG7-induced ATG12-ATG5 complex21. We therefore developed mice with a floxed Atg16l1 allele that would allow for IEC-specific deletion via V-Cre (‘Atg16l1ΔIEC’; Extended Data Fig. 4g-i). Paneth cells in Atg16l1ΔIEC mice demonstrated a reduction in their overall size and number of granules, similar to gene-trap-targeted Atg16l1HM mice2,3 (Extended Data Fig. 4j-n). IECs from Atg16l1ΔIEC, compared to Wt mice, exhibited reduced expression of ATG7 and the ATG12-ATG5 conjugate (Extended Data Fig. 5a), along with disruption of the secretory pathway with a distended ER, reduced size and number of secretory granules, a loss of homeostatic autophagy (Fig. 2d and Extended Data Fig.5b, c) and increased p62 immunoreactivity in crypts (Extended Data Fig. 5d). To address the role of ATG16L1 under ER stress conditions, we generated V-Cre+;Atg16l1fl/fl;Xbp1fl/fl (‘Atg16l1/Xbp1ΔIEC’) mice. Atg16l1/Xbp1ΔIEC mice, which lacked UPR-induced autophagy (Extended Data Fig. 5b, c), developed severe spontaneous ileitis compared to Xbp1ΔIEC or Atg16l1ΔIEC mice, with discontinuous submucosal or transmural inflammation in >70% of 18-week old animals (Fig. 2b, e and Extended Data Fig. 5e, f) with features similar to those observed in Atg7/Xbp1ΔIEC mice, and present in two distinct animal facilities (Fig. 2b, e and Extended Data Fig. 5g). This phenotype highlights the important compensatory role played by autophagy and in particular ATG16L1 in defending against inflammation arising from unabated ER stress precisely in the small intestinal epithelium as a consequence of XBP1-deficiency.
ATG7 hypofunction in hepatocytes can induce ER stress22, raising the possibility that cross-talk between the UPR and autophagy may be bi-directional in IECs. Isolated Atg16l1ΔIEC crypts exhibited increased Xbp1 splicing compared to Wt (Fig. 2f, g) and increased grp78 expression (Extended Data Fig. 5h) localized to the crypt bottom (Fig. 2h). Dextran sodium sulfate, a colitis model involving ER stress in IECs that can be treated with ER stress-relieving chaperones23,24, induced more inflammation in Atg16l1ΔIEC compared to Wt mice (Extended Data Fig. 5i-l), similar to Atg16l1HM mice3. Thus, disturbances in autophagy within IECs also affect the UPR, and autophagy-associated factors such as ATG16L1 endow IECs with the ability to mitigate ER stressors that are commonplace at the mucosal surface25.
We next turned our attention to mechanisms by which autophagy counteracts ER stress and synergistic increase in intestinal inflammation when absent. Increased numbers of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)+ cells in Atg16l1/Xbp1ΔIEC and Atg7/Xbp1ΔIEC mice (Extended Data Fig. 6a, b) correlated with enteritis severity in double-mutant, in contrast to Xbp1ΔIEC mice (Extended Data Fig. 6b), and, as demonstrated for Atg16l1/Xbp1ΔIEC mice, concomitantly with increasing age (Extended Data Fig. 6c and Video 1). Silencing of Atg16l1 in shXbp1 MODE-K cells significantly increased the proportion of apoptotic cells in vitro (Fig. 3a and Extended Data Fig. 6d), indicating that increased apoptosis could function as an initial event in intestinal inflammation. Further, IEC-associated NFκB activation, a critical player in intestinal inflammation26, was absent in primary IECs from Wt mice and gradually increased in Atg16l1ΔIEC, Xbp1ΔIEC, and Atg16l1/Xbp1ΔIEC mice (Fig. 3b and Extended Data Fig. 6e, f). shXbp1 MODE-K cells exhibited increased NFκB activation when stimulated with tumor necrosis factor (TNF) (Fig. 3c and Extended Data Fig. 6g, h) or Toll-like receptor ligands (data not shown) relative to shCtrl MODE-K cells, demonstrating increased sensitivity of XBP1-deficient IECs to inflammatory and environmental stimuli. Inhibition of NFκB by treatment with BAY11-7082 decreased IEC death in Xbp1T-ΔIEC mice (Fig. 3d and Extended Data Fig. 6i) and protected from enteritis in both Xbp1T-ΔIEC and Xbp1ΔIEC mice (Fig. 3e and Extended Data Fig. 6j) relative to vehicle treated mice. A progressive increase in IECs of total and phosphorylated IRE1α (encoded by Ern1), the sensor of ER stress upstream of XBP1 and known to control NFκB27 was observed in Atg16l1ΔIEC, Xbp1ΔIEC and Atg16l1/Xbp1ΔIEC in comparison to Wt mice (Fig. 3f) and mirrored the escalating elevations in NFκB (Fig. 3b and Extended Data Fig. 6e, f). Indeed, increased NFκB activity and ileitis were governed by IRE1α as the increased epithelial NFκB phosphorylation observed in Xbp1ΔIEC mice was abrogated in Ern1/Xbp1ΔIEC mice (Fig 3g), Ern1 co-silencing of shXbp1 MODE-K cells abolished increased expression of the NFκB target gene Nfkbia in response to TNF stimulation relative to shCtrl MODE-K cells (Extended Data Fig. 6k), and enteritis was diminished in Ern1/Xbp1ΔIEC compared to Xbp1ΔIEC mice (Fig. 3h). Enteritis was also reversed by germline deletion of Tnfrsf1a in Xbp1ΔIEC mice (Fig. 3i), and re-derivation of Xbp1ΔIEC mice into a germ-free environment (Fig. 3j), which was associated with reduced p-IκBα immunoreactivity in Xbp1-deficient IECs compared to mice housed under SPF conditions (Fig. 3k). These studies together demonstrate that enteritis in this model is driven by TNF, the cytokine targeted by the most potent therapeutics of human CD25, and microbes in a pathway that derives from IEC death and IRE1α-dependent activation of NFκB with ATG16L1-dependent autophagy serving to restrain the inflammatory nature of the latter likely through its removal.
Figure 3. Autophagy restrains IRE1α-mediated NFκB activation in Xbp1-deficient epithelium.

a, shCtrl or shXbp1 MODE-K cells were co-silenced for Atg16l1 (siAtg16l1) or with scrambled siRNA (siCtrl), and analyzed by flow cytometry for annexin V and propidium iodide (PI; one-way ANOVA with post-hoc Bonferroni). b, Immunoblot of primary IEC scrapings (n=3). c, Immunoblot of cytoplasmic extracts from shCtrl or shXbp1 MODE-K cells after TNF stimulation. d, TUNEL+ IECs per 100 crypts after BAY11-7082 or vehicle treatment (n=3/4/4; one-way ANOVA with post-hoc Holm’s-corrected unpaired Student’s t-test). e, Enteritis histology score of mice treated with BAY11-7082 or vehicle (n=10/10/9; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). f, Immunoblot of IEC scrapings for (p-)IRE1α after IRE1α immunoprecipitation (IP). β-actin, loading control of whole lysates. g, Immunoblot of primary IEC scrapings (n=4). h, i, Enteritis histology score of indicated genotypes (h, n=15/16/14/15; i, n=5/10/12; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). j, Enteritis histology score of specific pathogen free (SPF) and germ free (GF) housed mice (n=10/9/7/7; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). (k) Representative images of p-IκBα immunoreactivity under conditions as in (j) (n=4). Bar, 20 μm. Results represent four (f), three (c) or two (a, b) independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Accordingly, treatment of Xbp1T-ΔIEC mice with the mTOR inhibitor rapamycin10 induced autophagy primarily in crypts (Extended Data Fig. 7a, b), diminished IEC-associated NFκB activation and number of TUNEL+ IECs in Xbp1ΔIEC mice (Fig. 4a and Extended Data Fig. 7c-e), and markedly reduced the severity of enteritis (Fig. 4b). These beneficial consequences of rapamycin in the setting of unabated ER stress in IECs were not observed in Atg7/Xbp1ΔIEC and Atg16l1/Xbp1ΔIEC mice (Extended Data Fig. 7e-i), demonstrating that these effects required intact autophagy within IECs.
Figure 4. ER stress-induced enteritis originates from Paneth cells and is alleviated through autophagy induction.

a, Immunoblot of primary IEC scrapings from mice treated with or without rapamycin for 14 consecutive days (n=3). b, Enteritis histology score for experiment as in (a) (n=4; median shown; Mann-Whitney U). c, Representative images of EYFP-Rosa26/D6-Cre+/− reporter mice and EYFP-Rosa26 (controls). Co-localization of Defa6 Cre-driven EYFP expression (yellow) with lysozyme expressing Paneth cells (red; n=3). DAPI, blue; bar, 50 μm. d, Immunoblots of crypt IECs from Xbp1ΔPC and Wt controls (n=2). e, f, Representative confocal images of lysozyme (green) expressing Paneth cells (e) with quantification of crypts with indicated number of lysozyme+ granulated dots in (f) (n=5; unpaired Student’s t-test). DAPI, blue; bar, 10 μm. g, Immunohistochemistry for p-eIF2α (n=3). Bar, 20 μm. h, Representative H&E images of Xbp1ΔPC and Wt mice scored in (i). Bar, 50 μm. (i) Enteritis scoring in Xbp1fl/fl (Wtfl), Defa6 Cre+ (WtCre) and Defa6 Cre+;Xbp1fl/fl (Xbp1ΔPC) mice (n=21/26/29; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). Results represent two (b, d) independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
The spatial convergence of the consequences of hypomorphic UPR and autophagy function in Paneth cells prompted us to test the hypothesis that these pathways were interdependent in this cell type. We developed a defensin 6 alpha promoter28-driven Cre transgenic line (D6-Cre), and confirmed the exclusive activity of Cre recombinase in lysozyme+ Paneth cells (Fig. 4c). Paneth cell-specific deletion of Xbp1 in D6-Cre;Xbp1fl/fl (‘Xbp1ΔPC’) mice resulted in autophagy activation (Fig. 4d and Extended Data Fig. 8a, b) and structural defects in granule morphology in Paneth cells (Fig. 4e, f). Crypts of Xbp1ΔPC compared to Wt mice exhibited increased GRP78 expression (Fig. 4d and Extended Data Fig. 8b) and p-eIF2α immunoreactivity (Fig. 4g), accompanied by increased conversion of LC3-I/II and reduced p62 levels (Fig. 4d and Extended Data Fig. 8b), demonstrating ER stress and autophagy induction. Strikingly, 75% of these mice developed spontaneous enteritis (Fig. 4h, i) that shared the characteristics with Xbp1ΔIEC mice (Fig. 2b). Xbp1ΔPC mice exhibited increased cell death in crypts (Extended Data Fig. 9a, b) and IEC turnover (Extended Data Fig. 9c-f), while goblet cells remained unaffected (Extended Data Fig. 9g, h). Notably, Atg7/Xbp1ΔPC mice exhibited transmural disease as early as 8 weeks of age (Extended Data Fig.9i). We conclude that deletion of Xbp1 specifically in Paneth cells results in unresolved ER stress, UPR activation and induction of autophagy which can serve as a nidus for the emergence of spontaneous intestinal inflammation that evolves into transmural disease in the absence of the compensation provided by autophagy.
