

Graphical abstract
Highlights
► Current leishmaniasis treatments are not adequate. ► The great need for better drugs is being addressed by research centers and NGOs. ► Recent developments in drug assays have greatly aided efforts in the leishmaniasis drug discovery field. ► The drug development process and a target product profile are discussed.
Keywords: Leishmania, Leishmaniasis, Drug discovery, High throughput screening
Abstract
Leishmaniasis is one of the most neglected tropical disease in terms of drug discovery and development. Most antileishmanial drugs are highly toxic, present resistance issues or require hospitalization, being therefore not adequate to the field. Recently improvements have been achieved by combination therapy, reducing the time and cost of treatment. Nonetheless, new drugs are still urgently needed.
In this review, we describe the current visceral leishmaniasis (VL) treatments and their limitations. We also discuss the new strategies in the drug discovery field including the development and implementation of high-throughput screening (HTS) assays and the joint efforts of international teams to deliver clinical candidates.
1. Leishmaniasis
Leishmaniasis is a group of diseases caused by trypanosomatids from the genus Leishmania. Transmission occurs in 88 tropical and subtropical countries where the sandfly vector is present, meaning that approximately 350 million people are at risk of contracting the disease (WHO, 1990; Piscopo and Mallia Azzopardi, 2007). It is one of the most neglected tropical diseases, with a major impact among the poorest.
Leishmania parasites live a dual-form life cycle (digenetic life cycle), as either a promastigote flagellar or an amastigote form. The promastigotes are found in the insect vector and are injected into the mammalian host during the vector’s blood meal. Then, they are phagocytised by macrophages, dendritic cells and/or neutrophils attracted to the biting site in the skin. Once inside the phagosome, promastigotes differentiate into amastigotes, multiply by simple division until bursting the host cell. In the mammalian host, these protozoa are obligate intracellular parasites of macrophage-dendritic cell lineages. This complex life cycle includes several facets that might be exploited for drug design optimization and development (Hammarton et al., 2003). The parasites can have different host cells and organs tropism, infecting either superficial cells or visceral cells, which results in hepatosplenomegaly and bone marrow infiltration. Depending on the tropism, the disease can be characterized by at least four syndromes: cutaneous leishmaniasis (CL), muco-cutaneous leishmaniasis (MCL), visceral leishmaniasis (VL) – also known as kala-azar in the Indian sub continent or black fever, which is the most severe form of the disease being fatal if untreated, and post-kala-azar dermal leishmaniasis (PKDL). Factors determining the kind of clinical manifestation depend upon the infecting species and host factors, such as general health, genetic and immune constitution (Locksley et al., 1999; Tripathi et al., 2007).
In non-endemic areas of the world, VL is mostly an opportunistic infection with up to 70% of adult leishmaniasis cases being related to human immunodeficiency virus (HIV) infection. The greatest prevalence of reported co-infection in non-endemic areas is in the Mediterranean area. Atypical presentations of VL are reported in HIV patients, and HIV/AIDS clinical development is promoted by VL (WHO, 2007).
The numbers of leishmaniasis cases are increasing worldwide. Some reasons are the lack of vaccines, difficulties in controlling vectors and the increasing number of parasites resistance to chemotherapy.
In the following sections we briefly review the currently available chemotherapy and discuss the recent developments on the drug discovery field of VL.
2. Current therapies
Over the past decades, few alternative drugs or new formulations of old ones have become available but, as yet, none of them are ideal for treatment due to high toxicity, resistance issues, prohibitive prices, long treatment length or inadequate mode of administration not adapted to the field (see Table 1). In addition, many patients are unable to complete the whole treatment, increasing the risk of drug resistance development. Drug combinations have demonstrated positive results and may be a short-term solution to delay or prevent the emergence of resistance, increasing efficacy, or shortening the course of treatment (Alvar et al., 2006; Sundar et al., 2011).
Table 1.
Current VL treatments and their main characteristics.
| Drugs | Administration | Regimen | Efficacy (⁎) | Resistance | Toxicity | Price |
|---|---|---|---|---|---|---|
| Pentavalent antimonials | IV, IM and IL | 30 days 20 mg/kg/day |
35–95% (depending on area) | Common (>60% in Bihar, India) | +++ Cardiotoxicity, pancreatitis, nephrotoxicity, hepatotoxicity | $50–70 |
| Amphotericin B | IV | 30 days 1 mg/kg (15 mg/kg total dose) |
>90% | Laboratory strains | +++ Nephrotoxicity | ∼$100 |
| Liposomal amphotericin B | IV | 5–20 mg/kg total dose 4–10 doses over 10–20 days |
>97% | Not documented | +/− Rigors and chills during infusion | $280 |
| Miltefosine | PO | 28 days 1.5–2.5 mg/day |
94–97% | Laboratory strains | + Gastrointestinal, nephrotoxicity, hepatotoxicity, teratogenicity | ∼$70 |
| Paromomycin sulfate | IM | 21 days 15 mg/kg/day |
94% (India) 46–85% (Africa, depending on dose) |
Laboratory strains | + Nephrotoxicity, ototoxicity, hepatotoxicity | ∼$10 |
IV = intravenous administration; IM = intramuscular administration; IL = intralymphatic administration; PO = oral administration.
Definitive cure at 6 months.
For many years, pentavalent antimonials have been the recommended drug for VL and CL. Pentavalent antimonials – meglumine antimoniate (Glucantime, Sanofi-Aventis) and sodium stibogluconate (Pentostan, GlaxoSmithKline) – have variable efficacy against VL and CL, and require injectable administration, that can be intravenous (IV), intramuscular (IM) or intralymphatic (IL). Due to side effects such as high cardiotoxicity (Matoussi et al., 2007), pancreatitis (Gasser et al., 1994; Shahian and Alborzi, 2009) and nephrotoxicity (Zaghloul and Al-Jasser, 2004), patients should be hospitalized and monitored, as treatment may need to be suspended. There are currently efforts to reduce toxicity and improve delivery of antimonials (Frezard et al., 2009), with attempts including liposome-based formulations for VL treatment (Schettini et al., 2006) and cyclodextrin-based formulation for oral delivery (Demicheli et al., 2004).
Antimonials seem to have a broad mechanism of action. Data suggest pentavalent antimony (SbV) enters the host cells, crosses the phagolysosomal membrane and is converted into trivalent antimony (SbIII). Then, SbIII acts against amastigotes by compromising the cells thiol redox potential by inducing efflux of intracellular thiols and consequently inhibiting trypanothione reductase (TR) (Wyllie et al., 2004). SbV reduction can be non-enzymatic, under acidic conditions such as those found in the phagolysosome, by glutathione (GSH), glycylcysteine and trypanothione, or enzymatic by thiol-dependent reductase (TDR1) (Denton et al., 2004) and antimonite reductase (ACR2) (Ashutosh et al., 2007). ACR2 also increases the sensitivity of Leishmania to SbV (Zhou et al., 2004). SbV may also kill parasites by indirect mechanisms, such as increasing cytokine levels (Pathak and Yi, 2001). Antimonials also act at the DNA level, inducing DNA damage in vivo (Lima et al., 2010), and inhibiting DNA topoisomerase I (Chakraborty and Majumder, 1988).
Amphotericin B deoxycholate (Fungizone) is a systemic antifungal and a highly active antileishmanial. Due to the increasing resistance to antimonials, it is used as an alternative drug for VL. It is highly toxic, requiring careful and slow IV administration. Amphotericin B complexes with 24-substituted sterols from the biological membrane, such as ergosterol. These complexes open pores which alter the ion balance and lead to cell death (Roberts et al., 2003).
