

Introduction
Cardiofaciocutaneous syndrome (OMIM 115150) is a rare genetic disorder characterized by cardiac abnormalities, distinctive craniofacial appearance and cutaneous abnormalities. Cognitive delay (mild to severe) is seen in all affected individuals. CFC syndrome is a member of a family of syndromes that includes the Noonan and Costello syndromes presenting with phenotypic similarities. All these syndromes are caused by mutations in genes belonging to RAS-ERK pathway that regulates cell differentiation, proliferation and apoptosis. Thus, the clinical overlap of these conditions, which often poses a problem of differential diagnosis, is explained by their pathogenic relatedness.1–3 We report one such case.
Case report
An 18-month-old male toddler born of third degree consanguinity presented with delayed developmental milestones, in that the child could only stand with support and could speak only bisyllables. The dysmorphic facies prompted the authors to carry out a complete examination and look for syndromic association. The child weighed 8.7 kg (<3rd percentile) with a length of 79 cm (10th–25th percentile). The head circumference was 50.9 cm (>97th percentile). Head to toe examination revealed a large anterior fontanel, bitemporal constriction, hypertelorism, epicanthal folds, antimongoloid slant, strabismus; low set ears; depressed nasal bridge with a short nose and short neck (Fig. 1). Teeth were decayed and dysplastic. The patient had hyperkeratotic skin with multiple patches of hyperpigmentation and fuzzy hair with slow growth. A pansystolic murmur, hepatosplenomegaly and generalized hypotonia were noted in the systemic examination. The above clinical features led to a differential diagnosis of Noonan, Costello, CFC syndrome, mucopolysaccharidosis and congenital hypothyroidism. Radiographs showed a normal bone age with no evidence of dysostosis multiplex. Echocardiography showed a large subaortic VSD. Thyroid profile and MRI brain were normal with a 46 XY karyotype. Urine was negative for glycosaminoglycans. In the present case, nearly all the described clinical features of CFC syndrome as described in the standard textbook as well as review of literature were present. Moreover, CFC index was calculated by adding the defined values for various traits present in the patient against a table for CFC index developed by Kavamura et al. We got a value of 14.199 that fell between 12.1 and 17.3 (mean ± 1 SD), indicating that the patient has CFC syndrome.
Fig. 1.
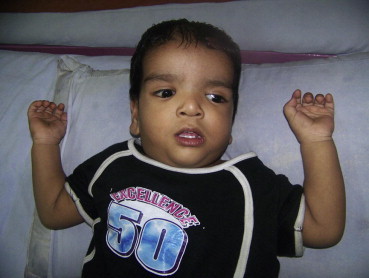
A photograph of the patient showing characteristic facies.
Discussion
A mental retardation syndrome having a combination of cardiac defects, distinct facial appearance, ectodermal abnormalities, growth retardation and mental retardation was reported by Reynolds et al in 1986 and designated the cardiofaciocutaneous syndrome.1 More than 100 cases have been reported and an estimated 200–300 persons with this condition live worldwide.4 To the best of the authors' knowledge, there is only one previous Indian report of CFC syndrome.5
Patients with CFC syndrome are defined by the following phenotype:
-
(a)
Short stature
-
(b)
Failure to thrive
-
(c)
Short webbed neck
-
(d)
Relative macrocephaly
-
(e)
Hypotonia
-
(f)
Developmental delay (most commonly in motor and speech)
-
(g)
Congenital cardiac defects (most frequently valvar pulmonary stenosis, hypertrophic cardiomyopathy and atrial septal defect)
-
(h)
Typical facies, with high forehead, bitemporal constriction, supra-orbital hypoplasia, short nose with depressed nasal bridge and anteverted nostrils, low implanted and posteriorly angulated ears, hypertelorism, ptosis, epicanthal folds, and downslanting of palpebral fissures
-
(i)
Ectodermal abnormalities such as sparse, slow growing and curly hair, sparse or absent eyelashes and eyebrows, follicular keratosis, icthyosis, hyperkeratotic skin, generalized hyperpigmentation, hyperelastic skin, and slow growth of nails.1,5–7