Our studies thus support a mechanistic model (Extended Data Fig. 10a-c) for how ATG16L1-associated genetic risk may convert into a disease phenotype. The competence of the UPR likely sets the threshold for susceptibility of the host with hypofunctional autophagy to interacting genetic and environmental factors capable of inducing inflammation. Consistent with our model, patients with CD carrying the ATG16L1T300A risk variant, which impairs autophagosome formation29, frequently exhibit ER stress in their Paneth cells, in contrast to those harbouring the normal variant30. Finally, our studies also unequivocally establish that these inflammatory susceptibilities can emerge directly from highly secretory Paneth cells and suggest that small intestinal CD may be a specific disorder of this cell type.
Methods
Mice
Xbp1fl/fl mice were crossed with V-Cre-ERT2+ (B6) mice kindly provided by Drs. Nicholas Davidson (Washington University, St. Louis) and Sylvie Robine (Institut Curie-CNRS, Paris) to generate mice with tamoxifen inducible Xbp1 deletion in the intestinal epithelium (‘Xbp1T-ΔIEC’). V-Cre-ERT2+ recombinase was activated by 3 or 5 daily intraperitoneal administrations of 1 mg tamoxifen (MP Biomedicals) as indicated in figure legends. V-Cre+;Xbp1fl/fl (‘Xbp1ΔIEC’) mice were crossed with Atg7fl/fl mice14 kindly provided by Dr. Komatsu, Tokyo Metropolitan Institute of Medical Science, Tokyo, to obtain V-Cre+;Atg7fl/fl;Xbp1fl/fl (‘Atg7/Xbp1ΔIEC’) mice. Mice with a floxed Atg16l1 allele were generated in collaboration with GenOway, France. Briefly, a proximal loxP site was introduced within the promoter region of Atg16l1 gene upstream of exon 1, a distal loxP site was introduced with an FRT flanked neomycin selection cassette within intron 1. The resultant mouse line was bred with deleter-mice constitutively expressing Flp-recombinase to remove the neomycin selection cassette, creating an Atg16l1fl/+ mouse in which Atg16l1 exon 1 was flanked by two loxP sites (Extended Data Fig. S4g). After backcrossing onto B6, these mice were crossed with V-Cre+ mice31 resulting in V-Cre;Atg16l1fl/fl mice with IEC-specific Atg16l1 deletion (‘Atg16l1ΔIEC’). Atg16l1fl/fl mice were crossed with Xbp1ΔIEC mice to develop V-Cre+;Atg16l1fl/fl;Xbp1fl/fl (‘Atg16l1/Xbp1ΔIEC’) mice. GFP-LC3 transgenic mice12, generous gift of Dr. Mizushima, Tokyo Medical and Dental University, Tokyo, were crossed with V-Cre-ERT2+;Xbp1fl/fl to generate GFP-LC3;V-Cre-ERT2+;Xbp1fl/fl mice (‘GFP-LC3;Xbp1T-ΔIEC’). For the generation of Paneth cell-specific Defa6-Cre;Xbp1fl/fl mice, a 1.1 kb cDNA fragment encoding improved Cre (iCre)32 recombinase was subcloned downstream of nucleotides −6500 to +34 of mouse cryptdin-2 gene (Defa6) in the BamHI site of the pCR2-TAg-hGH plasmid33,34 to replace the DNA fragment containing the simian virus 40 large antigen (SV40). A linearized 10.2 kb fragment containing the Defa6 promoter, iCre and hGH (Defa6-iCre-hGH) was removed by EcoRI digestion, agarose gel-electrophoresed and purified with QIAEX Gel Extraction Kit (Qiagen), and used for pronuclear injection of BL/6 mice. Six founders from 22 live born mice were identified by screening tail DNA using iCre-specific primers, and two lines were further characterized and crossed to Xbp1fl/fl and EYFP-Rosa26 reporter mice35, kindly provided by K. Rajewsky. Gadd34−/− mice16 were crossed with V-Cre;Xbp1fl/fl mice to generate V-Cre;Xbp1fl/fl;Gadd34+/− (‘Xbp1ΔIEC;Gadd34+/−’) mice. Ern1fl/f 36 and Tnfrsf1a−/− (Jackson) mice were crossed with V-Cre;Xbp1fl/fl mice to generate V-Cre;Ern1fl/fl;Xbp1fl/fl (‘Ern1/Xbp1ΔIEC’) and V-Cre;Xbp1fl/fl;Tnfrsf1a−/− (‘Xbp1ΔIEC;Tnfrsf1a−/−’) mice, respectively. Cre transgenes were maintained in the hemizygous state in all experimental strains with a floxed allele to generate littermate controls. Xbp1ΔIEC mice were re-derived in a germ-free environment and housed in sterile isolators at the Taconic Farms breeding facility (Germantown, NY, USA). Tail or ear biopsy genomic DNA was used for genotyping of respective mouse strains as described previously4. Primer sequences are available upon request. Mice were housed in specific pathogen free (SPF) barrier facilities at Harvard Medical School (Atg7/Xbp1ΔIEC mice, Atg7/Xbp1ΔPC, Xbp1ΔPC, GFP-LC3;Xbp1T-ΔIEC, Ern1/Xbp1ΔIEC and their respective controls), University of Cambridge (Atg16l1/Xbp1ΔIEC, Xbp1ΔIEC/Gadd34+/−, Ern1/Xbp1ΔIEC mice and their respective controls), Innsbruck Medical University (Atg16l1/Xbp1ΔIEC, Xbp1ΔIEC;Tnfrsf1a−/−, Xbp1T-ΔIEC mice and their respective controls), and Christian-Albrechts-Universität zu Kiel (Atg16l1ΔIEC mice and their respective controls). Colonies maintained at Boston and Innsbruck were murine norovirus (MNV) positive by Taqman qRT-PCR (Extended Data Fig. 9j). Xbp1ΔIEC, Atg16l1ΔIEC, Ern1ΔIEC and their associated double-mutant strains were re-derived from the Innsbruck colony into the MNV-free enhanced barrier Cambridge facility, and colonies confirmed MNV-negative by PCR (Extended Data Fig. 9j) and serology (data not shown), as were Atg16l1ΔIEC mice held at the Kiel facility. MNV Taqman qRT-PCR was performed as described37. The phenotype of single- and double-mutant colonies that had been re-derived from the MNV+ Innsbruck facility into the MNV− Cambridge facility were indistinguishable, in particular relating to qualitative and quantitative measures of enteritis and the reciprocal induction of autophagy and ER stress. Mice were handled and all experiments performed in accordance with institutional guidelines and with the approval of the relevant authorities. 8-10 week old mice were used for all experiments unless stated otherwise in the figure legend, and were randomly allocated into treatment groups.
Antibodies and reagents
The following antibodies and reagents were used for immunoblotting: Sigma Aldrich: anti-LC3B (L7543). Cell Signaling Technology: anti-β-actin (4970; 13E5) anti-GAPDH (2118; 14C10), anti-eIF2α (9722), anti-phospho-eIF2α (3597; 119A11), anti-PERK (3192; C33E10), anti-phospho-PERK (3179; 16F8), anti-JNK (9252), anti-phospho-JNK (4668; 81E11), anti-ATG5 (8540; D1G9), anti-Beclin-1 (3495; D40C5), anti-ATG7 (8558; D12B11), anti-CHOP (5554; D46F1), anti-ATG12 (4180; D88H11), anti-p62 (5114), anti-IKK1 (2682), anti-IKK2 (2370; 2C8), anti-phospho-IKK1/2 (2697; 16A6), anti-IRE1α (3294; 14C10), anti-phospho-NFκB p65 (33033; 93H1), anti-NFκB p65 (4764; C22B4) and anti-rabbit/mouse HRP antibodies (7074, 7076). Abcam: anti-phospho-IRE1α (48187). MBL: anti-ATG16L1 (M150-3; 1F12). Stressgen: anti-heme-oxygenase-1 (ADI-SPA-895). Novus Biologicals: anti-GRP78 (NBP1-06274). Santa Cruz Biotechnology: anti-ATF4 (sc-200; C20). Immunoprecipitation antibody: anti-IRE1α (Santa Cruz Biotechnology, 20790; H190). Immunohistochemistry antibodies: Santa Cruz Biotechnology: anti-Lysozyme (27958; C19), MBL: anti-ATG16L1 (M150-3; 1F12). Cell Signaling Technology: anti-ATG16L1 (8089; D6D5), anti-phospho-IκBα (2859; 14D4), anti-phospho-eIF2α (3597; 119A11). Abcam: anti-Ki67 (15580), anti-GRP78 (21685). Progen: anti-p62 (GP62-C). BD Bioscience: anti-BrdU (551321).