Lipid formulations of amphotericin B have been developed in order to improve its bioavailability and pharmacokinetic (PK) properties, considerably reducing side effects (Gangneux et al., 1996; Yardley and Croft, 1997; Torchilin, 2005). The larger lipid particles are rapidly assimilated by the mononuclear phagocyte system (hepatic macrophages), where Leishmania donovani parasites accumulate and VL develops. Another advantage is that smaller liposomes stay in the blood stream longer than the free drug (Yardley and Croft, 1997). The liposomal formulation (AmBisome) is an approved treatment for VL that besides the reduced toxicity has a better half-life and a high level of efficacy, with 90% cure rate. In experimental VL models, AmBisome has hepatic accumulation, reaches therapeutic levels faster than antimonials and has a longer half-life (Yardley and Croft, 1997; Berman et al., 1998; Sundar et al., 2004). The main limitations are its high cost, administration route and lack of stability at high temperature (cold chain is needed).
Miltefosine (Impavido), also known as hexadecylphosphocholine, was simultaneously discovered as an anticancer and antileishmanial drug (Croft et al., 1987; Scherf et al., 1987). It is the most recent antileishmanial drug on the market and the first effective oral treatment against VL, being recommended as first line drug for childhood VL (Bhattacharya et al., 2004). Although its toxicity is not very high, its teratogenicity is a problem (Sundar, 2007). The mechanism of action of miltefosine can be a direct action against the parasite by impairing the lipid metabolism (Croft et al., 1987) and causing parasite apoptosis (Paris et al., 2004). Miltefosine was also shown to act at the host cell level stimulating the production of inducible nitric oxide synthetase 2 (iNOS2) that catalyzes the generation of nitric oxide (NO) to kill the parasite within the macrophage (Wadhone et al., 2009).
A combination therapy of miltefosine with amphotericin B or paromomycin is very efficient and could be helpful to treat antimony-resistant VL infections in India (Seifert and Croft, 2006).
Paromomycin is an aminoglycoside antibiotic with antileishmanial activity. It is used in topical treatment for CL and as an IV drug for VL (Scott et al., 1992). It is off-patent and has been considered an orphan drug (Alvar et al., 2006). Paromomycin impairs the mitochondrial membrane potential, inhibits protein synthesis, and leads to respiratory dysfunction. It also alters membrane fluidity and lipid metabolism (Jhingran et al., 2009).
Pentamidine was used as second-line drug in antimony-resistant VL treatment. High toxicity combined with decreased efficacy in treatment of patients suggesting resistance (Sundar, 2001), drove to a complete abandonment of this drug to treat VL in India (Ouellette et al., 2004). However, this compound is still valuable for combined therapies (Croft and Coombs, 2003). The cellular target of pentamidine is unknown, but it seems to play a role in the mitochondria, as it accumulates in this organelle (Basselin et al., 2000).
Sitamaquine is the second oral drug in development for leishmaniasis treatment, after miltefosine, and was recently in clinical trials (Jha et al., 2005). It is an 8-amino-quinoline and preliminary clinical studies in Kenya and Brazil showed satisfactory efficacy against different species of Leishmania (Sherwood et al., 1994; Dietze et al., 2001). A recent study showed that it targets succinate dehydrogenase causing oxidative stress in L. donovani promastigotes (Carvalho et al., 2011).
As indicated, all the main drugs being individually used for the treatment of leishmaniasis have drawbacks (Table 1). Recent clinical studies have shown the potential benefit of antileishmanial drug combination for VL treatment in India, comparing short-course multidrug treatment with standard therapy (reviewed by Olliaro, 2010). The World Health Organization (WHO), following a meeting in 2010, encourages countries to adopt policies based on combination therapies when available (WHO, 2010). Nevertheless, the lack of adequacy for administration in the field, toxicity and resistance issues of the current therapies highlights the need of new drugs for VL patients.
3. VL drug discovery: new treatments on the way
During the past years several novel drugs or treatments for tropical diseases have been developed or carried out through drug repositioning. A sense of urgency to treat these diseases has led to pragmatic approaches with short and medium-term objectives, favoring the assessment of combination therapy and repurposing of known drugs. For example, amphotericin B was primarily developed as an antimycotic (Oura et al., 1955) and paromomycin is an antibiotic designed against cryptosporidiosis and amoebiasis (Courtney et al., 1959). This strategy reduces considerably the time and cost to have an approved treatment after efficacy confirmation, with most of the toxicological data and clinical tests already available (Nwaka and Hudson, 2006). However, this approach is limited to the number of approved drugs and the probability of finding a completely new mechanism of action against the parasite is very low. Given the needs for development of effective and better antileishmanials, this strategy is acceptable on the short-term, but for a mid or long-term solution, a different approach has to be considered to find optimal and specific antileishmanials.
Discovery of new drugs for VL treatment should therefore be seen as a mid or long-term objective. As such and as for any other drug discovery programs, it is coupled with risk, has to deal with attrition rate and needs adequate resources, both human and financial. This implies the need of optimized drug discovery processes, infrastructures, tools and teams, as well as the adequate funding to be possible to deliver candidates that will be the basis of the new treatment for VL in the near future.
The target product profile (TPP) is an essential feature of the drug discovery management tools and plays a central role in the discovery process, since it depicts the desired outcome of the research and development (R&D) program (Curry and Brown, 2003). With VL being the targeted disease, its specificities, such as patients living in poor and remote areas with no adequate health care, imply that the TPP needs to be designed through consultation with expert committees that include stakeholders such as physicians, local disease control programs, regulators, patient representatives, and local and global public health organizations (see Table 2 for a proposed TPP for VL drug development).
Table 2.
VL drug target product profile (TPP).
| Target label | VL and PKDL |
|---|---|
| Specificity | All species |
| Distribution | All areas |
| Target population | Immunocompetent and immunosuppressed |
| Clinical efficacy | >95% |
| Resistance | Active against resistant strains |
| Safety and tolerability | No adverse event requiring monitoring |
| Contraindications | None |
| Interactions | None – compatible for combination therapy |
| Formulation | Oral (PO)/intramuscular (IM) |
| Treatment regimen | 1/day for 10 days PO/3 shots over 10 days⁎ |
| Stability | 3 years in tropical areas |
| Cost | <$10/course |
This is for primary VL only. PKDL, HIV co-infection and relapse case treatments may require longer treatment durations.
The drug discovery process from the identification of active compounds (hit or lead) through lead optimization to the clinical candidate nomination is depicted in Fig. 1. This process is an iterative loop that proceeds through several “go/no-go” decisions from the identification of an active compound to a clinical candidate. Decision gates are usually predefined criteria described in the TPP that a compound should meet in order to be moved forward along the development chain.
Fig. 1.
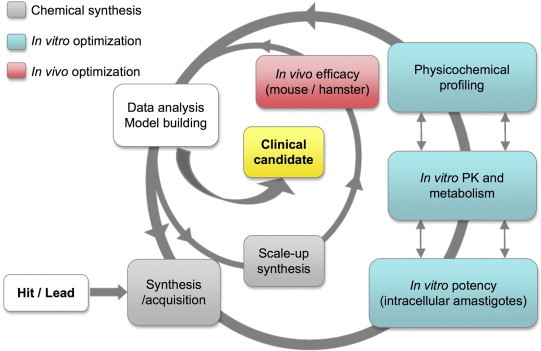
Hit-to-lead and lead optimization interactive model. Proposed workflow for an optimal process to generate a drug candidate against VL.
4. Compound screening: rapidly assessing hits
To identify starting points for drug discovery for VL nowadays two approaches can be undertaken, the molecular target approach (usually referred to as target-based) and the phenotypic approach (also called target-free), both having their pros and cons (Gilbert et al., 2011).
