

Abstract
A direct method to synthesize lignan cyclobutanes and analogs via photoinduced electron transfer is presented. A variety of oxygenated alkenes are employed to furnish terminal or substituted cyclobutane adducts with complete regiocontrol, yielding cycloadducts with trans stereochemistry. Key to minimizing competing cycloreversion is the inclusion of an aromatic electron relay (ER). This method has been adapted to the synthesis of the natural products magnosalin and pellucidin A.
Introduction
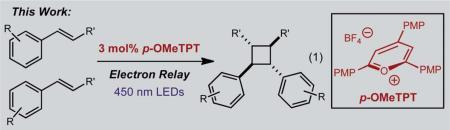
Cyclobutane rings are prominent structural features in a number of natural products including terpenes, steroids, fatty acids, as well as the more modest lignan and neolignan compounds (Scheme 1).1 The chemically and architecturally diverse lignan family features over two-dozen cyclobutane-containing members, which are comprised of two fused phenylpropanoid subunits.2 Many of these cyclobutane lignans and neolignans have significant bioactivity and are often considered as lead compounds for the development of antifungal, antiviral, and anticancer drugs.3 Several cyclobutane lignan natural products have notable inhibitory activity toward nitric oxide synthase (NOS) enzymes,4 as well as potent binding to the glucocorticoid receptor (GR).5 Despite the biological importance of this class of compounds, there have been few efforts devoted to the stereoselective synthesis of cyclobutane lignans and potentially valuable analogs. Herein, we report a method for the stereoselective synthesis of C2-symmetric cyclobutane alkene dimers (eqn (1)) that has been applied to the total syntheses of the lignans magnosalin,6 endiandrin A,3,7 and the lignan-like cyclobutane, pellucidin A,8 of which we propose a revised structure.
 |
Scheme 1.

Several examples of bioactive cyclobutane lignan natural products and derivatives.
Cyclobutane lignan biosynthesis has been proposed to occur via the homodimerization of the requisite styrene.9 The intermolecular head-to-head dimerization of such styrenes has been attempted via direct photolytic sensitization using ultraviolet light, however, this method typically results in the exclusive formation of meso isomers.10 Olefin isomerization can also compete with cycloaddition and the subsequent alkene isomers can go on to form multiple stereoisomeric products that can often make isolation of single isomers challenging.11 We hypothesized that a single electron transfer (SET) approach12 could provide a biomimetic route to the all trans-cyclobutane core found in many bioactive lignan natural products.
Seminal work by Ledwith,13 Bauld14 and others15 has demonstrated that catalytic quantities of single electron oxidants promote [2 + 2] alkene cycloaddition manifolds. Lewis11 and others16 have also seen success by implementing photooxidants activated by ultraviolet light. However, the photooxidants were employed in stoichiometric amounts,11 and reactions were hampered by three competing pathways: olefin isomerization, back electron transfer, and cycloreversion.17 Furthermore, notable amounts of decomposition and byproducts were observed when the photooxidant was excited with ultraviolet light. During the preparation of this manuscript, Yoon and coworkers reported an alkene heterodimerization strategy using a ruthenium photooxidant that largely prevents this competing cycloreversion process.18 By judicious selection of a photooxidant with an excited state reduction potential that is higher than the alkene substrate, but is lower than the subsequent cycloadduct, Yoon was able to successfully realize a number of alkene [2 + 2] heterodimerizations with starting alkenes close to the redox potential of anethole. Chiba and coworkers have also successfully realized the heterodimerization between unactivated alkenes and 3,4-dihydro-2H-pyran by electrochemical methods.19 Although a very unique and effective redox tag mechanism is proposed, reactions required the unactivated alkene to be present in high excess.