The following reagents were used: TNF (Peprotech, 315-01A), Bafilomycin A1 (Sigma Aldrich, B1793), rapamycin (LC Laboratories, R-5000), JNK inhibitor (Sigma Aldrich, SP600125), NFκB inhibitor (Calbiochem, BAY11-7082) and salubrinal (Alexis Biochemicals, ALX-270-428) were dissolved in DMSO as recommended. N-acetyl cysteine (Sigma Aldrich, A9165) and glutathione (Calbiochem, NOVG3541) were used at final concentration of 1mM. Ambion siRNA for Atg16l1 (94892, sense: GAACUGUUAGGGAAGAUCATT, antisense: UGAUCUUCCCUAACAGUUCCA), Perk (65405, sense: CCCGAUAUCUAACAGAUUUTT, antisense: AAAUCUGUUAGAUAUCGGGAT; 65406, sense: CGAAGAAUACAGUAAUGGUTT, antisense: ACCAUUACUGUAUUCUUCGTG), Gadd34 (70230, sense: CCAUAGCUCCGGGAUACAATT, antisense: UUGUAUCCCGGAGCUAUGGAA; 70231, sense: AGACAACAGCGAUUCGGAUTT, antisense: AUCCGAAUCGCUGUUGUCUTC), Ern1 (95857, sense: GUUUGACCCUGGACUCAAATT, antisense: UUUGAGUCCAGGGUCAAACTT; 95858, sense: GGAUGUAAGUGACCGAAUATT, antisense: UAUUCGGGUCACUUACAUCCTG; 95859, sense: GCUCGUGAAUUGAUAGAGATT, antisense: UCUCUAUCAAUUCACGAGCAA) and scrambled control were used at a final concentration of 10μM.
Chromatin Immunoprecipitation (ChIP)
ChIP with anti-ATF4 and control IgG rabbit antibody was performed in Xbp1 and control silenced MODE-K cells according to ChIP protocol by Agilent. To determine the presence of ATF4 binding sites in the Atg7 promoter, a 4-kb region proximal to transcription start site identified with the Eukaryotic Promoter Database Primers was analyzed using MatInspector (Genomatix). Immunoprecipitated DNA was subject to quantitative PCR (qPCR) to determine enrichment of ATF4 binding to respective promoters and results were normalized to input chromatin DNA. Primers used for qPCR were as follows for Map1LC3b13, for Atg7 F 5’-GCGCTTCCGCGTTTGTGTGG and R 5’CTGCTCCGCAACCACGGCTT.
Salubrinal, rapamycin and BAY11-7082 treatment in vivo
Salubrinal [1mg/kg/d], rapamycin [1.5mg/kg/d] or vehicle (DMSO) was administered intraperitoneally (i.p.) 24h prior to the first tamoxifen administration to GFP-LC3;V-CreERT2;Xbp1fl/fl (GFP-LC3;Wt) and GFP-LC3;Xbp1T-ΔIEC mice. 3-day treatment was used for evaluation of accumulation of GFP-LC3 punctae in the intestinal epithelium, while a 5-day combined tamoxifen and salubrinal treatment followed by two daily salubrinal injections was used in experiments with enteritis assessment as an end point (Extended Data Fig. 2a and 3g). To assess the effects of rapamycin on ER stress-induced intestinal inflammation in XBP1 deficiency, V-Cre;Xbp1fl/fl (Xbp1ΔIEC), V-Cre;Atg16l1fl/fl;Xbp1fl/fl or V-Cre;Atg7fl/fl;Xbp1fl/fl and the respective control mice were treated with rapamycin or vehicle for 14 consecutive days i.p. and inflammation was evaluated. BAY11-708238 or vehicle (DMSO) was administered intraperitoneally (i.p.) every other day at 5mg/kg for 14 consecutive days in Xbp1ΔIEC mice, or for 5 consecutive days at 20mg/kg in Xbp1T-ΔIEC mice concomitant with i.p. tamoxifen.
Transmission electron microscopy
Small intestinal tissue from mice was handled by standard methods to be fixed with 1.25% glutaraldehyde, 4% formaldehyde in 0.1 M cacodylate buffer at pH 7.4 at room temperature for electron microscopy. The detailed procedures for electron microscopy was previously described39 and the tissue was observed with a JEOL 1400 transmission electron microscope at 120 kV operating voltage. For quantification of autophagy, number of autophagic vacuoles was manually counted by TEM expert (J.H.) blinded to sample identity in 10 consecutive Paneth cells per sample. ImageJ software was used to measure average size of autophagic vacuoles.
Histology
Formalin-fixed and paraffin-embedded intestinal tissue was sectioned and stained with hematoxylin and eosin (H&E) as previously described4. A semi-quantitative composite scoring system was used for the assessment of spontaneous intestinal inflammation, computed as a sum of five histological subscores, multiplied by a factor based on the extent of the inflammation. Histological subscores (for each parameter: 0, absent; 1, mild; 2, moderate; 3, severe): mononuclear cell infiltrate (0-3), crypt hyperplasia (0-3), epithelial injury/erosion (0-3), polymorphonuclear cell infiltrates (0-3) and transmural inflammation (0, absent; 1, submucosal; 2, one focus extending into muscularis and serosa; 3 up to five foci extending into muscularis and serosa; 4, diffuse). Extent factor was derived according to the fraction of bowel length involved by inflammation: 1, < 10%, 2, 10-25%, 3, 25-50% and 4, >50%. Ileal inflammation was assessed by an expert gastrointestinal pathologist (J.N.G.) who was blinded to the genotype and experimental conditions of the samples. No spontaneous colonic inflammation was detected in any of the reported genotypes.
Reactive oxygen species (ROS), cell death detection by flow cytometry and NFκB activity assays
To evaluate oxidative stress, Xbp1- and control-silenced MODE-K cells40 were incubated with 5 μM 5-(and-6)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Molecular Probes) for 30 min41. After washing with PBS, cells were further incubated with complete medium for 2 hours. ROS generation was determined using flow cytometry. To evaluate cell death, shXbp1 and shCtrl cells were co-silenced for Atg16l1 using siRNA or scrambled control (Ambion). After 4 days, cells were harvested and stained for Annexin V (Biolegend, UK) in staining buffer (Biolegend, UK) and mode of cell death was determined by flow cytometry after addition of propidium iodide (PI). To assess NFκB signaling pathway activation, Xbp1- or control-silenced MODE-K cells were stimulated with 50ng/ml TNF for indicated periods of time, followed by immunoblotting (using NE-PER [Thermo Scientific] isolated cytoplasmic extracts), qRT-PCR, and chemiluminescent detection of NFκB consensus sequence binding activity with the NFκB p65 transcription factor assay kit (Thermo Scientific). Ern1 or scrambled siRNA (Ambion) was used for co-silencing as indicated.
Immunohistochemistry, bromodeoxyuridine (BrdU) and terminal deoxinucleotidyl transferase (TUNEL) labeling
Formalin fixed paraffin embedded sections were stained according to standard immunohistochemistry protocols and manufacturer’s recommendations as described previously4. Cell death was assessed by TUNEL labeling of formalin fixed paraffin embedded slides of the respective genotypes using the TUNEL cell death detection kit (Roche). Entire slides were analyzed for TUNEL+ cells and numbers normalized to intestinal length on the slide. Proliferation of the intestinal epithelium was assessed after a 24h pulse with BrdU (BD Pharmingen) and incorporated BrdU was detected by the BrdU in situ detection kit (BD Pharmingen). BrdU+ nuclei per total IECs along the crypt villus axis are shown. Toluidine blue staining and Periodic acid-Schiff (PAS) reaction was performed according to standard protocols.
Confocal microscopy for detection of GFP-LC3 and EYFP
For detection of GFP-LC3 or EYFP, mice were euthanized, followed by transcardiac perfusion with PBS (2-3 minutes) and 3.7 % formaldehyde (3-4 minutes). Small intestine was dissected and promptly washed with PBS. The tissue was fixed in formalin for an additional 12-18 hours. Fixed tissue was embedded in OCT and sectioned on a cryotome into 5μm sections. Slides were washed with PBS and Prolong Gold Antifade reagent with DAPI (Invitrogen) with application of an attached coverglass. Images of the sections were collected using Olympus semi-confocal system. MetaMorph software was used for image analysis. For the detection of autophagosome formation in vitro, Xbp1 and control silenced MODE-K cells were transfected with a GFP-LC3 plasmid12 (a kind gift of Dr. Noboru Mizushima, Tokyo Medical and Dental University) with use of Lipofectamine LTX (Invitrogen) following the manufacturer’s instructions. Accumulation of GFP-LC3 punctae or EYFP signal was assessed using LSM510 META Confocal Microscopy (Carl Zeiss).
Intestinal epithelial cell purification and crypt isolation
Mice were euthanized and the intestine was washed with ice-cold PBS after being cut open longitudinally. Peyer’s patches were removed and the intestine was cut into small pieces. Mucus was removed by shaking the intestine in 1x HBSS containing 1mM DTT for 10 min at RT. After washing with PBS, pieces were digested with Dispase (1U/ml in RPMI with 2% FCS) for 30 min at 37 degree with shaking (250 rpm). Cells were collected and debris removed with a 100μm cell strainer, and centrifuged for 5 min at 1500rpm. IECs were collected in the top layer after 40% - 100% Percoll gradient centrifugation. Purity of the population was determined by staining with anti-EpCAM antibody and flow cytometry analysis. IECs were lysed with RIPA buffer and equal amounts of protein were used for western blot analysis as indicated in figure legends. To isolate small intestinal crypts, the intestine was flushed, cut open longitudinally and incubated on ice for 30 minutes in 2mM EDTA/PBS. Two sedimentation steps and application of a cell strainer separated crypts from villi42, which were then used for RNA isolation (Qiagen), Xbp1 splicing assay, and for protein lysis with RIPA buffer and subsequent immunoblotting.
Intestinal epithelial scrapings
Mice were euthanized, intestines collected and longitudinally opened, and immediately washed with ice-cold PBS. Intestinal epithelium was collected by scraping with glass slides and snap frozen into liquid nitrogen for further analysis. For protein analysis, intestinal epithelial scrapings were homogenized in RIPA buffer using a 25G needle with a syringe. Lysates were cleared by centrifugation and aliquots of protein were used for protein assessment using standard western blot or immunoprecipitation protocols as indicated. Isolation of mRNA from intestinal epithelial scrapings or MODE-K lysates and RT-qPCR was performed as described4 using following pairs of primers. grp78 5-ACTTGGGGACCACCTATTCCT and 5-ATCGCCAATCAGACGCTCC; Gadd34 5-CCCGAGATTCCTCTAAAAGC and 5-CCAGACAGCAAGGAAATGG; LC3b 5-GCGCCATGCCGTCCGAGAAG and 5-GCTCCCGGATGAGCCGGACA; Xbp1 5-ACACGCTTGGGAATGGACAC and 5-CCATGGGAAGATGTTCTGGG; Atg7 5-CCTTCGCGGACCTAAAGAAGT and 5-CCCGGATTAGAGGGATGCTC; Nfkbia 5-TGAAGGACGAGGAGTACGAGC and 5-TTCGTGGATGATTGCCAAGTG.