For the target-based discovery approach, the initial step would be the identification and validation of potential target(s). Biochemical studies or genome mining are some of the available tools to search for and define targets. Specifically in the case of Leishmania parasites, the genome sequences of different species are publicly available (Ivens et al., 2005; Peacock et al., 2007) and a number of potential targets have been speculated. Some of these targets were identified and proved to be essential for parasite survival (Croft and Coombs, 2003), including the parasite dihydrofolate reductase (DHFR) (Cunningham and Beverley, 2001; Gilbert, 2002), cyclin-dependent kinases (Doerig et al., 2002) and the well characterized sterol biosynthesis pathway. The latter is a classic anti-fungal therapeutic target and has been pursued by several laboratories for antileishmanial and antitrypanosomal chemotherapy, as both Trypanosoma and Leishmania require specific sterols for growth and cell viability (reviewed by Urbina, 1997; Lepesheva and Waterman, 2011). Although significant progress on the identification and in the validation of these targets have been made, so far no drug focusing on these targets is being developed for VL.
Recently, efforts were made to select potential parasite targets, starting with the TDR targets database (https://www.tdrtargets.org). This database was created based on genetic, biochemical and pharmacological data related to tropical disease pathogens in addition to computationally predicted druggability (Aguero et al., 2008). In silico methods applied for the prioritization of these targets from the TDR database (using criteria such as sequence-derived information, functional data on expression, essentiality, metabolic pathways and assay development feasibility) ranked cysteine peptidase C, trypanothione reductase and DHFR as top-three targets for Leishmania (Crowther et al., 2010).
Another target that has been pursued for antileishmanial drug discovery is the cyclin-dependent kinase CRK3/CYC6 cyclin complex. Inhibitors of CRK3-CYC6 were discovered on a target-based high-throughput screening (HTS) recently performed, showing in vitro activity against promastigotes and intramacrophagic amastigotes (Walker et al., 2011). Another example of effort targeting Leishmania kinases is the Leishdrug Consortium – a Framework Program (FP7, 2007–2013) – created in 2008 for the development of novel antiparasitic strategies, involving different institutes and universities from Asia, Europe, Middle-East, South America and Africa (Dujardin et al., 2010).
In spite of these efforts, the target-based screening approach for antileishmanial drug discovery stays in the early discovery phase and has not moved forward to development. Other factors that preclude target-based drug discovery for VL are the low number of targets well characterized and validated both genetically and chemically. As for many other fields of discovery, the criteria and requirements for target validation in VL are loosely defined. Often it is assumed that protein essentiality per se equals a valid drug target; however it is crucial to provide evidence of the in vivo role of the putative target, or at least an essentiality in the intracellular amastigote form. Due to the particularities of Leishmania parasite – hiding in the parasitophorous vacuole in the macrophage where the pH is lower than in the surrounding environment, with several membranes to cross through a pH gradient, and the intricate aspects of the host immunity that influence the development of leishmaniasis – it is not difficult to imagine why an effect on a target does not translate to an effect on the parasite. Indeed target-based approaches do not seem to be the most successful strategy for drug discovery for infectious diseases (Payne et al., 2007).
5. The target-free screening approach
Instead of focusing on a specific enzyme, or other individual molecule, targeting a pathway might be a promising solution (Hellerstein, 2008). The robustness of critical cellular metabolism relies on a complex and highly linked adjustable network with redundant or alternate pathways to maintain a usual flow of molecules and materials in the cell (Fischer and Sauer, 2005). Therefore, the inhibition of an individual molecule would seldom generate the expected outcome, a premise in the target-based drug discovery approach. On the other hand, if a pathway itself can be defined as the target, it would be a way to select chemical compounds that are able to interfere in the pathway, no matter the individual pathway components.
In a more holistic approach, a target-free screening would not define any specific molecule or even pathways as a target, but simply select compounds that are able to eliminate the parasite. Although viability-based approaches are not applicable to every case in drug discovery, it is a simple albeit efficient solution for several infectious diseases including VL. The utmost advantage of this approach is that in theory a compound can become a drug if it is potent against the parasite in the disease context without affecting the host, independently of the target. It has been historically successful for the discovery of most of the current protozoan chemotherapy, and also for other fields of discovery and development: from 1999 and 2008, for the 75 first-in-class drugs with new molecular mechanisms of action approved by the Food and Drug Administration (FDA), 28 were discovered in phenotypic screenings, 17 were from target-based approaches and the other 30 based on other methodologies (Swinney and Anthony, 2011). A phenotypic screening is typically a target-free high-content screening (HCS), a versatile technology collecting information on several parameters during the screening, enabling thus the identification and categorization of complex cellular phenomena (phenotypes). This information is commonly interpreted by automated software that classifies and quantifies the cellular components and characteristics (Haney et al., 2006; Korn and Krausz, 2007; WHO, 2007; Bickle, 2010). Once a HCS is miniaturized and adapted to microtiter plate format, the assay can be automated and become compatible in high-throughput scales.
Besides some activities in academic laboratories, target-free phenotypic screening efforts to identify compounds active against Leishmania parasite started under the auspices of the Special Program for Research and Training in Tropical Diseases (TDR/WHO). TDR/WHO accessed compound libraries and tested their activity in vitro in parasitology laboratories, including the Swiss Tropical and Public Health Institute (STPH) in Basel, the Laboratory of Microbiology, Parasitology and Hygiene (LMPH) in Antwerp and the London School of Hygiene & Tropical Medicine (LSHTM) in London. However, these assays were not performed in an automated fashion and the low-throughput enabled only the screening of a small number of compounds, reducing thereby the odds of identifying promising series.
Historically the lack of high throughput capacity of phenotypic screening assays for Leishmania has prevented the access to a critical mass of quality hits needed for further development considering the high attrition rate occurring during the drug discovery process. To address this, numerous attempts to increase the throughput were made, using either different stages of the parasite, transgenic parasites, different readouts, different formats, different host cells, but all had their drawbacks (see Table 3) (Singh and Dube, 2004; Ashutosh et al., 2005; Buckner and Wilson, 2005; Lang et al., 2005; Mandal et al., 2009; Sharlow et al., 2009; Siqueira-Neto et al., 2010; Osorio et al., 2011). In vitro susceptibilities of L. donovani promastigote and amastigote stages to antileishmanial reference drugs were also addressed by the scientific community, indicating the practical relevance of stage-specific differences (Vermeersch et al., 2009; De Muylder et al., 2011). In a recent study, we found that only 4% of the hits found in a promastigote screening translated into anti-amastigote hits, while about 50% of hits from an intracellular amastigotes screening were confirmed to have activity on promastigotes (J Siqueira-Neto and L Freitas-Junior, unpublished data). Altogether these studies stress the importance of utilizing intracellular amastigotes as the stage of study for drug discovery for VL.
Table 3.
Antileishmanial screening formats.
| Form of screening | Advantages | Disadvantages |
|---|---|---|
| Transgenic Leishmania | Facilitates screening development and permits fast and sensitive readouts | Requires genetic manipulation of the parasite that may interfere with biological functions |
| HTS with promastigotes | Easy parasite handling | Insect stage of the parasite, poor hit confirmation rate in amastigote assays |
| HTS with axenic amastigotes | Easy parasite handling | Questionable similarity with intracellular amastigotes |
| HTS with intracellular amastigotes infecting human macrophage | Most relevant form of the parasite representing the human disease model | Technically challenging, laborious automation, usually expensive |
| Ex vivo splenic explants | Simulates pathophysiological infection environment | Limited throughput and the use of hamster cells, not human |
Of all the recent efforts to identify new antileishmanials, the most massive was the HTS of 700,000 compounds by Genomic Institute of the Novartis Research Foundation (GNF). The assay used axenic amastigotes in a 1536 well plates format (Bustamante et al., 2011). Although it is recognized that this form of the parasite is not fully representative of the intracellular amastigote (Rochette et al., 2009), some starting points for drug discovery were identified. The lack of activity translation between axenic amastigote and intracellular amastigotes has been reported (De Muylder et al., 2011).
The development of a robust HCS/HTS method using a clinically relevant form of the parasite – intracellular amastigotes infecting human macrophage – was firstly reported in 2010 as a secondary assay to confirm activity of compounds selected as actives against the promastigote stage (Siqueira-Neto et al., 2010). In 2011, it was adapted to a 96 well plate format to screen a few hundred compounds (De Muylder et al., 2011).