Results and discussion
To access the more variably oxygenated cyclobutane lignans and derivatives, we sought to develop a method whereby tuning the photooxidation catalyst was not necessary. We proposed that a single photooxidation catalyst could be used in conjunction with an arene or polyarene acting as an additive would expand the cycloaddition substrate scope to include lignan cyclobutanes. Although the addition of similar reagents have been observed as co-sensitizers in the past, we anticipated that oxidation of the electron relay (ER) by the organic photooxidant in its triplet excited state would instead result in the formation of a cation radical capable of oxidizing the alkene starting material. The relay would be required to have a marginally higher oxidation potential than the alkene substrate of interest to minimize the unproductive cycloreversion process by inhibiting the oxidation of the cycloadduct.20
In the selection of a single electron photooxidant, we focused our attention on triaryloxopyrylium salts as potential candidates due to their excitation in the visible region (hv > 400 nm).21,22 The use of triaryloxopyrylium salts is also potentially advantageous due to minimization of Coulombic attraction between catalyst and substrate after single electron transfer that ultimately is believed to hinder unproductive back-electron transfer. Of the triaryloxopyrylium salts considered, we elected to employ 2,4,6-tris(4-methoxyphenyl)pyrylium tetrafluoroborate ( p-OMeTPT) as it has an excited state reduction potential significantly higher than all of the styrenes we planned to investigate that could be attenuated with an appropriate ER.
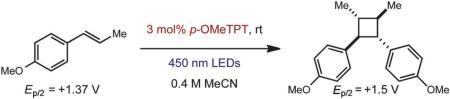
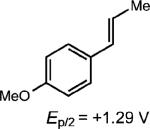
We first examined the homodimerization of anethole as a test case for this proposal. Subjecting anethole in acetonitrile to 3 mol% of p-OMeTPT in the presence of 450 nm LEDs afforded only starting material (Table 1, entry 1). However, in the presence of electron relays such as anthracene or naphthalene, appreciable quantities of the cyclobutane adduct were observed (entries 2–4). Since the triplet energy of the excited photocatalyst (51 kcal mol–1) is below the triplet energy of naphthalene (60 kcal mol–1) we propose the polyarenes behave as an electron-relay, similar to methyl viologen.23 Unfortunately, long reaction times (5 days) were required for higher levels of conversion (54% yield, entry 4). If the oxidation potential of the additive was vastly lower than the starting material, no cyclobutane adduct was attained (entry 6). Control experiments confirmed that cyclodimerization was not possible in the absence of light or the p-OMeTPT catalyst.
Table 1.
Effect of electron relays as an additive in the [2 + 2] dimerization of anetholea
| |||
|---|---|---|---|
| Entry | Electron relayb | Equivalents | Yield |
| 1 | None | None | 0% |
| 2 | Anthracene (+1.21 V) | 0.25 | 13% |
| 3 | Naphthalene (+1.61 V) | 0.25 | 16% |
| 4 | Naphthalene | 0.5 | 18% |
| 5 c | Naphthalene | 0.5 | 54% |
| 6 | NPh3 (+0.91 V) | 0.25 | 0% |
| 7d | Naphthalene | 0.5 | 0% |
| 8e | Naphthalene | 0.5 | 0% |
Reactions were carried out for 24 h, unless otherwise noted. 1H NMR yields are reported.
Peak potentials of electron relay in parenthesis.
Reaction time was 5 days.
Reaction in the dark.
Reaction in the absence of p-OMeTPT.
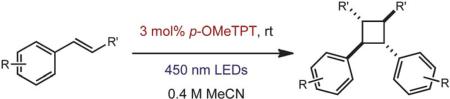
To test the viability of this method, we next turned our attention to additional styrene derivatives. First, we investigated the oxidative dimerization of 2,4-dimethoxy-β-methylstyrene (entry 2, Table 2). An additive with a lower oxidation potential was implemented since the oxidation potential of this styrene is significantly lower than anethole. By including anthracene as the additive, the dimer of 2,4-dimethoxy β-methyl styrene was obtained in 46%. Derivatives of anethole bearing an alkene (entry 3) and a protected amine (entry 4) furnished synthetically useful amounts of the cycloadducts (51% and 42% yields, respectively).
Table 2.