Dextran Sodium Sulfate induced colitis
Acute colitis was induced by adding 4% DSS (TdB Consultancy) to drinking water ad libitum for 5 consecutive days. Daily disease activity index (DAI) was assessed evaluating weight loss, stool consistency and rectal bleeding according to Table S1. A high resolution mouse endoscopic system (Hopkins, Germany) was used. Colitis severity was assessed by a semiquantitative score consisting of two subscores;endoscopic tissue damage (0-3, where 0 - no damage, 1 – lymphoepithelial lesions, 2- surface mucosal erosion or focal ulceration and 3 - extensive mucosal damage with expansion into deeper structures of the bowel wall) and inflammatory infiltration (0-3, where 0 – occasional inflammatory cells in the lamina propria, 1 – increased numbers on inflammatory cells in lamina propria, 2 – confluence of inflammatory cells extending into the submucosa and 3 – transmural extension of the infiltrate).
Xbp1 splicing assay and densitometric quantification
Xbp1 splicing was assessed as described4. Briefly, RNA was isolated, reverse transcribed and amplified by RT-PCR with the following primers: Xbp1 sp F: ACACGCTTGGGAATGGACAC; Xbp1 sp R: CCATGGGAAGATGTTCTGGG. The PCR product of 171 (unspliced) and 145 (spliced) bp were resolved on a 2% agarose gel. Densitometric analysis for splicing assay and immunoblots was performed with ImageJ.
Statistical methods
Statistical significance was calculated as appropriate using an unpaired two-tailed Student’s t-test or a Mann-Whitney U test and considered significant at p < 0.05. In experiments where more than two groups were compared, Kruskal-Wallis test followed by Mann-Whitney U and post-hoc Bonferroni Holm’s correction or one-way ANOVA/Bonferroni was performed. Grubb’s test was used as appropriate to identify outliers. Data was analyzed using GraphPad Prism software. Experimental group sizes were based upon the goal of achieving desired effect sizes typically of ≤2.0 standard deviations and a power of 0.9 on the assumption of a normal distribution, and therefore typically involved n=6-10.
Supplementary Material
a, Xbp1 expression in shCtrl and shXbp1 MODE-K cells (n=7/6; unpaired Student’s t-test). b, c, Immunoblot of shCtrl and shXbp1 MODE-K cells. d, Immunoblot after autophagosome-lysosome fusion inhibition via bafilomycin in silenced MODE-K IECs. e, Silenced MODE-K IECs after GFP-LC3 reporter transfection (bar, 5μm) with green punctae per cell quantification in (f; n=14; unpaired Student’s t-test). g, TEM of shCtrl and shXbp1 cells. Note double-membraned structure with engulfed contents characteristic of autophagosomes (white arrows; n=10). Bar, 0.5 μm. h, Quantification of occupied area and average size of autophagic vacuoles from (g) (n=10; unpaired Student’s t-test). i, j, Densitometry of Fig. 1a (i; n=5/4; unpaired Student’s t-test) and Fig. 1b (j; n=3; unpaired Student’s t-test). k, Low magnification (1380×, original magnification here and in the remainder of this legend) TEM image of Paneth cells from Wt mice (1), demonstrating the abundant endoplasmic reticulum (ER) and characteristic secretory granules at the apical, lumenally (‘L’) oriented side. (2) Higher magnification (5520×) of inset ‘2’ in (1) demonstrating typical secretory granules in Paneth cells from Wt mice. (3) High magnification (20700×) of inset ‘3’ in (1) illustrating a double membrane structure characteristic of an autophagosome (white arrow) in close proximity to the ER and a mitochondrion. (4) Low magnification (2160×) TEM image of Paneth cell remnants present in Xbp1ΔIEC small intestinal crypts, which lack expansion of the ER and exhibit only minuscule granule remnants. (5) Higher magnification (9000×) of inset ‘5’ in (4), demonstrating degradative autophagic vacuoles (black arrows), in close proximity to mitochondria, and the virtual absence of ER membranes. (6) High-power (14400×) magnification of inset ‘6’ in (4), illustrating a double-membrane structure (white arrow) characteristic of autophagosomes, and a degradative autophagic vacuole (black arrow). L, lumen; M, mitochondrion; ER, endoplasmic reticulum; N nucleus; as indicated, bar represents 2μm, 0.5μm and 200nm, respectively. Results represent three (b, c) or two (d, k) independent experiments. *P< 0.05, ***P< 0.001.
{kind=link}
a, Deletion of Xbp1 in the intestinal epithelium, specifically in Paneth cells, leads to ER stress and activation of the PERK/eIF2α branch of the UPR. ATF4, a transcriptional mediator of this pathway, transactivates genes essential for autophagosome formation, such as Map1lc3b (LC3b) and Atg7 which catalyzes the creation of the ATG12-ATG5 conjugate that stabilizes ATG16L1 through complex formation21. UPR-induced autophagy in the intestinal epithelium is essential for restoration of homeostasis and restraint of ER-stress induced intestinal inflammation due to XBP1-deficiency. Activation of the UPR in the setting of XBP1-deficiency results in activation of IRE1α, resulting in the recruitment of TRAF2 and activation of IKK2 leading to IκBα degradation 4,27,45,46. As shown here, UPR-mediated autophagy however serves an important role in restraining NFκB activation, conceivably by removing hyperinflammatory ER membranes containing activated IRE1α. Pharmacological augmentation of this compensatory autophagy-dependent mechanism via inhibition of eIF2α dephosphorylation through salubrinal, or via the mTOR inhibitor rapamycin results in amelioration of UPR-induced enteritis, which is driven by the commensal microbiota, NFκB, and TNF-RI signaling in IECs and myeloid cells, whereby the ligand TNF can originate from XBP1- deficient IECs4. b, ATG16L1-deficiency in IECs leads to ER stress as revealed through upregulation of the chaperone GRP78 in IECs, increased expression of GRP78 protein in Paneth cells, increased IRE1α expression and increased splicing of Xbp1 mRNA in intestinal crypts as well as increased IEC death. This leads to increased sensitivity of the epithelium to environmental triggers (e.g. dextran sodium sulfate) that further challenge the UPR and its compensatory pathways. c, Deficiency of ATG16L1 or ATG7 in the intestinal epithelium results in abrogation of the compensatory autophagic mechanism that restrains IRE1α activity, conceivably via removal of hyperinflammatory ER membranes, and further fosters IEC death in the context of ER stress due to Xbp1 deficiency, resulting in spontaneous transmural small intestinal inflammation that is associated with further increases in NFκB activation and cell death via the mechanisms described in (a). The UPR allows for responses to a variety of signals that impact on protein folding, including genetic (e.g. rare XBP1 variants, ORMDL3 as risk factor of IBD4,47), environmental (e.g. low O2 tension in the intestinal tract) and microbial factors (e.g. microbial toxins such as trierixin48) which determines the level of ER stress in the intestinal epithelium. UPR-induced autophagy function provides a buffer to cope with different levels of ER stress and vice-versa. However, in the presence of genetic risk variants, such as ATG16L117,49,50 or IRGM51, which are relatively prevalent in the general population, this compensatory mechanism is impaired, resulting in development and/or exacerbation of intestinal inflammation in the setting of unabated ER stress.
{kind=link}
a, Timeline for salubrinal experiment shown in Fig. 1j. b, Representative indirect immunofluorescence images of GFP-LC3 punctae accumulation (green) in small intestinal sections co-stained with antilysozyme antibody (red) treated as in (a) (n=3). DAPI, blue. Bar, 10 μm. c, Immunoblot of crypt lysates after 3 days of tamoxifen or vehicle administration (n=3). d, e, Immunoblot of primary IEC scrapings (d), densitometry in (e) (n=3; unpaired Student’s t-test). f, g, Immunoblot of shXbp1 and shCtrl MODE-K cells. ER stress-induced Jun N-terminal kinase-1 (JNK1) has previously been connected in other cellular model systems to autophagy activation through phosphorylation of B cell leukemia 2 (Bcl-2) and its dissociation from Beclin-143, as have oxidative stress/free radicals and heme oxygenase-1 (HO-1) activation44. h, Intracellular reactive oxygen species (ROS) determined by dichlorofluorescein assay and mean fluorescent intensity (MFI) after vehicle or dichlorofluorescein diacetate (DCF-DA) treatment. i, Immunoblot of shXbp1 and shCtrl MODE-K cells after administration of the JNK inhibitor SP600125 (0, 5 or 25 μM) for 4h. Note the absence of an effect of SP600125 treatment on the conversion of LC3-I to LC3-II or the levels of p-eIF2α, thereby excluding a major contribution of the JNK pathway to autophagy induction in the presence of IEC-associated XBP1-deficiency. j, Immunoblot of shXbp1 and shCtrl MODE-K cells after N-acetylcysteine (NAC), glutathione (GSH) or vehicle for 16h. Note the absence of an effect of the free radical scavengers on either of these markers of UPR-induced autophagy (LC3-II or p-eIF2α). Results represent three (f-j) independent experiments. *P < 0.05.