The Institut Pasteur Korea (IPK), together with the Drugs for Neglected Diseases initiative (DNDi) developed a HTS/HCS visual screen in a 384 well plate format in order to find promising new chemical compound classes active against Leishmania. The human macrophage cell line THP-1 (human acute monocytic leukemia) was used as the host cell and infected with L. donovani metacyclic promastigotes. The readout of the assay was image-based, and a customized software was developed for image analysis, allowing access to information such as infection ratio, number of cells per well, number of parasites per infected cell, and cell cytoplasm area among others. This assay was used for screening over 300,000 compounds and more than 350 hits were identified with EC50 lower than 10 μM against intracellular parasites, and no cytotoxicity at 30 μM based on host cell counting (J. Siqueira-Neto and L. Freitas-Junior, unpublished data). This is a key milestone in the field of leishmaniasis drug discovery and is expected to change dramatically the hit generation dynamic for VL.
Another promising approach for the discovery of new antileishmanial compounds was developed by Peter Melby and collaborators based on ex vivo explant culture system from hamsters infected with luciferase-transfected L. donovani to screen chemical compounds for antileishmanial activity (Osorio et al., 2011). The advantage of this system is that it simulates the complex pathophysiological environment of the infected spleen in a well plate, and the compounds are tested in this condition with a diverse host cell population. While amenable to HTS conditions, this assay requires infected hamster’s explants, implying that it is more time and cost consuming than the assays using THP-1 infected with L. donovani, thus better suited in secondary screenings for hit prioritization and holds promises for lead optimization stages, when tests of several series of analogues are rapidly needed to advance potential candidates through the discovery process (see below).
Because of the variety of assays to assess the activity and potency of compounds against the Leishmania parasite in vitro it is important to have reference assays, standardized and validated methods to be able to compare data and select good hits and/or series among the wealth of described active compounds and screens datasets. After the selection of hit compounds or series of compounds coming from any screening method, (i) structure–activity relationship studies are performed to prioritize chemical classes, (ii) derivatives are synthesized, and (iii) the active scaffolds are identified (lead).
6. Hit-to-lead & lead optimization
Major gaps in the drug discovery process for novel antileishmanial, once active compounds against Leishmania have been identified, are twofold. On the one hand, lack of structures adequate for assessing the potential and identifying possible liabilities of good hit compounds (hit-to-lead process with synthesis of few analogues in order to profile hits and assess which can be considered leads) and the further optimization of leads (lead optimization). On the other hand the lack of systematic studies undertaken to have a proper understanding of the pharmacokinetic/pharmacodynamic (PK/PD) relationships for the disease: in short, how can one relate the exposure of a compound in the body to its efficacy and safety.
There are rather numerous articles depicting the activity of a compound against the Leishmania parasite in vitro – whatever the model – or a Leishmania target, but very few have been further assessed for in vivo models of the disease. Moreover, the follow-up of compounds active in vitro and their further optimization through the synthesis of analogues remains very scarce. Recognizing this gap, DNDi has put together with partners a lead optimization structure that includes not only synthetic chemistry capacity but also PK/PD and efficacy assessment. This integrated process allows profiling active compounds according to specific criteria, having always in mind the TPP (Chatelain and Ioset, 2011). Once a lead is introduced into a lead optimization program, it enters a critical path (Woosley and Cossman, 2007), which promises development through to the patient access, unless the compound series fails because of liabilities that cannot be optimized. A proposed criteria for a lead optimization endpoint and selection of a clinical candidate for VL treatment is presented in the Table 4. To guarantee rapid turnaround of data and allow iterative cycles of optimization until getting to this endpoint, sufficient resources are allocated to the lead optimization program; in general a team consists of 5–6 chemists, 2–3 pharmacologists and dedicated screening facilities to assess in vitro potency and in vivo efficacy with guaranteed infrastructures to support medicinal chemistry, in vitro and in vivo distribution-metabolism-pharmacokinetic (DMPK) and toxicology.
Table 4.
Proposed criteria for clinical candidate selection of a VL drug candidate.
| Efficacy | Cell-baseda activity IC50 ⩽ 1 μM |
| Selectivity ⩽ 50 fold (mammalian isoforms) | |
| Hamster (chronic; liver and spleen): cure after 4 days (PO) | |
| Mouse (acute; liver): cure after 4 days (PO) | |
| Chemistry properties and profile | Solubility: >0.1 mg/ml (at pH 7.4) |
| Chirality/bioconversion characterized | |
| COGs amenable to cost effective scale up: <6 steps for synthesis preferred, and <10$/treatment | |
| >2 kg available for GLP studies | |
| DMPK | Metabolic stable in S9 liver fraction and liver microsomes of mouse, rat, hamster, dog and human |
| Major metabolites identified and characterized qualitatively | |
| No induction and no irreversible inhibition of Cytochrome P450 (5 major isoforms) | |
| Protein binding <85% | |
| Plasma, whole blood and GI fluid stability >80% after 4 h. | |
| Oral BA in rats and dogs >30% (in aqueous solutions or clinical accepted formulations) | |
| Elimination half life (oral) rat >120 min | |
| Elimination half life (oral) dog >180 min | |
| Toxicity | Evaluation on selected panel of receptors, enzymes and ion channels. |
| hERG assay IC50 < 10 μM and ratio of plasma Cmax/hERG IC20 | |
| 7 days exploratory toxicology/dose ranging (rats) and identify any organ toxicities: GLP studies | |
| Pharm.chemistry/development | API formulation with preliminary stability, suitable for preclinical tests |
| Human pharmacology/safety | Determined starting dose for FIM and projected clinical doses |
| Preparation of clinical development plan | |
| Intellectual property status | Freedom to operate |
PO = oral administration; COG = cost of goods; GLP = good laboratory practices; BA = bioavailable; API = active pharmaceutical ingredient; FIM = first in man; QC = quality control.
Cell-based refers to intracellular amastigotes infecting macrophages.
Indeed, going through rounds of optimization (Fig. 1), looking at what the body does to the drug (PK) through classical in vitro and in vivo drug metabolism and pharmacokinetics (DMPK) assays and what the drug does to the body (pharmacodynamics – PD) through various in vivo animal models, one ends in an optimized lead that has the desired properties of a potential future drug (see Table 2 for predefined properties of the ideal optimized lead for VL) and is ready to enter through the classical safety assessment in animals.
To test the in vivo efficacy and potency of the leads and optimized leads, there are different animal models, including mouse, rat, hamster, dog and non-human primate models. None of these models, however, completely reproduces the pathology and immunology of the human disease. The in vivo efficacy models for VL have been shortly reviewed by Gupta and Nishi (2011). Although dog (Chapman et al., 1979; Keenan et al., 1984) and non-human primate (Chapman and Hanson, 1981; Chapman et al., 1981; Dube et al., 1999) models of the disease have been described, their use remains low and is not really compatible with early stage of drug discovery as the amount of compound needed for testing in these models is high.
Rodent models remain, therefore, the standard for drug discovery. Mouse models of leishmaniasis are being extensively used to get preliminary information of the potential of compounds as they are fast, reproducible and need relative low amount of compound. The Balb/c mouse is commonly used for the so-called acute model and the endpoint of the assay is the parasite load in the liver of the animals (amastigotes per liver cell nuclei) following 5 days of treatment (Croft et al., 2006).
According to the classical screening cascade for VL, compounds for which 80% or more reduction of the liver parasite load in the mouse model is obtained are then put through more stringent and relevant chronic hamster model (Melby et al., 2001; Nieto et al., 2011). The most common strain used is the outbred Syrian golden hamster (Mesocricetus auratus), defined as a model for chronic non-cure VL, more similar to the pathology in human with a more synchronous infection in the liver and spleen (Farrell, 1976; Gifawesen and Farrell, 1989). The clinic-pathological features of the hamster model of VL mimic active human disease. Systemic infection of the hamster with L. donovani results in a relentless increase in visceral parasite burden, progressive cachexia, hepatosplenomegaly, pancytopenia, hypergammaglobulinemia and ultimately death (Farrell, 1976). Following 5 days treatment and at different time points up to 50–60 days after end of treatment parasite burden in spleen, liver and bone marrow is determined. In this model, AmBisome at 5 mg/kg IV is 100% effective against liver stages but clearance from the spleen and bone marrow was not achieved (Maes et al., 2004). This model gives us an idea of the target to achieve to consider the potential of a compound to move forward in development.