[2 + 2] Dimerization of aromatic alkenes via photoinduced electron transfera
| ||||
|---|---|---|---|---|
| Entry | Substrate | Electron relay | Product | Yieldb,c |
| 1 |
|
0.5 equiv. naphthalene |
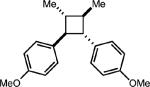
|
54% (0%) |
| 2 |
|
0.25 equiv. anthracene |
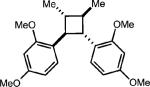
|
46% (3%) |
| 3 |
|
0.5 equiv. naphthalene |
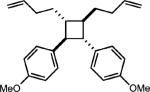
|
51% (30%) |
| 4 |
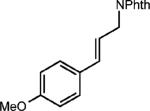
|
0.5 equiv. naphthalene |
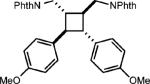
|
42% (27%) |
| 5d |
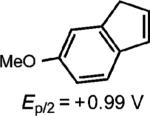
|
3 equiv. propylene oxide |
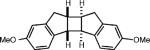
|
72% (0%) |
| 6e |
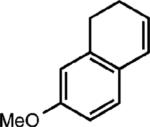
|
None |
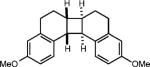
|
(62%) |
| 7 |
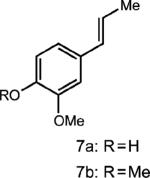
|
0.25 equiv. anthracene |
|
0% (0%) |
| 8 |
|
None |
|
(31%) |
| 9f |
|
0.25 equiv. naphthalene |
|
78% (14%) |
Reactions carried out in 0.4 M degassed acetonitrile.
Average of two isolated yields on a 100 mg scale.
Parenthetical product yield in absence of electron relay.
Reaction carried out at 0 °C in sparged dichloromethane.
Reaction carried out at –10 °C in sparged dichloromethane.
Reaction carried out at –10 °C in acetonitrile.
Homodimerization of 6-methoxyindene gave the highest yields in the presence of propylene oxide as the additive (entry 5). At this time, we do not have definitive mechanistic data to determine the role of the propylene oxide, however it is possible that the epoxide forms a Lewis base complex with the cation radical to prevent polymerization, which is the predominant reactivity observed in its absence.
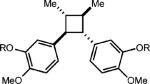
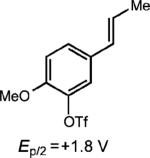
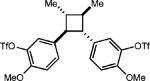
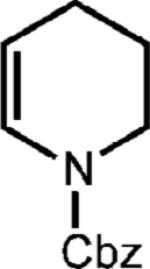
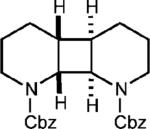
Curiously, our system did not seem amenable to styrenes with electron-rich substitution at the meta position, despite the presence of another electron rich substituent in the para position (entry 7). It is conceivable that due to the presence of the meta donating substituent, the charge density of the cation radical is mainly located on the arene ring rather than the alkene, preventing productive reactivity.24 To support this notion, a derivative in which the meta substituent possessed an electron withdrawing group furnished modest amounts of the desired cyclodimer (31% yield, entry 8). As the oxidation potential of this substrate is closest to the photooxidant, it is plausible that the oxidation potential of the cyclodimer is higher than the photooxidant. This method was also tolerant of cyclic enamines such as the Cbz protected 3,4-dihydropyridine, giving the cycloadduct in good yield (78%, entry 9). It should be noted that (1) significantly higher yields of the cyclobutane adducts were obtained in all cases where electron relays were employed and (2) the nature of the ER remains unchanged upon completion of the reaction, and is easily removed during purification.
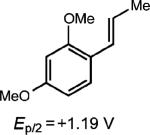
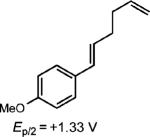
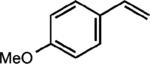
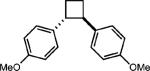
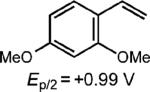
To extend this method, we next considered the head-to-head dimerization of terminal styrenes (Table 3). Not surprisingly, unsubstituted styrenes were extremely susceptible to polymerization when subjected to the previously developed conditions for β-substituted substrates. In an attempt to inhibit the polymerization pathway, we carried out the reactions at cryogenic temperatures in the presence of an electron relay. Using anthracene as the electron relay, we were able to obtain the [2 + 2] cycloadducts of 4-methoxystyrene and 2,4-dimethoxystyrene in good yield (83% and 73%, entries 1 and 2). In order to use the more electron-rich 2,4,5-trimethoxystyrene as a substrate, diethylaniline (Ep/2 = +0.66 V vs. Ag/AgCl) was employed to prevent cycloreversion and subsequent polymerization (entry 3). This furnished a cyclobutane adduct that matched the spectral data of the natural product pellucidin A. According to Bamya and coworkers, the structure of the head-to-head dimer was proposed to be the meso isomer.8 To verify the stereochemistry, X-ray quality crystals were obtained, which unambiguously revealed the trans geometry of the cyclobutane substituents. Lastly, the dimerization of N-vinyl carbazole, which had been previously reported by Bauld, furnished the desired cycloadduct in a quantitative yield (entry 4).