{kind=link}
a, Immunoblot of shXbp1 and shCtrl MODE-K cells co-silenced with Perk (siPerk) or scrambled siRNA. b, Cumulative densitometry of the immunoblot in Fig. 1g and two additional experiments (not shown, n=10; unpaired Student’s t-test). c, p-eIF2α immunohistochemistry of small intestinal epithelium. Bar, 50 μm. d, mRNA expression levels of Map1lc3b (LC3b) and Atg7 relative to Gapdh in shCtrl and shXbp1 MODE-K cells (n=3; unpaired Student’s t-test). e, Accumulation of GFP-LC3 punctae after salubrinal and 3-day tamoxifen treatment according to timeline in Extended Data Fig. 2a (n=5). Bar, 5 μm. f, Immunoblot of primary epithelial scrapings from small intestine upon vehicle or salubrinal (Salu) treatment according to the timeline (g) (n=2). Note the increased relative levels of LC3 conversion and CHOP, a transcriptional target of ATF4, in salubrinal treated mice. g, Timeline of salubrinal experiment. Results are reported in (f) and Fig. 1k. h, Gadd34 expression in shCtrl and shXbp1 MODE-K cells co-silenced for Gadd34 (siGadd34) and control (siCtr) (one-way ANOVA with post-hoc Holm’s corrected unpaired Student’s t-test). i, Cells as in (h) were immunoblotted. j, Immunoblot of crypt lysates (n=2/4/2). Results represent three (a, h, i) independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
{kind=link}
a, Immunoblot of primary IEC scrapings (n=4/3/3/3). Note the loss of ATG5, 12 and 16 proteins in ATG7-deficient mice. b, Representative high magnification TEM images (n=2). Note intact ER in Wt mice; severe distortion of ER in Atg7ΔIEC mice; autophagic vacuoles with the characteristic double membrane (white arrow) in Xbp1ΔIEC mice; and the absence of ER and autophagic vacuoles in Atg7/Xbp1ΔIEC mice. Bar, 200nm. ER, endoplasmic reticulum. c, Quantification of autophagic vacuoles (n=10; unpaired Student’s t-test). d, High magnification (400x) H&E images. Note knifelike extension of inflammation along blood vessels through whole thickness of muscularis propria into the serosa in the Atg7/Xbp1ΔIEC mice. Bar, 10 μm. e, Linear regression analysis for the correlation of inflammation with age. Each dot represents a single animal. (R2 and P value for deviation from 0 are shown). f, Deep inflammation score (as described in materials and methods) in Atg7/Xbp1ΔIEC mice plotted at indicated ages in weeks. (n=13/17/9; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). g, Atg16l1 IEC specific knockout design. Exon 1 of Atg16l1 gene was flanked by two loxP sites and Atg16l1 deletion was mediated by Cre recombinase (Cre) under the control of Villin (V-) promoter (V-Cre+;Atg16l1fl/fl or ‘Atg16l1ΔIEC’). Detailed description in the methods section. h, Immunohistochemical staining (brown) with anti-ATG16L1 antibody (MBL) (n=2). Note cytoplasmic IEC specific ATG16L1 immunostaining in Wt but not Atg16l1ΔIEC mice with retained ATG16L1 expression in lamina propria mononuclear cells in Atg16l1ΔIEC mice. Bar, 50μm. i, Indirect immunofluorescence microscopy (green) with anti-ATG16L1 antibody (Cell Signaling Technology) (n=2). Note the similar staining pattern as in (h) with cytoplasmic IEC-specific ATG16L1 immunoreactivity in Wt but not Atg16l1ΔIEC mice and retained ATG16L1 expression in lamina propria mononuclear cells in Atg16l1ΔIEC mice. Dashed line denotes crypt unit. Bar, 20μm. j, Schematic representation of lysozyme+ granule allocation patterns (green): normal (D0), disordered (D1), depleted (D2), and diffuse (D3)2. k, l, Lysozyme immunofluorescence (green) in crypts of Atg16l1ΔIEC and Wt mice (k), quantified in (l; n=3; unpaired Student’s t-test) according to granule allocation patterns shown in (j). Dashed line denotes crypt unit. DAPI, blue. Bar, 5μm. m, n, Quantification of Paneth cells based on the number of granular vesicles stained with Toluidine blue (m) as shown in (n) (n=4; unpaired Student’s t-test). Bar, 5μm. *P< 0.05, **P< 0.01, ***P< 0.001.
{kind=link}
a, Immunoblot of primary IEC scrapings. Note identical GAPDH loading control as in figure 3b (n=3). b, Representative high magnification TEM images (n=2). Note intact ER in Wt mice; severe distortion of ER in Atg16l1ΔIEC mice; autophagic vacuoles with the characteristic double membrane (arrow) in Xbp1ΔIEC mice; and absence of ER and autophagic vacuoles in Atg16l1/Xbp1ΔIEC mice. Bar, 200 nm. ER, endoplasmic reticulum. c, Quantification of autophagic vacuoles (n=10 unpaired Student’s t-test). d, Representative images of p62 immunostaining on small intestinal sections (n=3). Brown staining indicates p62 with maximal signal intensity in the region of Paneth cells. Bar, 20 μm. e, High resolution H&E images. Note the extension of the inflammation into muscularis propria (arrow) in Atg16l1/Xbp1ΔIEC mice. Bar, 20μm. f, Deep inflammation score (as described in materials and methods) in mice assessed at 18 weeks of age (n=11; Kruskal-Wallis with post-hoc Dunn’s correction). g, Enteritis histology score (n=14; 7-10 weeks old mice housed in Innsbruck; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). h, qRT-PCR of grp78 in primary IEC scrapings (n=7; unpaired Student’s t-test). i, Disease activity index during DSS colitis (n=7/4, two-way ANOVA with post-hoc Bonferroni). j, Representative endoscopic images from the colon at day 5 of DSS treatment (n=3). k, Representative H&E staining of colonic sections from 5-day DSS- treated and untreated mice (n=7/4). Bar, 50μm. l, Colitis score after 5 days of DSS administration (n=7/4, Mann-Whitney U). *P < 0.05, **P < 0.01, ***P < 0.001.
{kind=link}
a, Representative images of TUNEL labeled intestinal epithelium (brown) (n=7). Bar, 50 μm. b, Correlation of TUNEL+ cells with the severity of inflammation. Linear regression analysis of Atg16l1/Xbp1ΔIEC (left) and Atg7/Xbp1ΔIEC (right) with significant R2 and P value for deviation from zero shown (left panel: n=5/5/7/7; right panel: n=5). c, Linear least square regression analysis for the correlation of enteritis histology score with cell death and age of animals by genotype. Each dot represents a single animal (grey, Wt; yellow, Atg16l1ΔIEC; blue, Xbp1ΔIEC; red, Atg16l1/Xbp1ΔIEC mice) and the plane represents the linear regression for the enteritis histology score as a function of age and TUNEL labeling for Atg16l1/Xbp1ΔIEC mice (n=6/13/12/12). Note that the severity of inflammation significantly correlates with numbers of TUNEL+ IECs and age only in Atg16l1/Xbp1ΔIEC mice (R2=0.602, p=0.016). The three-dimensional (3D) plot is also available online in video-format. Regression analysis was performed using the R package lessR (http://cran.r-project.org/web/packages/lessR/index.html), last accessed May 2013. d, shCtrl or shXbp1 MODE-K cells were co-silenced for Atg16l1 (siAtg16l1) or with scrambled siRNA (siCtrl), and analyzed by flow cytometry for Annexin V and propidium iodide (PI) uptake (one-way ANOVA with post-hoc Bonferroni). e, Densitometry of the immunoblot shown in Fig. 3b (n=3; one-way ANOVA with post-hoc Holm’s corrected unpaired Student’s t-test). f, Immunohistochemical staining for p-IκBα in the small intestinal epithelium (n=3). Bar, 20 μm. g, NFκB consensus sequence binding assay after stimulation of shCtrl and shXbp1 MODE-K cells for 5 and 20 minutes with indicated concentrations of TNF. h, Nfkbia expression, a prototypic NFκB-transactivated gene, after TNF stimulation of shCtrl and shXbp1 MODE-K cells. i, Representative images of TUNEL labeled sections after administration of BAY11-7082 or vehicle (n=3/4/4). Bar, 50 μm. j, Enteritis histology score of mice treated with the NFκB inhibitor BAY11-7082 or vehicle (n=7/9/8; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). k, Expression of Nfκbia in Ern1- and control-silenced shXbp1 and control MODE-K cells after TNF stimulation. Results represent three (h, k) or two (d, g) independent experiments. *P< 0.05, **P< 0.01, ***P<0.001.
{kind=link}
a, b, Quantification of GFP-LC3 punctae accumulation after 3-day tamoxifen treatment and induction by rapamycin (a; n=10; one-way ANOVA with post-hoc Bonferroni) and representative images in (b; n=3). Bar, 5 μm. c, Densitometry of the immunoblot in Fig. 4a (n=3; one-way ANOVA with post-hoc Holm’s-corrected unpaired Student’s t-test). d, Quantification of TUNEL+ IECs per cm of gut in mice treated with rapamycin or vehicle (n=5; unpaired Student’s t-test). e, Representative images of TUNEL labeled sections from rapamycin or vehicle treated Wt, Atg16l1ΔIEC, Xbp1ΔIEC, Atg16l1/Xbp1ΔIEC mice or Wt, Atg7ΔIEC, Xbp1ΔIEC, Atg7/Xbp1ΔIEC mice. Bar, 50 μm. f, g, Quantification of TUNEL labeling in Wt, Atg16l1ΔIEC, Atg16l1/Xbp1ΔIEC mice (f; n=4/4/5/5/5/5) or Wt, Atg7ΔIEC, Atg7/Xbp1ΔIEC mice (g; n=5; one-way ANOVA with post-hoc Holm’s-corrected unpaired Student’s t-test). h, i, Enteritis histology scores of rapamycin or vehicle treated mice of respective genotypes (h; n=8/9/6/9/12/13) (i; n=8/7/6/7/7/6) (median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). *P < 0.05, **P < 0.01, ***P < 0.001.