In spite of extensive physiopathological characterization, the two most used models, the mouse and the hamster, have not been yet characterized as good predictors of compound clinical relevancy for VL, especially considering critical aspects as PK/PD and drug efficacy in these models. This issue arises, in great part, due to the neglected nature of VL and the lack thereof of drug discovery and development – in other words, there is no correlation between clinical and pre-clinical data since there are no pre-clinical candidates being advanced into clinical phases. This situation is unfortunately not exclusive of VL; indeed if one look at the PK/PD issues in the field of parasitic infections, with the exception of malaria, very little is known or undertaken for other diseases (Edwards and Krishna, 2004). In analogy to antimicrobial research (Drusano, 2004), PK/PD data are critical to better understand the pharmacokinetics of a compound and its effect on the parasite.
For a late stage drug development, the dog model infected with L. infantum or L. chagasi, should be considered an important model. First, because in some endemic areas, the dogs are natural reservoir of the parasite and second, because the infection process and development is similar to the one occurring in human (Rioux et al., 1969).
From early discovery by HTS to animal models for pre-clinical development, several tools are already available for the discovery and development of a new and more efficient treatment against VL. The expectations are that promising drug candidates will be in clinical phase by 2013.
7. Conclusion
Current therapy for leishmaniasis is far from ideal. In recent years substantial progress has been made in the in search for novel chemotherapeutics – from the initiative of individual academic labs to consortia and public–private partnerships. Cutting-edge technologies have been combined with optimized assays that enabled the screening of a total of near half million compounds for VL. This and other notorious efforts have generated several potential starting points for drug development. The downstream processes of drug discovery is being established with lead optimization consortia with the critical mass necessary to a successful lead optimization program and the potential delivery of clinical candidates in the near future. The number of players has also increased dramatically answering to various incentives.
However, there is still a long way to go. Development of new technologies together with new models to perform lead optimization, as well as information sharing and further partnerships should pave the way to the identification and development of new candidates during the following couple of years. It is important to highlight that the late discovery process, such as lead generation and optimization steps, and the drug development process, with the pre-clinical animal models for drug efficacy and PK/PD, are still in its infancy for leishmaniasis, given that more systematic approaches to develop new chemical entities for this important but neglected disease have started only relatively recently. Therefore still much about VL chemotherapy is going to be learnt on the way of development of new drugs to treat leishmaniasis, and the real measure of success will be the delivery of these future drugs to the patients in need.
Acknowledgements
The authors would like to thank Dr. Manica Balasegaram and Ivan Scandale for providing material used in the preparation of this manuscript. Ideas in this article represent the views of the authors but not necessarily those of their respective institutions.
References
- Aguero F., Al-Lazikani B., Aslett M., Berriman M., Buckner F.S., Campbell R.K., Carmona S., Carruthers I.M., Chan A.W., Chen F., Crowther G.J., Doyle M.A., Hertz-Fowler C., Hopkins A.L., McAllister G., Nwaka S., Overington J.P., Pain A., Paolini G.V., Pieper U., Ralph S.A., Riechers A., Roos D.S., Sali A., Shanmugam D., Suzuki T., Van Voorhis W.C., Verlinde C.L. Genomic-scale prioritization of drug targets: the TDR targets database. Nat. Rev. Drug Discov. 2008;7:900–907. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvar J., Croft S., Olliaro P. Chemotherapy in the treatment and control of leishmaniasis. Adv. Parasitol. 2006;61:224–261. doi: 10.1016/S0065-308X(05)61006-8. [DOI] [PubMed] [Google Scholar]
- Ashutosh, Gupta S., Ramesh, Sundar S., Goyal N. Use of Leishmania donovani field isolates expressing the luciferase reporter gene in in vitro drug screening. Antimicrob. Agents Chemother. 2005;49:3776–3783. doi: 10.1128/AAC.49.9.3776-3783.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashutosh, Sundar S., Goyal N. Molecular mechanisms of antimony resistance in Leishmania. J. Med. Microbiol. 2007;56:143–153. doi: 10.1099/jmm.0.46841-0. [DOI] [PubMed] [Google Scholar]
- Basselin M., Coombs G.H., Barrett M.P. Putrescine and spermidine transport in Leishmania. Mol. Biochem. Parasitol. 2000;109:37–46. doi: 10.1016/s0166-6851(00)00234-6. [DOI] [PubMed] [Google Scholar]
- Berman J.D., Badaro R., Thakur C.P., Wasunna K.M., Behbehani K., Davidson R., Kuzoe F., Pang L., Weerasuriya K., Bryceson A.D. Efficacy and safety of liposomal amphotericin B (AmBisome) for visceral leishmaniasis in endemic developing countries. Bull. World Health Organ. 1998;76:25–32. [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S.K., Jha T.K., Sundar S., Thakur C.P., Engel J., Sindermann H., Junge K., Karbwang J., Bryceson A.D.M., Berman J.D. Efficacy and tolerability of miltefosine for childhood visceral leishmaniasis in India. Clin. Infect. Dis. 2004;38:217–221. doi: 10.1086/380638. [DOI] [PubMed] [Google Scholar]
- Bickle M. The beautiful cell: high-content screening in drug discovery. Anal. Bioanal. Chem. 2010;398:219–226. doi: 10.1007/s00216-010-3788-3. [DOI] [PubMed] [Google Scholar]
- Buckner F.S., Wilson A.J. Colorimetric assay for screening compounds against Leishmania amastigotes grown in macrophages. Am. J. Trop. Med. Hyg. 2005;72:600–605. [PubMed] [Google Scholar]
- Bustamante J.M., Park H.J., Tarleton R.R. Report of the 2nd chagas drug discovery consortium meeting, held on 3 Novermber 2010. Expert Opin. Drug Discov. 2011;6:965–973. doi: 10.1517/17460441.2011.602063. [DOI] [PubMed] [Google Scholar]
- Carvalho L., Luque-Ortega J.R., López-Martín C., Castanys S., Rivas L., Gamarro F. The 8-aminoquinoline analogue sitamaquine causes oxidative stress in Leishmania donovani promastigotes by targeting succinate dehydrogenase. Antimicrob. Agents Chemother. 2011;55:4202–4210. doi: 10.1128/AAC.00520-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A.K., Majumder H.K. Mode of action of pentavalent antimonials: specific inhibition of type I DNA topoisomerase of Leishmania donovani. Biochem. Biophys. Res. Commun. 1988;152:605–611. doi: 10.1016/s0006-291x(88)80081-0. [DOI] [PubMed] [Google Scholar]
- Chapman W.L., Jr., Hanson W.L., Waits V.B., Kinnamon K.E. Antileishmanial activity of selected compounds in dogs experimentally infected with Leishmania donovani. Rev. Inst. Med. Trop. Sao Paulo. 1979;21:189–193. [PubMed] [Google Scholar]
- Chapman W.L., Jr., Hanson W.L. Visceral leishmaniasis in the squirrel monkey (Saimiri sciurea) J. Parasitol. 1981;67:740–741. [PubMed] [Google Scholar]
- Chapman W.L., Jr., Hanson W.L., Hendricks L.D. Leishmania donovani in the owl monkey (aotus trivirgatus) Trans. R. Soc. Trop. Med. Hyg. 1981;75:124–125. doi: 10.1016/0035-9203(81)90032-8. [DOI] [PubMed] [Google Scholar]
- Chatelain E., Ioset J.R. Drug discovery and development for neglected diseases: the DNDi model. Drug Des. Dev. Ther. 2011;5:175–181. doi: 10.2147/DDDT.S16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney K.O., Thompson P.E., Hodgkinson R., Fitzsimmons J.R. Paromomycin as a therapeutic substance for intestinal amebiasis and bacterial enteritis. Antibiot. Annu. 1959;7:304–309. [PubMed] [Google Scholar]
- Croft S.L., Neal R.A., Pendergast W., Chan J.H. The activity of alkyl phosphorylcholines and related derivatives against Leishmania donovani. Biochem. Pharmacol. 1987;36:2633–2636. doi: 10.1016/0006-2952(87)90543-0. [DOI] [PubMed] [Google Scholar]
- Croft S.L., Coombs G.H. Leishmaniasis – current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003;19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Croft S.L., Seifert K., Yardley V. Current scenario of drug development for leishmaniasis. Indian J. Med. Res. 2006;123:399–410. [PubMed] [Google Scholar]
- Crowther G.J., Shanmugam D., Carmona S.J., Doyle M.A., Hertz-Fowler C., Berriman M., Nwaka S., Ralph S.A., Roos D.S., Van Voorhis W.C., Aguero F. Identification of attractive drug targets in neglected-disease pathogens using an in silico approach. PLoS Negl. Trop. Dis. 2010;4:e804. doi: 10.1371/journal.pntd.0000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham M.L., Beverley S.M. Pteridine salvage throughout the Leishmania infectious cycle: implications for antifolate chemotherapy. Mol. Biochem. Parasitol. 2001;113:199–213. doi: 10.1016/s0166-6851(01)00213-4. [DOI] [PubMed] [Google Scholar]
- Curry, A., Brown, R., 2003. The target product profile as a planning tool in drug discovery research. PharmaTech, 67–71.