Table 3.
[2 + 2] dimerization of monosubstituted alkenes via photoinduced electron transfera

| ||||
|---|---|---|---|---|
| Entry | Substrate | Electron relay | Product | Yieldb,c |
| 1 |
|
0.75 equiv. anthracene |
|
83% (0%) |
| 2 |
|
0.5 equiv. anthracene |
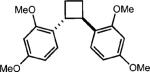
|
73% (0%) |
| 3 |
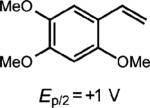
|
15 mol% diethylaniline |
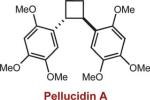
|
34% (0%) |
| 4d |
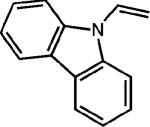
|
None |
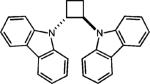
|
(99%) |
Reactions carried out in 0.4 M of sparged acetone.
Average of two isolated yields of 100 mg scale.
Parenthetical product yield in absence of electron relay.
Reaction was carried out at 0 °C.
Most importantly, we were able to utilize this method to synthesize two lignan cyclobutane natural products. Treatment of E-asarone to the oxidative reaction conditions in the presence of 0.5 equiv. of anthracene furnished magnosalin in 50% yield. This result is in contrast to the direct irradiation (hv > 300 nm) of E-asarone, where Yamamura observed the formation of the meso isomer, heterotropan, as the sole adduct in 15% yield.10 Attempts at synthesizing endiandrin A via a [2 + 2] cycloaddition of meta-oxygenated anethole derivatives were not successful, presumably for the aforementioned reasons. However, we were able to further elaborate the anethole cyclobutane dimer 1 (Table 1, entry 1) by a bromination, demethylation and methoxylation25 sequence to furnish endiandrin A in 59% yield over 3 steps. This represents the first synthesis of the bioactive cyclobutane lignan.
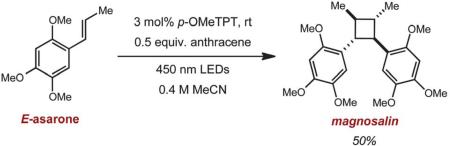 |
(2) |
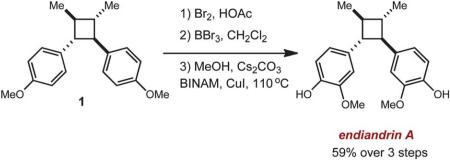 |
(3) |
To probe the role of the ER additive and its effect on cycloreversion, we resubmitted the cyclodimer 2 in entry 2, Table 2 to the standard conditions. In the absence of the electron relay, the cyclodimer was recovered in 65% yield and significant quantities (11%) of the alkene monomer were observed. The remainder of the mass balance was attributed to unidentifiable decomposition products. Conversely, when 2 was resubmitted to the reaction conditions containing 0.5 equiv. of anthracene, 83% of 2 was recovered and only trace amounts of the styrene monomer were observed. These results suggest that the presence of anthracene is critical to impeding the cycloreversion process (Fig. 1).26
Fig. 1.

Cycloreversion was prominent when an additive was excluded (eqn(4)) and circumvented when the additive was employed (eqn(5)).
Based on these results, we propose the following mechanistic hypothesis (Fig. 2). Following excitation of the p-OMeTPT, oxidation of the electron relay (ER) provides the active oxidant (ER•+), which oxidizes the alkene substrate. After cycloaddition with another molecule of alkene starting material, the ER or 3 most likely reduces cyclobutane cation radical 4. In the absence of the ER, the cycloaddition is reversible, most likely due to single electron oxidation of the cyclobutane by p-MeOTPT. Presently, we propose that the ER suppresses cycloreversion by shielding the cyclobutane products from oxidative degradation by the oxopyrylium salt.
