

Highlights
► Hypercalcemia is an extremely rare paraneoplastic syndrome in children. ► Small cell carcinoma is the commonest ovarian tumor associated with hypercalcemia. ► Small cell carcinoma must be ruled out because of poor prognosis. ► We report the only third case of JGCT associated with paraneoplastic hypercalcemia.
Keywords: Juvenile granulosa cell tumor, Hypercalcemia, Ovarian cancer
Introduction
Hypercalcemia complicating cancers are common in adults, but are seen very infrequently in children (Tsunematsu et al., 2000). It occurs in 5% of ovarian malignancies, with small cell and clear cell carcinoma being the commonest ovarian tumors associated with hypercalcemia and historically carries a poor prognosis (Martinez-Borges et al., 2009; McDonald et al., 2012). Juvenile granulosa cell tumors (JGCTs) of ovary are rare neoplasms that typically occur in children and young women (Bouffet et al., 1997; Calaminus et al., 1997). The majority of children with this tumor are frequently characterized by hormonal symptoms like sexual precocity or present with signs of abdominal distension, and/or a palpable pelvic tumor. The majority of JGCTs are in FIGO (International Federation of Gynecology and Obstetrics) stage I and have a less aggressive behavior than that of adult type (Calaminus et al., 1997). It has a favorable prognosis when treated with surgery alone while advanced FIGO stages of this tumor entity are frequently associated with early relapses. Two cases of ovarian JGCT with hypercalcemia have previously been documented in the literature (Piura et al., 2008; Daubenton and Sinclair-Smith, 2000). The histological features and the differential diagnosis of the ovarian cancer with hypercalcemia in children are discussed.
Case presentation
A 15-year-old white girl was admitted with abdominal pain and vomiting for 2 days. Two days prior, she was seen by her primary physician for vomiting. She was prescribed intravenous metoclopramide without improvement. She was referred to the pediatric emergency unit for appendicitis suspicion. The girl was febrile with a body temperature of 38 °C. On physical examination, signs of puberty were appropriate for her age (she had menarche at age 14). A mass that was painful on palpation was located in the right lower quadrant. Headache was reported without alterations in level of consciousness, or other neurologic symptoms. Her vital signs were significant with respiratory rate of 24 breaths per minute, oxygen saturation of 96%, heart rate of 120 beats per min indicating tachycardia, and blood pressure of 136/85 mm Hg. Laboratory examination showed normal urea and creatinine, but corrected serum calcium was 14.9 mg/dl (3.70 mmol/l), phosphorus 0.50 mmol/l, LDH 348 U/l, and C-reactive protein level 259 mg/l. Serum parathyroid hormone-intact (PTH-i) level was 10 pg/ml (normal 10–65). Alpha fetoprotein and beta human chorionic gonadotropin were both normal at < 10 IU/ml. Serum CA-125 was 204 (normal, 0–35) U/ml, while serum CA-15-3 was normal. Serum inhibin was not ever checked. Electrocardiograph was normal. Abdominal ultrasonography demonstrated semisolid pelvic mass measuring 125 mm × 105 mm × 65 mm. An abdominal contrast-enhanced computed tomography scan revealed a right ovarian mass with pelvic ascites, without involvement of pelvic structures (Fig. 1). She was referred to intensive care unit for an antihypercalcemic treatment. Isotonic saline hydration was started together with intravenous infusion of pamidronate (1 mg/kg) and furosemide. An isotopic (99 mTc-HMDP-“Osteocis”) bone scanning did not detect bone metastasis.
Fig. 1.
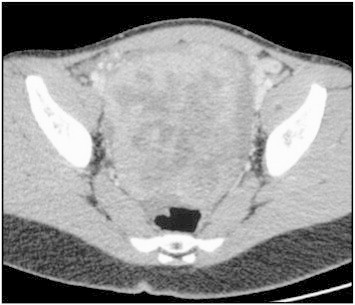
Enhanced pelvis scan shows semisolid pelvic mass.
On the six day after starting the management of hypercalcemia, she underwent a median infraumbilical laparotomy with right salpingo-oophorectomy, omentectomy and drainage of ascites. Her serum calcium remained normal.
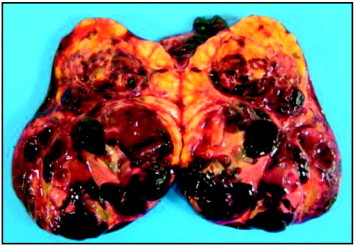
The tumor measured 120 mm × 90 mm × 65 mm with soft consistency. Its external surface was smooth and intact. The cut surface was heterogeneous, showing bright yellow nodular areas alternating with hemorrhagic areas and small cystic structures (Fig. 2). The right fallopian tube and the omentum were normal.