- De Muylder G., Ang K.K.H., Chen S., Arkin M.R., Engel J.C., McKerrow J.H. A screen against Leishmania intracellular amastigotes: comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Negl. Trop. Dis. 2011;5:e1253. doi: 10.1371/journal.pntd.0001253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demicheli C., Ochoa R., da Silva J.B., Falcao C.A., Rossi-Bergmann B., de Melo A.L., Sinisterra R.D., Frezard F. Oral delivery of meglumine antimoniate-beta-cyclodextrin complex for treatment of leishmaniasis. Antimicrob. Agents Chemother. 2004;48:100–103. doi: 10.1128/AAC.48.1.100-103.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton H., McGregor J.C., Coombs G.H. Reduction of anti-leishmanial pentavalent antimonial drugs by a parasite-specific thiol-dependent reductase, TDR1. Biochem. J. 2004;381:405–412. doi: 10.1042/BJ20040283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietze R., Carvalho S.F., Valli L.C., Berman J., Brewer T., Milhous W., Sanchez J., Schuster B., Grogl M. Phase 2 trial of WR6026, an orally administered 8-aminoquinoline, in the treatment of visceral leishmaniasis caused by Leishmania chagasi. Am. J. Trop. Med. Hyg. 2001;65:685–689. doi: 10.4269/ajtmh.2001.65.685. [DOI] [PubMed] [Google Scholar]
- Doerig C., Meijer L., Mottram J.C. Protein kinases as drug targets in parasitic protozoa. Trends Parasitol. 2002;18:366–371. doi: 10.1016/s1471-4922(02)02321-8. [DOI] [PubMed] [Google Scholar]
- Drusano G.L. Antimicrobial pharmacodynamics: critical interactions of “bug and drug”. Nat. Rev. Microbiol. 2004;2:289–300. doi: 10.1038/nrmicro862. [DOI] [PubMed] [Google Scholar]
- Dube A., Srivastava J.K., Sharma P., Chaturvedi A., Katiyar J.C., Naik S. Leishmania donovani: cellular and humoral immune responses in Indian langur monkeys, Presbytis entellus. Acta Trop. 1999;73:37–48. doi: 10.1016/s0001-706x(99)00007-8. [DOI] [PubMed] [Google Scholar]
- Dujardin J.C., Gonzalez-Pacanowska D., Croft S.L., Olesen O.F., Spath G.F. Collaborative actions in anti-trypanosomatid chemotherapy with partners from disease endemic areas. Trends Parasitol. 2010;26:395–403. doi: 10.1016/j.pt.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Edwards G., Krishna S. Pharmacokinetic and pharmacodynamic issues in the treatment of parasitic infections. Eur. J. Clin. Microbiol. Infect. Dis. 2004;23:233–242. doi: 10.1007/s10096-004-1113-9. [DOI] [PubMed] [Google Scholar]
- Farrell J.P. Leishmania donovani: acquired resistance to visceral leishmaniasis in the golden hamster. Exp. Parasitol. 1976;40:89–94. doi: 10.1016/0014-4894(76)90069-2. [DOI] [PubMed] [Google Scholar]
- Fischer E., Sauer U. Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism. Nat. Genet. 2005;37:636–640. doi: 10.1038/ng1555. [DOI] [PubMed] [Google Scholar]
- Frezard F., Demicheli C., Ribeiro R.R. Pentavalent antimonials: new perspectives for old drugs. Molecules. 2009;14:2317–2336. doi: 10.3390/molecules14072317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangneux J.P., Sulahian A., Garin Y.J., Derouin F. Lipid formulations of amphotericin b in the treatment of experimental visceral leishmaniasis due to Leishmania infantum. Trans. R. Soc. Trop. Med. Hyg. 1996;90:574–577. doi: 10.1016/s0035-9203(96)90330-2. [DOI] [PubMed] [Google Scholar]
- Gasser R.A., Jr., Magill A.J., Oster C.N., Franke E.D., Grogl M., Berman J.D. Pancreatitis induced by pentavalent antimonial agents during treatment of leishmaniasis. Clin. Infect. Dis. 1994;18:83–90. doi: 10.1093/clinids/18.1.83. [DOI] [PubMed] [Google Scholar]
- Gifawesen C., Farrell J.P. Comparison of T-cell responses in self-limiting versus progressive visceral Leishmania donovani infections in golden hamsters. Infect. Immun. 1989;57:3091–3096. doi: 10.1128/iai.57.10.3091-3096.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert I.H. Inhibitors of dihydrofolate reductase in Leishmania and trypanosomes. Biochim. Biophys. Acta. 2002;1587:249–257. doi: 10.1016/s0925-4439(02)00088-1. [DOI] [PubMed] [Google Scholar]
- Gilbert I.H., Leroy D., Frearson J.A. Finding new hits in neglected disease projects: target or phenotypic based screening? Curr. Top Med. Chem. 2011;11:1284–1291. doi: 10.2174/156802611795429176. [DOI] [PubMed] [Google Scholar]
- Gupta S., Nishi Visceral leishmaniasis: experimental models for drug discovery. Indian J. Med. Res. 2011;133:27–39. [PMC free article] [PubMed] [Google Scholar]
- Hammarton T.C., Mottram J.C., Doerig C. The cell cycle of parasitic protozoa: potential for chemotherapeutic exploitation. Prog. Cell Cycle Res. 2003;5:91–101. [PubMed] [Google Scholar]
- Haney S.A., LaPan P., Pan J., Zhang J. High-content screening moves to the front of the line. Drug Discov. Today. 2006;11:889–894. doi: 10.1016/j.drudis.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Hellerstein M.K. A critique of the molecular target-based drug discovery paradigm based on principles of metabolic control: advantages of pathway-based discovery. Metab. Eng. 2008;10:1–9. doi: 10.1016/j.ymben.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Ivens A.C., Peacock C.S., Worthey E.A., Murphy L., Aggarwal G., Berriman M., Sisk E., Rajandream M.A., Adlem E., Aert R., Anupama A., Apostolou Z., Attipoe P., Bason N., Bauser C., Beck A., Beverley S.M., Bianchettin G., Borzym K., Bothe G., Bruschi C.V., Collins M., Cadag E., Ciarloni L., Clayton C., Coulson R.M., Cronin A., Cruz A.K., Davies R.M., De Gaudenzi J., Dobson D.E., Duesterhoeft A., Fazelina G., Fosker N., Frasch A.C., Fraser A., Fuchs M., Gabel C., Goble A., Goffeau A., Harris D., Hertz-Fowler C., Hilbert H., Horn D., Huang Y., Klages S., Knights A., Kube M., Larke N., Litvin L., Lord A., Louie T., Marra M., Masuy D., Matthews K., Michaeli S., Mottram J.C., Muller-Auer S., Munden H., Nelson S., Norbertczak H., Oliver K., O’Neil S., Pentony M., Pohl T.M., Price C., Purnelle B., Quail M.A., Rabbinowitsch E., Reinhardt R., Rieger M., Rinta J., Robben J., Robertson L., Ruiz J.C., Rutter S., Saunders D., Schafer M., Schein J., Schwartz D.C., Seeger K., Seyler A., Sharp S., Shin H., Sivam D., Squares R., Squares S., Tosato V., Vogt C., Volckaert G., Wambutt R., Warren T., Wedler H., Woodward J., Zhou S., Zimmermann W., Smith D.F., Blackwell J.M., Stuart K.D., Barrell B., Myler P.J. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha T.K., Sundar S., Thakur C.P., Felton J.M., Sabin A.J., Horton J. A phase II dose-ranging study of sitamaquine for the treatment of visceral leishmaniasis in India. Am. J. Trop. Med. Hyg. 2005;73:1005–1011. [PubMed] [Google Scholar]