Fig. 2.

Working mechanism for the alkene cyclodimerization reaction.
Conclusions
We have devised a simple and direct method for the synthesis of C2-symmetric cyclobutanes including magnosalin, various lignan analogs, and 1,1 unsubstituted cyclobutane compounds. By including an electron relay additive, we have offered a protocol for preventing cycloreversion and polymerization of β-substituted styrenes and terminal styrenes, respectively. This method sheds light on a possible biosynthesis of lignans via a single electron oxidation pathway.
Supplementary Material
Acknowledgements
The project described was supported by The University of North Carolina at Chapel Hill, generous funding from NIGMS (R01 GM098340) and an Eli Lilly New Faculty Award.
Footnotes
Electronic supplementary information (ESI) available. CCDC 932605. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc50643f
Notes and references
- 1.a Sergeiko A, Poroikov V, Hanus LO, Dembitsky V. Open. Med. Chem. J. 2008;2:26–37. doi: 10.2174/1874104500802010026. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hansen TV, Stenstrøm Y. Naturally Occurring Cyclobutanes. In: Hudlicky LT, editor. Organic Synthesis: Theory and Applications. Vol. 5. Elsevier; Oxford, UK: 2001. pp. 1–38. [Google Scholar]; c Dembitsky VM. J. Nat. Med. 2008;62:1. doi: 10.1007/s11418-007-0166-3. [DOI] [PubMed] [Google Scholar]
- 2.Dictionary of Natural Products on CD-ROM, Version 15.2. Chapman and Hall Electronic Publishing Division; 2007. [Google Scholar]
- 3.a Apers S, Vlietinch A, Pieters L. Phytochem. Rev. 2003;2:201–217. [Google Scholar]; b Davis R, Barnes E, Longden J, Avery V, Healy P. Bioorg. Med. Chem. 2009;17:1387. doi: 10.1016/j.bmc.2008.12.030. [DOI] [PubMed] [Google Scholar]
- 4.Rhu J, Son H, Lee S, Sohn D. Bioorg. Med. Chem. Lett. 2002;12:649. doi: 10.1016/s0960-894x(01)00812-5. [DOI] [PubMed] [Google Scholar]
- 5.Davis R, Carroll A, Duffy S, Avery V, Guymer G, Forster P, Quinn R. J. Nat. Prod. 2007;70:1118. doi: 10.1021/np070073x. [DOI] [PubMed] [Google Scholar]
- 6.Magnosalin was initially misassigned. For isolation and characterization of Magnosalin: Malhotra S, Koul S, Taheja S, Pushpangadan P, Dhar K. Phytochemistry. 1990;29:2733.Mahindru RN, Taneja SC, Dhar KL, Brown RT. Phytochemistry. 1999;32:1073.Kikuchi T, Kadota S, Yanada K, Tanaka K, Watanabe K, Yoshizaki M, Yokoi T, Shingu T. Chem. Pharm. Bull. 1983;31:1112.Yamamura S, Niwa M, Nonoyama M, Terada Y. Tetrahedron Lett. 1978;49:4891.
- 7.Kuntzsch D, Bergann T, Dames P, Fromm A, Fromm M, Davis RA, Melzig MF, Schulzke JD. PLoS One. 2012;7:e49426. doi: 10.1371/journal.pone.0049426. DOI: 10.1371/journal.pone.0049426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Bamya J, Arruda M, Muller A, Aruda A, Canto W. Phytochemistry. 2000;55:779–782. doi: 10.1016/s0031-9422(00)00224-7. [DOI] [PubMed] [Google Scholar]; b Ngadjui B, Lontsi D, Lontsi JF, Ayafor J, Sondengam BL. Phytochemistry. 1989;28:231. [Google Scholar]
- 9.Wang Q, Terreaux C, Marston A, Tan R, Hostettmann K. Phytochemistry. 2000;54:909. doi: 10.1016/s0031-9422(00)00105-9. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 10.Yamamura S, Niwa M, Yukimasa T, Nonoyama M. Bull. Chem. Soc. Jpn. 1982;55:3573. [Google Scholar]
- 11.Lewis F, Kojima M. J. Am. Chem. Soc. 1988;110:8664. [Google Scholar]
- 12.For a reviews, see: Waske P, Tzvetkov NT, Mattay J. Synlett. 2007;5:669–685.Fagnoni M, Dondi D, Ravelli D, Albini A. Chem. Rev. 2007;107:2725. doi: 10.1021/cr068352x.