As with other dysmorphic syndromes the diagnosis of CFC syndrome depends upon a characteristic picture rather than any specific diagnostic test. Demonstration of the underlying genetic abnormality requires sophisticated methodology beyond the scope of a standard clinical or genetic laboratory. Till recently, controversy existed concerning the delineation of CFC syndrome from Noonan syndrome and Costello syndrome with which it has a considerably overlapping phenotypic expression. Several authors have suggested that they were different phenotypes of the same condition. The presence of the six characteristic features of characteristic facies, mental retardation, hyperkeratotic skin lesions, sparse and thin hair, congenital heart disease and sporadic occurrence led the authors to the diagnosis of CFC syndrome. Further, the characteristic high forehead, bitemporal constriction, downslanting eyes and the overall facial ‘gestalt’ resembling that of a large molecular weight storage disorder which had led to evaluations for mucopolysaccharidosis were further supportive of the diagnosis.2
In 2002, Kavamura et al proposed the use of CFC index for conformation of CFC diagnosis and to differentiate CFC from other phenotypically similar genetic conditions. They did a mathematical multifactorial correlation study using 82 clinical traits on 54 patients with CFC diagnosis to calculate the frequency of these anomalies. The CFC index is calculated by adding the allocated value for each of the 82 characteristics in a given patient. The normal distribution of CFC index curve allows the following interpretations – 68% of the CFC population has indices between 12.1 and 17.3 (mean ± 1SD), 95% between 9.5 and 19.9 (mean ± 2SD) and 99% between 6.9 and 22.5 (mean ± 3SD). Score obtained with other related syndromes is below the second SD. In our case the calculated CFC index was 14.199, which is again a strong indicator that the patient is really a case of CFC syndrome.8
CFC syndrome is the result of a mutation in the RAS/mitogen-activated protein kinase (MAPK) genes BRAF, MAP2K1, MAP2K2 and KRAS. Treatment of a patient with CFC syndrome requires a multidisciplinary team of healthcare providers with surveillance for possible secondary complications. Risk to the sibs of proband is small since all individuals with CFC syndrome have been due to a de novo dominant mutation. To date, no individuals with CFC syndrome have been known to reproduce.4
Conflicts of interest
All authors have none to declare.
References
- 1.Reynolds J.F., Neri G., Herrmann J.P. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement – the CFC syndrome. Am J Med Genet. 1986;25:413–427. doi: 10.1002/ajmg.1320250303. [DOI] [PubMed] [Google Scholar]
- 2.Neri G., Zollino M., Reynolds J.F. The Noonan-CFC controversy [editorial comment] Am J Med Genet. 1991;39:367–370. doi: 10.1002/ajmg.1320390323. [DOI] [PubMed] [Google Scholar]
- 3.Fryer A.E., Holt P.J., Hughes H.E. The cardio-facio-cutaneous (CFC) syndrome and Noonan syndrome: are they the same? Am J Med Genet. 1991;38:548–551. doi: 10.1002/ajmg.1320380410. [DOI] [PubMed] [Google Scholar]
- 4.Rauen KA. Cardiofaciocutaneous syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, eds. Gene Reviews. Online Edition. Seattle: University of Washington. Available from http://www.ncbi.nlm.nih.gov/books/NBK1186/ (cited 2011 Sep 17).
- 5.Nanda S., Rajpal M., Reddy B.S.N. Cardio-facio-cutaneous syndrome: report of a case with a review of the literature. Int J Dermatol. 2004;43:447–450. doi: 10.1111/j.1365-4632.2004.02102.x. [DOI] [PubMed] [Google Scholar]
- 6.Ward K.A., Moss C., McKeown C. The cardio-facio-cutaneous syndrome: a manifestation of the Noonan syndrome? Br J Dermatol. 1994;131:270–274. doi: 10.1111/j.1365-2133.1994.tb08504.x. [DOI] [PubMed] [Google Scholar]
- 7.Jones K.L. 6th ed. Elsevier Saunders; Philadelphia, Pennysylvania: 2006. Smith's Recognizable Patterns of Human Malformation. [Google Scholar]
- 8.Kavamura M.I., Peres C.A., Alchorne M.M., Brunoni D. CFC index for the diagnosis of cardiofaciocutaneous syndrome. Am J Med Genet. 2002;112:12–16. doi: 10.1002/ajmg.10681. [DOI] [PubMed] [Google Scholar]