- Jhingran A., Chawla B., Saxena S., Barrett M.P., Madhubala R. Paromomycin: uptake and resistance in Leishmania donovani. Mol. Biochem. Parasitol. 2009;164:111–117. doi: 10.1016/j.molbiopara.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan C.M., Hendricks L.D., Lightner L., Webster H.K., Johnson A.J. Visceral leishmaniasis in the German shepherd dog. I. Infection, clinical disease, and clinical pathology. Vet. Pathol. 1984;21:74–79. doi: 10.1177/030098588402100113. [DOI] [PubMed] [Google Scholar]
- Korn K., Krausz E. Cell-based high-content screening of small-molecule libraries. Curr. Opin. Chem. Biol. 2007;11:503–510. doi: 10.1016/j.cbpa.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Lang T., Goyard S., Lebastard M., Milon G. Bioluminescent Leishmania expressing luciferase for rapid and high throughput screening of drugs acting on amastigote-harbouring macrophages and for quantitative real-time monitoring of parasitism features in living mice. Cell Microbiol. 2005;7:383–392. doi: 10.1111/j.1462-5822.2004.00468.x. [DOI] [PubMed] [Google Scholar]
- Lepesheva G.I., Waterman M.R. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr. Top Med. Chem. 2011;11:2060–2071. doi: 10.2174/156802611796575902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima M.I., Arruda V.O., Alves E.V., de Azevedo A.P., Monteiro S.G., Pereira S.R. Genotoxic effects of the antileishmanial drug Glucantime. Arch. Toxicol. 2010;84:227–232. doi: 10.1007/s00204-009-0485-0. [DOI] [PubMed] [Google Scholar]
- Locksley R.M., Pingel S., Lacy D., Wakil A.E., Bix M., Fowell D.J. Susceptibility to infectious diseases: Leishmania as a paradigm. J. Infect. Dis. 1999;179(Suppl. 2):S305–308. doi: 10.1086/513843. [DOI] [PubMed] [Google Scholar]
- Maes L., Germonprez N., Quirijnen L., Van Puyvelde L., Cos P., Vanden Berghe D. Comparative activities of the triterpene saponin maesabalide III and liposomal amphotericin B (AmBisome) against Leishmania donovani in hamsters. Antimicrob. Agents Chemother. 2004;48:2056–2060. doi: 10.1128/AAC.48.6.2056-2060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S., Maharjan M., Ganguly S., Chatterjee M., Singh S., Buckner F.S., Madhubala R. High-throughput screening of amastigotes of Leishmania donovani clinical isolates against drugs using a colorimetric beta-lactamase assay. Indian J. Exp. Biol. 2009;47:475–479. [PMC free article] [PubMed] [Google Scholar]
- Matoussi, N., Ameur, H.B., Amor, S.B., Fitouri, Z., Becher, S.B., 2007. Cardiotoxicity of n-methyl-glucamine antimoniate (Glucantime). A case report. Med. Mal. Infect. 37 (Suppl 3), S257–S259. [DOI] [PubMed]
- Melby P.C., Chandrasekar B., Zhao W., Coe J.E. The hamster as a model of human visceral leishmaniasis: progressive disease and impaired generation of nitric oxide in the face of a prominent Th1-like cytokine response. J. Immunol. 2001;166:1912–1920. doi: 10.4049/jimmunol.166.3.1912. [DOI] [PubMed] [Google Scholar]
- Nieto A., Dominguez-Bernal G., Orden J.A., De La Fuente R., Madrid-Elena N., Carrion J. Mechanisms of resistance and susceptibility to experimental visceral leishmaniosis: BALB/c mouse versus Syrian hamster model. Vet. Res. 2011;42:39. doi: 10.1186/1297-9716-42-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaka S., Hudson A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. 2006;5:941–955. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- Olliaro P.L. Drug combinations for visceral leishmaniasis. Curr. Opin. Infect. Dis. 2010;23:595–602. doi: 10.1097/QCO.0b013e32833fca9d. [DOI] [PubMed] [Google Scholar]
- Osorio Y., Travi B.L., Renslo A.R., Peniche A.G., Melby P.C. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop. Dis. 2011;5:e962. doi: 10.1371/journal.pntd.0000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette M., Drummelsmith J., Papadopoulou B. Leishmaniasis: drugs in the clinic, resistance and new developments. Drug Resist. Updat. 2004;7:257–266. doi: 10.1016/j.drup.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Oura M., Sternberg T.H., Wright E.T. A new antifungal antibiotic, amphotericin B. Antibiot. Annu. 1955;3:566–573. [PubMed] [Google Scholar]
- Paris C., Loiseau P.M., Bories C., Breard J. Miltefosine induces apoptosis-like death in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 2004;48:852–859. doi: 10.1128/AAC.48.3.852-859.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak M.K., Yi T. Sodium stibogluconate is a potent inhibitor of protein tyrosine phosphatases and augments cytokine responses in hemopoietic cell lines. J. Immunol. 2001;167:3391–3397. doi: 10.4049/jimmunol.167.6.3391. [DOI] [PubMed] [Google Scholar]
- Payne D.J., Gwynn M.N., Holmes D.J., Pompliano D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Peacock C.S., Seeger K., Harris D., Murphy L., Ruiz J.C., Quail M.A., Peters N., Adlem E., Tivey A., Aslett M., Kerhornou A., Ivens A., Fraser A., Rajandream M.A., Carver T., Norbertczak H., Chillingworth T., Hance Z., Jagels K., Moule S., Ormond D., Rutter S., Squares R., Whitehead S., Rabbinowitsch E., Arrowsmith C., White B., Thurston S., Bringaud F., Baldauf S.L., Faulconbridge A., Jeffares D., Depledge D.P., Oyola S.O., Hilley J.D., Brito L.O., Tosi L.R., Barrell B., Cruz A.K., Mottram J.C., Smith D.F., Berriman M. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscopo T.V., Mallia Azzopardi C. Leishmaniasis. Postgrad. Med. J. 2007;83:649–657. doi: 10.1136/pgmj.2006.047340corr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux J.A., Golvan Y.J., Croset H., Houin R. Leishmanioses in the Mediterranean “Midi”: results of an ecologic survey. Bull. Soc. Pathol. Exot. Filiales. 1969;62:332–333. [PubMed] [Google Scholar]
- Roberts C.W., McLeod R., Rice D.W., Ginger M., Chance M.L., Goad L.J. Fatty acid and sterol metabolism: potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Parasitol. 2003;126:129–142. doi: 10.1016/s0166-6851(02)00280-3. [DOI] [PubMed] [Google Scholar]