- 13.Ledwith A. Acc. Chem. Res. 1972;4:133. [Google Scholar]
- 14.a Bauld N. Tetrahedron. 1989;45:5307. [Google Scholar]; b Bauld N, Pabon R. J. Am. Chem. Soc. 1983;105:633. [Google Scholar]; c Bauld N, Bellville D, Harirchian B, Lorenz K, Pablon R, Reynolds D, Worth D, Chiou H, Marsh B. Acc. Chem. Res. 1987;20:371. [Google Scholar]
- 15.a Schepp N, Johnston L. J. Am. Chem. Soc. 1994;116:6895. [Google Scholar]; b Schepp N, Johnston L. J. Am. Chem. Soc. 1994;116:10330. [Google Scholar]; c Muller F, Mattay J. Chem. Rev. 1993;93:99. [Google Scholar]; d Tojo S, Toki S, Takamuku S. J. Org. Chem. 1991;56:6240. [Google Scholar]; e Nozki H, Noyori O, Kawanisi M. Tetrahedron. 1967;24:2183. [Google Scholar]
- 16.Kadota S, Tsubono K, Makino K, Takeshita M, Kikuchi T. Tetrahedron Lett. 1987;28:2857. [Google Scholar]
- 17.Bauld NL, Gao D. Electron transfer in Chemistry. In: Balzani V, Mattay J, editors. Organic, Organometallic, and Inorganic Molecules (Part 1) Vol. 2. Wiley-VCH; Weinheim: 2001. pp. 133–205. [Google Scholar]
- 18.a Ischay M, Yoon T. Chem. Sci. 2012;3:2807. doi: 10.1039/C2SC20658G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ischay M, Yoon T. Eur. J. Org. Chem. 2012:3359. [Google Scholar]; c Lu Z, Yoon Y. Angew. Chem., Int. Ed. 2012;51:10329. doi: 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okada Y, Nishimoto A, Akaba R, Chiba K. J. Org. Chem. 2011;76:3470. doi: 10.1021/jo200490q. [DOI] [PubMed] [Google Scholar]
- 20.The use of electron relays in single electron processes have been utilized before, albeit in a different context: Majima T, Pac C, Nakasone A, Sakurai H. J. Am. Chem. Soc. 1981;103:4499.Tamai T, Ichinose N, Tanaka T, Sasuga T, Hashida I, Mizuno K. J. Org. Chem. 1998;63:3204–3212.Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2011;40:102. doi: 10.1039/b913880n.
- 21.a Miranda M, Garcia H. Chem. Rev. 1994;94:1063. [Google Scholar]; b Martiny M, Steckhan E, Esch T. Chem. Ber. 1993;126:1671. [Google Scholar]
- 22.Farid S, Shealer SE. 2,4,6-Triphenylpyrylium tetrafluoroborate has been previously used to cyclodimerize indene and 1,1-dimethylindene, though yields were not reported. J. Chem. Soc., Chem. Commun. 1973:677. [Google Scholar]
- 23.Darwent JR, Kalyanasundaram K. J. Chem. Soc., Faraday Trans. 2. 1981;77:373. [Google Scholar]
- 24.Yamashita T, Yasuda M, Isami T, Tanabe K, Shima K. Tetrahedron. 1994;50:9275. [Google Scholar]
- 25.Naidu AB, Sekar G. Tetrahedron Lett. 2008;49:3147. [Google Scholar]
- 26.We cannot completely rule out a triplet quenching mechanism involving anthracene and *p-OMeTPT. However, given the regio-and diastereocontrol observed in the forward reaction, this mechanism seems unlikely.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.