{kind=link}
a, Representative TEM images of small intestinal crypts from Defa6Cre+ (Wt) and Defa6Cre+;Xbp1fl/fl (Xbp1ΔPC) mice. (1) Low magnification TEM image of Paneth cells at the base of a small intestinal crypt in Wt mice, demonstrating abundant ER and characteristic secretory granules at the apical, lumenally oriented side. Bar, 5 μm. (2) Higher magnification of inset ‘2’ in (1) with numerous secretory granules. Bar, 1μm. (3) High magnification of inset ‘3’ in (1) with intact ER. Bar, 200 nm. (4) Low magnification TEM image of Paneth cells in an Xbp1ΔPC small intestinal crypt. Note the reduced number of secretory granules, the exfoliating hypomorphic Paneth cell into crypt lumen (‘E’, black dashed outline) and a transmigrating polymorphonuclear cell through the mucosa (white dashed outline). Bar, 5μm. (5) Higher magnification of inset ‘5’ in (4), demonstrating abnormalities in secretory granule maturation with numerous hypodense granules (*) and a distorted ER. Note the accumulation of electron dense cargo indicative of degradative autophagic vacuoles (black arrow) in close proximity to the nucleus. Bar, 1μm. (6) High magnification of the inset ‘6’ in (4), demonstrating degradative autophagic vacuoles filled with electron dense material (black arrow). Note the double membrane structures characteristic for autophagosomes (white arrow heads). Bar, 200 nm. (7) Low magnification TEM image of Paneth cells in an Xbp1ΔPC small intestinal crypt. Note the disorganized ER, hypodense secretory granules (*), empty area after exfoliation of a Paneth cell into the crypt lumen (‘E’, black dashed outline) similar to image (4). (8) High magnification of inset ‘8’ identified in (7), demonstrating an autophagic vacuole (black arrow) surrounded by a markedly distorted ER. Bar, 200 nm. (9) High magnification of the inset ‘9’ in (7), with additional examples of degradative autophagic vacuoles accompanied by a distorted ER. Representative of 2 independent experiments. Bar, 200 nm. L, lumen; M, mitochondrion; ER, endoplasmic reticulum; N nucleus. b, Densitometry of the immunoblot in Fig. 4d (n=2; unpaired Student’s t-test). *P< 0.05.
{kind=link}
a, b, TUNEL labeled ileal sections (a) with quantification of TUNEL+ cells per cm gut shown in (b; n=7/11; unpaired Student’s t-test). Bar, 50 μm. c, d, Ki67 immunohistochemical staining of ileal sections (c) with quantification of Ki67+ cells per total IEC count along the crypt-villus axis shown in (d; n=5; unpaired Student’s t-test). Bar, 20 μm. e, f, Bromodeoxyuridine (BrdU) labeled ileal sections (e) after a 24h with quantification of BrdU+ cells per total IEC count along the crypt-villus axis in (f; n=5/3; unpaired Student’s t-test). Bar, 50μm. g, h, Periodic acid-Schiff (PAS)-stained ileal sections (g) with quantification of PAS+ cells per total IEC count along the crypt-villus axis shown in (h; n=5; unpaired Student’s t-test). Bar, 50μm. i, Enteritis histology score of indicated genotypes (n=3; median shown; Kruskal-Wallis). j, Taqman qRT-PCR for MNV of fecal samples in three different animal facilities (n=3). Note detectable MNV genome copy numbers in Boston (Atg7/Xbp1ΔIEC, Fig. 2c) and Innsbruck (Atg16l1/Xbp1ΔIEC, Extended Data Fig. 5g) colonies, and absence of MNV after re-derivation of colonies into the MNV-free Cambridge facility (Atg16l1/Xbp1ΔIEC, Fig 2e), yet similar levels of enteritis in Xbp1ΔIEC and Atg16l1/Xbp1ΔIEC mice maintained in Innsbruck (MNV+) and Cambridge (MNV−) (Fig. 2b, e and Extended Data Fig. 5g) and relatively more severe disease in Xbp1ΔIEC mice from Boston (Fig. 2c). Sentinel mice from the Cambridge colony also tested negative for MNV by serological analysis (data not shown). These observations suggest that the inflammatory phenotypes observed are not necessarily dependent upon MNV. *P < 0.05.
{kind=link}
Acknowledgments
The authors wish to thank L. Glimcher, A. Goldberg, J. Yuan, M. Parkes and C.L. Bevins for thoughtful discussion of the project and are grateful to J. Gordon, L. Hooper and K. Rajewsky for providing critical reagents. This work was supported by NIH grants DK044319, DK051362, DK053056, DK088199, the Harvard Digestive Diseases Center (HDDC) (DK0034854) (R.S.B.); the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013) / ERC Grant agreement n° 260961 (A.K.); the National Institute for Health Research Cambridge Biomedical Research Centre (A.K.); the Austrian Science Fund and Ministry of Science P21530-B18 and START Y446-B18 (A.K.); the Addenbrooke’s Charitable Trust (A.K.); BMBF NGFN Animal Model grant (P.R.), the DFG Cluster of Excellence Inflammation at Interfaces; DFG grants RO2994/5-1 (S.S. & P.R.) and SFB 877 project B9 (P.R.); fellowships from Inflammatory Bowel Disease Working Group (M.F.T.), European Crohn’s and Colitis Organization (T.E.A.), Crohn’s in Childhood Research Association (L.N.), National Research Foundation of Korea funded by the Korean government KRF-2008-357-E00022 /No. 2011-0009018 (H.J.-K.).
Footnotes
Author Contributions T.E.A., M.F.T., L.N. and H.-J.K. performed most experiments, together with J.B., E.M.N., M.T., S.H., M.B.F., S.B.-B., T.R., R.B. and M.N.K. J.L.C. helped preparing the manuscript. S.J.H. and J.H. contributed electron microscopic analysis, A.C. provided expertise in autophagy assessment, and R.L. in histology of Paneth cells. S.S. and P.R. designed, generated and analyzed an essential mouse strain. K.K., T.I. and S.J.M. provided an essential mouse strain. J.N.G. assessed intestinal inflammation. A.K. und R.S.B. devised and coordinated the project, and together with T.E.A. and M.F.T. wrote the manuscript and designed the experiments.
The authors declare no competing financial interests.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Jostins L, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cadwell K, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaser A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Treton X, et al. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology. 2011;141:1024–1035. doi: 10.1053/j.gastro.2011.05.033. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi KS, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 7.Zheng W, et al. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun. 2006;7:11–18. doi: 10.1038/sj.gene.6364263. [DOI] [PubMed] [Google Scholar]
- 8.Wehkamp J, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 10.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarkar S, Ravikumar B, Rubinsztein DC. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol. 2009;453:83–110. doi: 10.1016/S0076-6879(08)04005-6. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouschop KM, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyce M, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 16.Novoa I, et al. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003;22:1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampe J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 18.Cleynen I, et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: results from the IBDchip European Project. Gut. 2012 doi: 10.1136/gutjnl-2011-300777. [DOI] [PubMed] [Google Scholar]
- 19.Fujita N, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima N, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 21.Fujita N, et al. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J Biol Chem. 2009;284:32602–32609. doi: 10.1074/jbc.M109.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell metabolism. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertolotti A, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001;107:585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao SS, et al. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology. 2013;144:989–1000. e1006. doi: 10.1053/j.gastro.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogler G, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–369. doi: 10.1016/s0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol Pharm Bull. 2003;26:931–935. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- 28.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deuring JJ, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut. 2013 doi: 10.1136/gutjnl-2012-303527. in press. [DOI] [PubMed] [Google Scholar]
- 31.Madison BB, et al. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 32.Shimshek DR, et al. Codon-improved Cre recombinase (iCre) expression in the mouse. Genesis. 2002;32:19–26. doi: 10.1002/gene.10023. [DOI] [PubMed] [Google Scholar]
- 33.Garabedian EM, Roberts LJ, McNevin MS, Gordon JI. Examining the role of Paneth cells in the small intestine by lineage ablation in transgenic mice. J Biol Chem. 1997;272:23729–23740. doi: 10.1074/jbc.272.38.23729. [DOI] [PubMed] [Google Scholar]
- 34.Bry L, et al. Paneth cell differentiation in the developing intestine of normal and transgenic mice. Proc Natl Acad Sci U S A. 1994;91:10335–10339. doi: 10.1073/pnas.91.22.10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci U S A. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baert L, et al. Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Appl Environ Microbiol. 2008;74:543–546. doi: 10.1128/AEM.01039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierce JW, et al. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 39.Satoh Y, Yamano M, Matsuda M, Ono K. Ultrastructure of Paneth cells in the intestine of various mammals. J Electron Microsc Tech. 1990;16:69–80. doi: 10.1002/jemt.1060160109. [DOI] [PubMed] [Google Scholar]
- 40.Vidal K, Grosjean I, evillard JP, Gespach C, Kaiserlian D. Immortalization of mouse intestinal epithelial cells by the SV40-large T gene. Phenotypic and immune characterization of the MODE-K cell line. J Immunol Methods. 1993;166:63–73. doi: 10.1016/0022-1759(93)90329-6. [DOI] [PubMed] [Google Scholar]
- 41.Hempel SL, Buettner GR, O’Malley YQ, Wessels DA, Flaherty DM. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2’,7’-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2’,7’-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic Biol Med. 1999;27:146–159. doi: 10.1016/s0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 42.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 43.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–951. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–2062. doi: 10.1002/hep.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 47.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tashiro E, et al. Trierixin, a novel Inhibitor of ER stress-induced XBP1 activation from Streptomyces sp. 1. Taxonomy, fermentation, isolation and biological activities. J Antibiot (Tokyo) 2007;60:547–553. doi: 10.1038/ja.2007.69. [DOI] [PubMed] [Google Scholar]
- 49.McKinney EF, et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. 2010;16:586–591. doi: 10.1038/nm.2130. 581p following 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rioux JD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parkes M, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a, Xbp1 expression in shCtrl and shXbp1 MODE-K cells (n=7/6; unpaired Student’s t-test). b, c, Immunoblot of shCtrl and shXbp1 MODE-K cells. d, Immunoblot after autophagosome-lysosome fusion inhibition via bafilomycin in silenced MODE-K IECs. e, Silenced MODE-K IECs after GFP-LC3 reporter transfection (bar, 5μm) with green punctae per cell quantification in (f; n=14; unpaired Student’s t-test). g, TEM of shCtrl and shXbp1 cells. Note double-membraned structure with engulfed contents characteristic of autophagosomes (white arrows; n=10). Bar, 0.5 μm. h, Quantification of occupied area and average size of autophagic vacuoles from (g) (n=10; unpaired Student’s t-test). i, j, Densitometry of Fig. 1a (i; n=5/4; unpaired Student’s t-test) and Fig. 1b (j; n=3; unpaired Student’s t-test). k, Low magnification (1380×, original magnification here and in the remainder of this legend) TEM image of Paneth cells from Wt mice (1), demonstrating the abundant endoplasmic reticulum (ER) and characteristic secretory granules at the apical, lumenally (‘L’) oriented side. (2) Higher magnification (5520×) of inset ‘2’ in (1) demonstrating typical secretory granules in Paneth cells from Wt mice. (3) High magnification (20700×) of inset ‘3’ in (1) illustrating a double membrane structure characteristic of an autophagosome (white arrow) in close proximity to the ER and a mitochondrion. (4) Low magnification (2160×) TEM image of Paneth cell remnants present in Xbp1ΔIEC small intestinal crypts, which lack expansion of the ER and exhibit only minuscule granule remnants. (5) Higher magnification (9000×) of inset ‘5’ in (4), demonstrating degradative autophagic vacuoles (black arrows), in close proximity to mitochondria, and the virtual absence of ER membranes. (6) High-power (14400×) magnification of inset ‘6’ in (4), illustrating a double-membrane structure (white arrow) characteristic of autophagosomes, and a degradative autophagic vacuole (black arrow). L, lumen; M, mitochondrion; ER, endoplasmic reticulum; N nucleus; as indicated, bar represents 2μm, 0.5μm and 200nm, respectively. Results represent three (b, c) or two (d, k) independent experiments. *P< 0.05, ***P< 0.001.