Microscopic examination revealed a solid nodular proliferation of medium-sized cells with hypocellular edematous areas, tumor cell necrosis and some pseudofollicular structures. The tumor cells had moderate to large amounts of pale or eosinophilic cytoplasm with focal features of luteinization. The nuclei were round to oval without groove. The cells were mitotically active, averaging 7 mitoses per 10 high power fields (Fig. 3A). Immunohistochemical profile was the following: the tumor cells demonstrated positive reactivity for alpha-inhibin (Fig. 3B) which confirms the histological suspicion of a sex-cord stromal process. The tumor also expressed reactivity to calretinin, estrogen and progesterone receptors, CD56 and smooth muscle actin. The cells were focally positive for cytokeratin KL1. The tumor was negative for TTF1, synaptophysin and chromogranin, pS100, EMA, and CD99.
Fig. 3.
B: Immunohistochemical staining for alpha-inhibin, showing strong cytoplasmic immunoreactivity (× 400).
These morphological and immunohistochemical features were the ones of a juvenile granulosa cell tumor, excluding the hypotheses of a neuroendocrine carcinoma or a clear cell carcinoma of the ovary which are however the most common ovarian tumors associated with hypercalcemia.
Discussion
Granulosa cell tumor (GCT) of the ovary is divided into adult and juvenile types. JGCTs predominantly occur in the first three decades of life and differ from adult granulosa cell tumors with regard to clinical and pathological features. A common symptom is abdominal pain associated with palpable mass. JGCTs in infants have been reported in association with hormonal secretion like sexual precocity, or recently a reported case with tuberous sclerosis. Calaminus et al. showed that abdominal distension with a palpable mass was the first sign of the disease reported in sixteen of 33 girls and signs of pseudo-precocious puberty for fourteen patients at the time of diagnosis (Calaminus et al., 1997).
Hypercalcemia complicating cancers (JGCTs) of ovary are exceedingly rare. To our knowledge, we present only the third case but the second one in children, with Daubenton et al. reporting a case of a girl treated by surgery and chemotherapy and died of the disease 4 months later (Table 1).
Table 1.
Patients' characteristics.
| Age (years) | Symptoms | Surgical approach | Stage (FIGO) | CT | Outcome | |
|---|---|---|---|---|---|---|
| Daubenton | 11 | Abdominal pain and abdominal mass | Salpingo-oophorectomy | III | Yes | Died of the disease |
| Piura | 25 | Vomiting and abdominal mass | Salpingo-oophorectomy and partial infra-colic omentectomy | IC | Yes | Remission (10 months) |
| Rod | 15 | Fever, vomiting and abdominal mass | Salpingo-oophorectomy and omentectomy | IA | No | Remission (24 months) |
CT: Chemotherapy.
Hypercalcemia is exceedingly rare in children. Clear cell adenocarcinoma of the ovary has been recognized as one of the most common histologic subtypes associated with hypercalcemia in adult malignancies (Martinez-Borges et al., 2009). In children, dysgerminoma of the ovary is the second most common ovarian malignancy after ovarian small cell carcinoma to be associated with hypercalcemia that is due to production of parathyroid hormone-related protein (PTH-rp) by the tumor (Piura et al., 2008). In our case, parathyroid hormone level was normal but like Daubenton et al. we did not have an assay for PTH-rp. Because hypercalcemia did not recur after removal of the tumor, we think that it was induced by PTH-rp which was secreted by the tumor.
Abdominal computed tomographic scan shows a large enhanced abdominopelvic solid mass that can sometimes be a solid and cystic mass. Piura et al. described a pelvic solid mass with moderate amount of ascites. Daubenton et al. described a solid and cystic mass in ultrasonography. Radiologic characteristics of JGCT are nonspecific and this tumor cannot be reliably distinguished from other ovarian neoplasms.
Surgery is the primary option for treatment of localized JGCTs. Stage IA has a favorable prognosis with a 3-year survival rate of 97% when treated with conservative surgery alone (unilateral oophorectomy) (Calaminus et al., 1997). In view of the excellent prognosis of stage IA JGCT, as it was in our case, additional chemotherapy was not proposed.
Surgery and additional chemotherapy are proposed in more advanced stages (stages IC and more) because the recurrence rate is higher (Calaminus et al., 1997). Stage is the most important prognostic factor in adult literature. Daubenton et al. reported an 11-year-old black girl treated with right oophorectomy, omentectomy and chemotherapy. The histologic diagnosis was a malignant juvenile granulosa cell tumor stage III and she died of progressive intraabdominal disease 4 months later. Piura et al. reported a 25-year-old treated with salpingo-oophorectomy and partial infra-colic omentectomy. Because of the ruptured ovarian capsule and presence of tumor cells in the ascites, the tumor was allocated as stage IC and chemotherapy was done. Ten months after the surgery and 7 months after completion of chemotherapy, the patient was alive and well and without evidence of recurrent disease.