- Rochette A., Raymond F., Corbeil J., Ouellette M., Papadopoulou B. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Mol. Biochem. Parasitol. 2009;165:32–47. doi: 10.1016/j.molbiopara.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Scherf H.R., Schuler B., Berger M.R., Schmahl D. Therapeutic activity of ET-18-OCH3 and hexadecylphosphocholine against mammary tumors in BD-VI rats. Lipids. 1987;22:927–929. doi: 10.1007/BF02535557. [DOI] [PubMed] [Google Scholar]
- Schettini D.A., Ribeiro R.R., Demicheli C., Rocha O.G., Melo M.N., Michalick M.S., Frezard F. Improved targeting of antimony to the bone marrow of dogs using liposomes of reduced size. Int. J. Pharm. 2006;315:140–147. doi: 10.1016/j.ijpharm.2006.01.048. [DOI] [PubMed] [Google Scholar]
- Scott J.A., Davidson R.N., Moody A.H., Grant H.R., Felmingham D., Scott G.M., Olliaro P., Bryceson A.D. Aminosidine (paromomycin) in the treatment of leishmaniasis imported into the United Kingdom. Trans. R. Soc. Trop. Med. Hyg. 1992;86:617–619. doi: 10.1016/0035-9203(92)90151-2. [DOI] [PubMed] [Google Scholar]
- Seifert K., Croft S.L. In vitro and in vivo interactions between miltefosine and other antileishmanial drugs. Antimicrob. Agents Chemother. 2006;50:73–79. doi: 10.1128/AAC.50.1.73-79.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahian, M., Alborzi, A., 2009. Effect of meglumine antimoniate on the pancreas during treatment of visceral leishmaniasis in children. Med. Sci. Monit. 15, CR290–293. [PubMed]
- Sharlow E.R., Close D., Shun T., Leimgruber S., Reed R., Mustata G., Wipf P., Johnson J., O’Neil M., Grogl M., Magill A.J., Lazo J.S. Identification of potent chemotypes targeting Leishmania major using a high-throughput, low-stringency, computationally enhanced, small molecule screen. PLoS Negl. Trop. Dis. 2009;3:e540. doi: 10.1371/journal.pntd.0000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood J.A., Gachihi G.S., Muigai R.K., Skillman D.R., Mugo M., Rashid J.R., Wasunna K.M., Were J.B., Kasili S.K., Mbugua J.M. Phase 2 efficacy trial of an oral 8-aminoquinoline (WR6026) for treatment of visceral leishmaniasis. Clin. Infect. Dis. 1994;19:1034–1039. doi: 10.1093/clinids/19.6.1034. [DOI] [PubMed] [Google Scholar]
- Singh N., Dube A. Short report: fluorescent Leishmania: application to anti-leishmanial drug testing. Am. J. Trop. Med. Hyg. 2004;71:400–402. [PubMed] [Google Scholar]
- Siqueira-Neto J.L., Song O.R., Oh H., Sohn J.H., Yang G., Nam J., Jang J., Cechetto J., Lee C.B., Moon S., Genovesio A., Chatelain E., Christophe T., Freitas-Junior L.H. Antileishmanial high-throughput drug screening reveals drug candidates with new scaffolds. PLoS Negl. Trop. Dis. 2010;4:e675. doi: 10.1371/journal.pntd.0000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S. Drug resistance in Indian visceral leishmaniasis. Trop. Med. Int. Health. 2001;6:849–854. doi: 10.1046/j.1365-3156.2001.00778.x. [DOI] [PubMed] [Google Scholar]
- Sundar S., Mehta H., Suresh A.V., Singh S.P., Rai M., Murray H.W. Amphotericin B treatment for Indian visceral leishmaniasis: conventional versus lipid formulations. Clin. Infect. Dis. 2004;38:377–383. doi: 10.1086/380971. [DOI] [PubMed] [Google Scholar]
- Sundar S. Miltefosine in the treatment of leishmaniasis: clinical evidence for informed clinical risk management. Ther. Clin. Risk Manag. 2007;3:733–740. [PMC free article] [PubMed] [Google Scholar]
- Sundar S., Sinha P.K., Rai M., Verma D.K., Nawin K., Alam S., Chakravarty J., Vaillant M., Verma N., Pandey K., Kumari P., Lal C.S., Arora R., Sharma B., Ellis S., Strub-Wourgaft N., Balasegaram M., Olliaro P., Das P., Modabber F. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet. 2011;377:477–486. doi: 10.1016/S0140-6736(10)62050-8. [DOI] [PubMed] [Google Scholar]
- Swinney D.C., Anthony J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- Torchilin V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- Tripathi P., Singh V., Naik S. Immune response to Leishmania: paradox rather than paradigm. FEMS Immunol. Med. Microbiol. 2007;51:229–242. doi: 10.1111/j.1574-695X.2007.00311.x. [DOI] [PubMed] [Google Scholar]
- Urbina J.A. Lipid biosynthesis pathways as chemotherapeutic targets in kinetoplastid parasites. Parasitology. 1997;114(Suppl):S91–99. [PubMed] [Google Scholar]
- Vermeersch M., da Luz R.I., Tote K., Timmermans J.P., Cos P., Maes L. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: practical relevance of stage-specific differences. Antimicrob. Agents Chemother. 2009;53:3855–3859. doi: 10.1128/AAC.00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhone P., Maiti M., Agarwal R., Kamat V., Martin S., Saha B. Miltefosine promotes IFN-gamma-dominated anti-leishmanial immune response. J. Immunol. 2009;182:7146–7154. doi: 10.4049/jimmunol.0803859. [DOI] [PubMed] [Google Scholar]
- Walker R.G., Thomson G., Malone K., Nowicki M.W., Brown E., Blake D.G., Turner N.J., Walkinshaw M.D., Grant K.M., Mottram J.C. High throughput screens yield small molecule inhibitors of Leishmania CRK3:CYC6 cyclin-dependent kinase. PLoS Negl. Trop. Dis. 2011;5:e1033. doi: 10.1371/journal.pntd.0001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Control of the leishmaniases. Report of a WHO expert committee. World Health Organ. Tech. Rep. Ser. 1990;793:1–158. [PubMed] [Google Scholar]
- WHO, 2007. Report of the Fifth Consultative Meeting on Leishmania/HIV Coinfection.
- WHO, 2010. Control of the leishmaniases. World Health Organ. Tech. Rep. Ser. xii–xiii, 1–186 (back cover). [PubMed]
- Woosley R.L., Cossman J. Drug development and the FDA’s critical path initiative. Clin. Pharmacol. Ther. 2007;81:129–133. doi: 10.1038/sj.clpt.6100014. [DOI] [PubMed] [Google Scholar]
- Wyllie S., Cunningham M.L., Fairlamb A.H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 2004;279:39925–39932. doi: 10.1074/jbc.M405635200. [DOI] [PubMed] [Google Scholar]
- Yardley V., Croft S.L. Activity of liposomal amphotericin B against experimental cutaneous leishmaniasis. Antimicrob. Agents Chemother. 1997;41:752–756. doi: 10.1128/aac.41.4.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghloul I.Y., Al-Jasser M. Effect of renal impairment on the pharmacokinetics of antimony in hamsters. Ann. Trop. Med. Parasitol. 2004;98:793–800. doi: 10.1179/000349804X3171. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Messier N., Ouellette M., Rosen B.P., Mukhopadhyay R. Leishmania major LmACR2 is a pentavalent antimony reductase that confers sensitivity to the drug pentostam. J. Biol. Chem. 2004;279:37445–37451. doi: 10.1074/jbc.M404383200. [DOI] [PubMed] [Google Scholar]