a, Deletion of Xbp1 in the intestinal epithelium, specifically in Paneth cells, leads to ER stress and activation of the PERK/eIF2α branch of the UPR. ATF4, a transcriptional mediator of this pathway, transactivates genes essential for autophagosome formation, such as Map1lc3b (LC3b) and Atg7 which catalyzes the creation of the ATG12-ATG5 conjugate that stabilizes ATG16L1 through complex formation21. UPR-induced autophagy in the intestinal epithelium is essential for restoration of homeostasis and restraint of ER-stress induced intestinal inflammation due to XBP1-deficiency. Activation of the UPR in the setting of XBP1-deficiency results in activation of IRE1α, resulting in the recruitment of TRAF2 and activation of IKK2 leading to IκBα degradation 4,27,45,46. As shown here, UPR-mediated autophagy however serves an important role in restraining NFκB activation, conceivably by removing hyperinflammatory ER membranes containing activated IRE1α. Pharmacological augmentation of this compensatory autophagy-dependent mechanism via inhibition of eIF2α dephosphorylation through salubrinal, or via the mTOR inhibitor rapamycin results in amelioration of UPR-induced enteritis, which is driven by the commensal microbiota, NFκB, and TNF-RI signaling in IECs and myeloid cells, whereby the ligand TNF can originate from XBP1- deficient IECs4. b, ATG16L1-deficiency in IECs leads to ER stress as revealed through upregulation of the chaperone GRP78 in IECs, increased expression of GRP78 protein in Paneth cells, increased IRE1α expression and increased splicing of Xbp1 mRNA in intestinal crypts as well as increased IEC death. This leads to increased sensitivity of the epithelium to environmental triggers (e.g. dextran sodium sulfate) that further challenge the UPR and its compensatory pathways. c, Deficiency of ATG16L1 or ATG7 in the intestinal epithelium results in abrogation of the compensatory autophagic mechanism that restrains IRE1α activity, conceivably via removal of hyperinflammatory ER membranes, and further fosters IEC death in the context of ER stress due to Xbp1 deficiency, resulting in spontaneous transmural small intestinal inflammation that is associated with further increases in NFκB activation and cell death via the mechanisms described in (a). The UPR allows for responses to a variety of signals that impact on protein folding, including genetic (e.g. rare XBP1 variants, ORMDL3 as risk factor of IBD4,47), environmental (e.g. low O2 tension in the intestinal tract) and microbial factors (e.g. microbial toxins such as trierixin48) which determines the level of ER stress in the intestinal epithelium. UPR-induced autophagy function provides a buffer to cope with different levels of ER stress and vice-versa. However, in the presence of genetic risk variants, such as ATG16L117,49,50 or IRGM51, which are relatively prevalent in the general population, this compensatory mechanism is impaired, resulting in development and/or exacerbation of intestinal inflammation in the setting of unabated ER stress.
a, Timeline for salubrinal experiment shown in Fig. 1j. b, Representative indirect immunofluorescence images of GFP-LC3 punctae accumulation (green) in small intestinal sections co-stained with antilysozyme antibody (red) treated as in (a) (n=3). DAPI, blue. Bar, 10 μm. c, Immunoblot of crypt lysates after 3 days of tamoxifen or vehicle administration (n=3). d, e, Immunoblot of primary IEC scrapings (d), densitometry in (e) (n=3; unpaired Student’s t-test). f, g, Immunoblot of shXbp1 and shCtrl MODE-K cells. ER stress-induced Jun N-terminal kinase-1 (JNK1) has previously been connected in other cellular model systems to autophagy activation through phosphorylation of B cell leukemia 2 (Bcl-2) and its dissociation from Beclin-143, as have oxidative stress/free radicals and heme oxygenase-1 (HO-1) activation44. h, Intracellular reactive oxygen species (ROS) determined by dichlorofluorescein assay and mean fluorescent intensity (MFI) after vehicle or dichlorofluorescein diacetate (DCF-DA) treatment. i, Immunoblot of shXbp1 and shCtrl MODE-K cells after administration of the JNK inhibitor SP600125 (0, 5 or 25 μM) for 4h. Note the absence of an effect of SP600125 treatment on the conversion of LC3-I to LC3-II or the levels of p-eIF2α, thereby excluding a major contribution of the JNK pathway to autophagy induction in the presence of IEC-associated XBP1-deficiency. j, Immunoblot of shXbp1 and shCtrl MODE-K cells after N-acetylcysteine (NAC), glutathione (GSH) or vehicle for 16h. Note the absence of an effect of the free radical scavengers on either of these markers of UPR-induced autophagy (LC3-II or p-eIF2α). Results represent three (f-j) independent experiments. *P < 0.05.
a, Immunoblot of shXbp1 and shCtrl MODE-K cells co-silenced with Perk (siPerk) or scrambled siRNA. b, Cumulative densitometry of the immunoblot in Fig. 1g and two additional experiments (not shown, n=10; unpaired Student’s t-test). c, p-eIF2α immunohistochemistry of small intestinal epithelium. Bar, 50 μm. d, mRNA expression levels of Map1lc3b (LC3b) and Atg7 relative to Gapdh in shCtrl and shXbp1 MODE-K cells (n=3; unpaired Student’s t-test). e, Accumulation of GFP-LC3 punctae after salubrinal and 3-day tamoxifen treatment according to timeline in Extended Data Fig. 2a (n=5). Bar, 5 μm. f, Immunoblot of primary epithelial scrapings from small intestine upon vehicle or salubrinal (Salu) treatment according to the timeline (g) (n=2). Note the increased relative levels of LC3 conversion and CHOP, a transcriptional target of ATF4, in salubrinal treated mice. g, Timeline of salubrinal experiment. Results are reported in (f) and Fig. 1k. h, Gadd34 expression in shCtrl and shXbp1 MODE-K cells co-silenced for Gadd34 (siGadd34) and control (siCtr) (one-way ANOVA with post-hoc Holm’s corrected unpaired Student’s t-test). i, Cells as in (h) were immunoblotted. j, Immunoblot of crypt lysates (n=2/4/2). Results represent three (a, h, i) independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
a, Immunoblot of primary IEC scrapings (n=4/3/3/3). Note the loss of ATG5, 12 and 16 proteins in ATG7-deficient mice. b, Representative high magnification TEM images (n=2). Note intact ER in Wt mice; severe distortion of ER in Atg7ΔIEC mice; autophagic vacuoles with the characteristic double membrane (white arrow) in Xbp1ΔIEC mice; and the absence of ER and autophagic vacuoles in Atg7/Xbp1ΔIEC mice. Bar, 200nm. ER, endoplasmic reticulum. c, Quantification of autophagic vacuoles (n=10; unpaired Student’s t-test). d, High magnification (400x) H&E images. Note knifelike extension of inflammation along blood vessels through whole thickness of muscularis propria into the serosa in the Atg7/Xbp1ΔIEC mice. Bar, 10 μm. e, Linear regression analysis for the correlation of inflammation with age. Each dot represents a single animal. (R2 and P value for deviation from 0 are shown). f, Deep inflammation score (as described in materials and methods) in Atg7/Xbp1ΔIEC mice plotted at indicated ages in weeks. (n=13/17/9; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). g, Atg16l1 IEC specific knockout design. Exon 1 of Atg16l1 gene was flanked by two loxP sites and Atg16l1 deletion was mediated by Cre recombinase (Cre) under the control of Villin (V-) promoter (V-Cre+;Atg16l1fl/fl or ‘Atg16l1ΔIEC’). Detailed description in the methods section. h, Immunohistochemical staining (brown) with anti-ATG16L1 antibody (MBL) (n=2). Note cytoplasmic IEC specific ATG16L1 immunostaining in Wt but not Atg16l1ΔIEC mice with retained ATG16L1 expression in lamina propria mononuclear cells in Atg16l1ΔIEC mice. Bar, 50μm. i, Indirect immunofluorescence microscopy (green) with anti-ATG16L1 antibody (Cell Signaling Technology) (n=2). Note the similar staining pattern as in (h) with cytoplasmic IEC-specific ATG16L1 immunoreactivity in Wt but not Atg16l1ΔIEC mice and retained ATG16L1 expression in lamina propria mononuclear cells in Atg16l1ΔIEC mice. Dashed line denotes crypt unit. Bar, 20μm. j, Schematic representation of lysozyme+ granule allocation patterns (green): normal (D0), disordered (D1), depleted (D2), and diffuse (D3)2. k, l, Lysozyme immunofluorescence (green) in crypts of Atg16l1ΔIEC and Wt mice (k), quantified in (l; n=3; unpaired Student’s t-test) according to granule allocation patterns shown in (j). Dashed line denotes crypt unit. DAPI, blue. Bar, 5μm. m, n, Quantification of Paneth cells based on the number of granular vesicles stained with Toluidine blue (m) as shown in (n) (n=4; unpaired Student’s t-test). Bar, 5μm. *P< 0.05, **P< 0.01, ***P< 0.001.