FOXL2 is one of the markers of ovarian differentiation and is required for the normal development of granulosa cells. This gene belongs to the large family of forkhead FOX transcription factors. Shah et al. identified a recurring somatic mutation in FOXL2: this mutation is described in nearly all adult GCT and is thus a specific marker of this diagnosis (Shah et al., 2009). In our case, we did not find FOXL2 mutation in the tumor, confirming that it is not an adult GCT. Kalfa et al. showed that FOXL2 is not expressed or is underexpressed in JGCT with an aggressive pattern of progression and it may be a prognostic factor for these tumors (Kalfa et al., 2007). These findings differ radically from those reported by D'Angelo et al. who found that patients with JGCT and higher FOXL2 expression had a worse disease-free survival and overall survival (D'Angelo et al., 2011). The prognostic significance of expression levels of FOXL2 is so far a controversial subject.
In our case, the patient's serum calcium levels were followed-up every 3–4 months, which were normal. There had been no recurrence of the symptoms, and there was normal ultrasonography 24 months after the surgery.
We present this unusual case of JGCT associated with hypercalcemia so that clinicians as well as pathologists are made aware of the rare paraneoplastic syndrome association of this tumor.
Conflict of interest statement
None.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary data
Fig. 2. Cross-section demonstrating solid yellow areas with gross appearance of 283 and blood-filled cystic cavities of the granulosa cell tumor.
Fig. 3. A: Diffuse growth of uniform-sized cells with eosinophilic cytoplasm (hematoxylin 285 and eosin × 400).
References
- Bouffet E., Basset T., Chetail N., Dijoud F., Mollard P., Brunat-Mentigny M., David M. Juvenile granulosa cell tumor of the ovary in infants: a clinicopathologic study of three cases and review of the literature. J. Pediatr. Surg. 1997;32(5):762–765. doi: 10.1016/s0022-3468(97)90029-4. (May) [DOI] [PubMed] [Google Scholar]
- Calaminus G., Wessalowski R., Harms D., Göbel U. Juvenile granulosa cell tumors of the ovary in children and adolescents: results from 33 patients registered in a prospective cooperative study. Gynecol. Oncol. 1997;65(3):447–452. doi: 10.1006/gyno.1997.4695. (Jun) [DOI] [PubMed] [Google Scholar]
- D'Angelo E., Mozos A., Nakayama D., Espinosa I., Catasus L., Munoz J., Prat J. Prognostic significance of FOXL2 mutation and mRNA expression in adult and juvenile granulosa cell tumors of the ovary. Mod. Pathol. 2011;24(10):1360–1367. doi: 10.1038/modpathol.2011.95. (Oct) [DOI] [PubMed] [Google Scholar]
- Daubenton J.D., Sinclair-Smith C. Severe hypercalcemia in association with a juvenile granulosa cell tumor of the ovary. Med. Pediatr. Oncol. 2000;34(4):301–303. doi: 10.1002/(sici)1096-911x(200004)34:4<301::aid-mpo22>3.0.co;2-d. (Apr) [DOI] [PubMed] [Google Scholar]
- Kalfa N., Philibert P., Patte C., Ecochard A., Duvillard P., Baldet P., Jaubert F., Fellous M., Sultan C. Extinction of FOXL2 expression in aggressive ovarian granulosa cell tumors in children. Fertil. Steril. 2007;87(4):896–901. doi: 10.1016/j.fertnstert.2006.11.016. (Apr) [DOI] [PubMed] [Google Scholar]
- Martinez-Borges A.R., Petty J.K., Hurt G., Stribling J.T., Press J.Z., Castellino S.M. Familial small cell carcinoma of the ovary. Pediatr. Blood Cancer. 2009;53(7):1334–1336. doi: 10.1002/pbc.22184. (Dec 15) [DOI] [PubMed] [Google Scholar]
- McDonald J.M., Karabakhtsian R.G., Pierce H.H., Iocono J.A., Desimone C.P., Bayliff S.L., Ueland F.R. Small cell carcinoma of the ovary of hypercalcemic type: a case report. J. Pediatr. Surg. 2012;47(3):588–592. doi: 10.1016/j.jpedsurg.2011.12.004. (Mar) [DOI] [PubMed] [Google Scholar]
- Piura B., Wiznitzer A., Shaco-Levy R. Juvenile granulosa cell tumor of the ovary associated with hypercalcemia. Arch. Gynecol. Obstet. 2008;277(3):257–262. doi: 10.1007/s00404-007-0459-5. (Mar) [DOI] [PubMed] [Google Scholar]
- Shah S.P., Köbel M., Senz J., Morin R.D., Clarke B.A., Huntsman D.G. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N. Engl. J. Med. 2009;360(26):2719–2729. doi: 10.1056/NEJMoa0902542. (Jun 25) [DOI] [PubMed] [Google Scholar]
- Tsunematsu R., Saito T., Iguchi H., Fukuda T., Tsukamoto N. Hypercalcemia due to parathyroid hormone-related protein produced by primary ovarian clear cell adenocarcinoma: case report. Gynecol. Oncol. 2000;76(2):218–222. doi: 10.1006/gyno.1999.5619. (Feb) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. 2. Cross-section demonstrating solid yellow areas with gross appearance of 283 and blood-filled cystic cavities of the granulosa cell tumor.
Fig. 3. A: Diffuse growth of uniform-sized cells with eosinophilic cytoplasm (hematoxylin 285 and eosin × 400).