a, Immunoblot of primary IEC scrapings. Note identical GAPDH loading control as in figure 3b (n=3). b, Representative high magnification TEM images (n=2). Note intact ER in Wt mice; severe distortion of ER in Atg16l1ΔIEC mice; autophagic vacuoles with the characteristic double membrane (arrow) in Xbp1ΔIEC mice; and absence of ER and autophagic vacuoles in Atg16l1/Xbp1ΔIEC mice. Bar, 200 nm. ER, endoplasmic reticulum. c, Quantification of autophagic vacuoles (n=10 unpaired Student’s t-test). d, Representative images of p62 immunostaining on small intestinal sections (n=3). Brown staining indicates p62 with maximal signal intensity in the region of Paneth cells. Bar, 20 μm. e, High resolution H&E images. Note the extension of the inflammation into muscularis propria (arrow) in Atg16l1/Xbp1ΔIEC mice. Bar, 20μm. f, Deep inflammation score (as described in materials and methods) in mice assessed at 18 weeks of age (n=11; Kruskal-Wallis with post-hoc Dunn’s correction). g, Enteritis histology score (n=14; 7-10 weeks old mice housed in Innsbruck; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). h, qRT-PCR of grp78 in primary IEC scrapings (n=7; unpaired Student’s t-test). i, Disease activity index during DSS colitis (n=7/4, two-way ANOVA with post-hoc Bonferroni). j, Representative endoscopic images from the colon at day 5 of DSS treatment (n=3). k, Representative H&E staining of colonic sections from 5-day DSS- treated and untreated mice (n=7/4). Bar, 50μm. l, Colitis score after 5 days of DSS administration (n=7/4, Mann-Whitney U). *P < 0.05, **P < 0.01, ***P < 0.001.
a, Representative images of TUNEL labeled intestinal epithelium (brown) (n=7). Bar, 50 μm. b, Correlation of TUNEL+ cells with the severity of inflammation. Linear regression analysis of Atg16l1/Xbp1ΔIEC (left) and Atg7/Xbp1ΔIEC (right) with significant R2 and P value for deviation from zero shown (left panel: n=5/5/7/7; right panel: n=5). c, Linear least square regression analysis for the correlation of enteritis histology score with cell death and age of animals by genotype. Each dot represents a single animal (grey, Wt; yellow, Atg16l1ΔIEC; blue, Xbp1ΔIEC; red, Atg16l1/Xbp1ΔIEC mice) and the plane represents the linear regression for the enteritis histology score as a function of age and TUNEL labeling for Atg16l1/Xbp1ΔIEC mice (n=6/13/12/12). Note that the severity of inflammation significantly correlates with numbers of TUNEL+ IECs and age only in Atg16l1/Xbp1ΔIEC mice (R2=0.602, p=0.016). The three-dimensional (3D) plot is also available online in video-format. Regression analysis was performed using the R package lessR (http://cran.r-project.org/web/packages/lessR/index.html), last accessed May 2013. d, shCtrl or shXbp1 MODE-K cells were co-silenced for Atg16l1 (siAtg16l1) or with scrambled siRNA (siCtrl), and analyzed by flow cytometry for Annexin V and propidium iodide (PI) uptake (one-way ANOVA with post-hoc Bonferroni). e, Densitometry of the immunoblot shown in Fig. 3b (n=3; one-way ANOVA with post-hoc Holm’s corrected unpaired Student’s t-test). f, Immunohistochemical staining for p-IκBα in the small intestinal epithelium (n=3). Bar, 20 μm. g, NFκB consensus sequence binding assay after stimulation of shCtrl and shXbp1 MODE-K cells for 5 and 20 minutes with indicated concentrations of TNF. h, Nfkbia expression, a prototypic NFκB-transactivated gene, after TNF stimulation of shCtrl and shXbp1 MODE-K cells. i, Representative images of TUNEL labeled sections after administration of BAY11-7082 or vehicle (n=3/4/4). Bar, 50 μm. j, Enteritis histology score of mice treated with the NFκB inhibitor BAY11-7082 or vehicle (n=7/9/8; median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). k, Expression of Nfκbia in Ern1- and control-silenced shXbp1 and control MODE-K cells after TNF stimulation. Results represent three (h, k) or two (d, g) independent experiments. *P< 0.05, **P< 0.01, ***P<0.001.
a, b, Quantification of GFP-LC3 punctae accumulation after 3-day tamoxifen treatment and induction by rapamycin (a; n=10; one-way ANOVA with post-hoc Bonferroni) and representative images in (b; n=3). Bar, 5 μm. c, Densitometry of the immunoblot in Fig. 4a (n=3; one-way ANOVA with post-hoc Holm’s-corrected unpaired Student’s t-test). d, Quantification of TUNEL+ IECs per cm of gut in mice treated with rapamycin or vehicle (n=5; unpaired Student’s t-test). e, Representative images of TUNEL labeled sections from rapamycin or vehicle treated Wt, Atg16l1ΔIEC, Xbp1ΔIEC, Atg16l1/Xbp1ΔIEC mice or Wt, Atg7ΔIEC, Xbp1ΔIEC, Atg7/Xbp1ΔIEC mice. Bar, 50 μm. f, g, Quantification of TUNEL labeling in Wt, Atg16l1ΔIEC, Atg16l1/Xbp1ΔIEC mice (f; n=4/4/5/5/5/5) or Wt, Atg7ΔIEC, Atg7/Xbp1ΔIEC mice (g; n=5; one-way ANOVA with post-hoc Holm’s-corrected unpaired Student’s t-test). h, i, Enteritis histology scores of rapamycin or vehicle treated mice of respective genotypes (h; n=8/9/6/9/12/13) (i; n=8/7/6/7/7/6) (median shown; Kruskal-Wallis with post-hoc Holm’s-corrected Mann-Whitney U). *P < 0.05, **P < 0.01, ***P < 0.001.
a, Representative TEM images of small intestinal crypts from Defa6Cre+ (Wt) and Defa6Cre+;Xbp1fl/fl (Xbp1ΔPC) mice. (1) Low magnification TEM image of Paneth cells at the base of a small intestinal crypt in Wt mice, demonstrating abundant ER and characteristic secretory granules at the apical, lumenally oriented side. Bar, 5 μm. (2) Higher magnification of inset ‘2’ in (1) with numerous secretory granules. Bar, 1μm. (3) High magnification of inset ‘3’ in (1) with intact ER. Bar, 200 nm. (4) Low magnification TEM image of Paneth cells in an Xbp1ΔPC small intestinal crypt. Note the reduced number of secretory granules, the exfoliating hypomorphic Paneth cell into crypt lumen (‘E’, black dashed outline) and a transmigrating polymorphonuclear cell through the mucosa (white dashed outline). Bar, 5μm. (5) Higher magnification of inset ‘5’ in (4), demonstrating abnormalities in secretory granule maturation with numerous hypodense granules (*) and a distorted ER. Note the accumulation of electron dense cargo indicative of degradative autophagic vacuoles (black arrow) in close proximity to the nucleus. Bar, 1μm. (6) High magnification of the inset ‘6’ in (4), demonstrating degradative autophagic vacuoles filled with electron dense material (black arrow). Note the double membrane structures characteristic for autophagosomes (white arrow heads). Bar, 200 nm. (7) Low magnification TEM image of Paneth cells in an Xbp1ΔPC small intestinal crypt. Note the disorganized ER, hypodense secretory granules (*), empty area after exfoliation of a Paneth cell into the crypt lumen (‘E’, black dashed outline) similar to image (4). (8) High magnification of inset ‘8’ identified in (7), demonstrating an autophagic vacuole (black arrow) surrounded by a markedly distorted ER. Bar, 200 nm. (9) High magnification of the inset ‘9’ in (7), with additional examples of degradative autophagic vacuoles accompanied by a distorted ER. Representative of 2 independent experiments. Bar, 200 nm. L, lumen; M, mitochondrion; ER, endoplasmic reticulum; N nucleus. b, Densitometry of the immunoblot in Fig. 4d (n=2; unpaired Student’s t-test). *P< 0.05.
a, b, TUNEL labeled ileal sections (a) with quantification of TUNEL+ cells per cm gut shown in (b; n=7/11; unpaired Student’s t-test). Bar, 50 μm. c, d, Ki67 immunohistochemical staining of ileal sections (c) with quantification of Ki67+ cells per total IEC count along the crypt-villus axis shown in (d; n=5; unpaired Student’s t-test). Bar, 20 μm. e, f, Bromodeoxyuridine (BrdU) labeled ileal sections (e) after a 24h with quantification of BrdU+ cells per total IEC count along the crypt-villus axis in (f; n=5/3; unpaired Student’s t-test). Bar, 50μm. g, h, Periodic acid-Schiff (PAS)-stained ileal sections (g) with quantification of PAS+ cells per total IEC count along the crypt-villus axis shown in (h; n=5; unpaired Student’s t-test). Bar, 50μm. i, Enteritis histology score of indicated genotypes (n=3; median shown; Kruskal-Wallis). j, Taqman qRT-PCR for MNV of fecal samples in three different animal facilities (n=3). Note detectable MNV genome copy numbers in Boston (Atg7/Xbp1ΔIEC, Fig. 2c) and Innsbruck (Atg16l1/Xbp1ΔIEC, Extended Data Fig. 5g) colonies, and absence of MNV after re-derivation of colonies into the MNV-free Cambridge facility (Atg16l1/Xbp1ΔIEC, Fig 2e), yet similar levels of enteritis in Xbp1ΔIEC and Atg16l1/Xbp1ΔIEC mice maintained in Innsbruck (MNV+) and Cambridge (MNV−) (Fig. 2b, e and Extended Data Fig. 5g) and relatively more severe disease in Xbp1ΔIEC mice from Boston (Fig. 2c). Sentinel mice from the Cambridge colony also tested negative for MNV by serological analysis (data not shown). These observations suggest that the inflammatory phenotypes observed are not necessarily dependent upon MNV. *P < 0.05.