

Abstract
Background
Although erythroid cells prepared from fetal liver, cord blood, or blood from β-thalassemia patients are known to express fetal hemoglobin at high levels, the underlying mechanisms remain elusive. We previously showed that cyclic nucleotides such as cAMP and cGMP induce fetal hemoglobin expression in primary erythroid cells. Here we report that cAMP signaling contributes to high-level fetal hemoglobin expression in erythroid cells prepared from cord blood and β-thalassemia.
Methods
The status of the cAMP signaling pathway was investigated using primary erythroid cells prepared from cord blood and the mononuclear cells of patients with β-thalassemia; erythroid cells from adult bone marrow mononuclear cells served as the control.
Results
We found that intracellular cAMP levels were higher in erythroid cells from cord blood and β-thalassemia than from adult bone marrow. Protein kinase A activity levels and cAMP-response element binding protein phosphorylation were higher in erythroid cells from cord blood or β-thalassemia than in adult bone marrow progenitors. Mitogen-activated protein kinase pathways, which play a role in fetal hemoglobin expression, were not consistently activated in cord blood or β-thalassemia erythroid cells. When cAMP signaling was activated in adult erythroid cells, fetal hemoglobin was induced at high levels and associated with reduced expression of BCL11A, a silencer of the β-globin gene.
Conclusion
These results suggest that activated cAMP signaling may be a common mechanism among erythroid cells with high fetal hemoglobin levels, in part because of downregulation of BCL11A. Activation of the cAMP signaling pathway with cAMP-elevating agents may prove to be an important signaling mechanism to reactivate fetal hemoglobin expression in erythroid cells.
Keywords: cAMP, signaling, fetal hemoglobin, β-globin disorders, cord blood, β-thalassemia
Introduction
Elevated fetal hemoglobin (HbF) expression ameliorates clinical symptoms of β-globin disorders such as sickle cell disease and β-thalassemia.1 Chemicals capable of elevating HbF synthesis could pave the way toward a treatment for these disorders.2 While there remain serious concerns about the long-term toxicity of HbF inducers, it is nevertheless of the utmost importance to develop potent and clinically safe HbF inducers for three reasons. First, HbF inducers will play a role in treating these disorders at least until gene therapy,3 cell therapy using induced pluripotent stem cells,4 and/or bone marrow transplantation5 become standard of care. Second, treatment with HbF inducers is more cost-effective than gene therapy or bone marrow transplantation, and HbF inducers are particularly beneficial for those without access to other treatment modalities. Third, treatment with HbF inducers is reversible; if proved ineffective, administration can be terminated at any time. Gene therapy and bone marrow transplantation, on the other hand, are irreversible therapies.
We and others have shown that chemicals or growth factors that induce HbF expression do so through intracellular signaling mechanisms.6–8 Pace et al reported that p38 kinase pathways use histone deacetylase inhibitors to induce HbF expression,7 while extracellular signal-regulated protein kinases, which constitute mitogen-activated protein (MAP) kinase pathways, are indispensable for HbF expression by growth factors such as stem cell factors.8 We previously reported that the soluble guanylate cyclase-cGMP pathway has a role in hydroxyurea-induced HbF expression.6,9 Cokic et al reached a similar conclusion.10 Our subsequent studies found that cAMP also induces HbF expression, and with greater potency.11,12
Considerable energy has been expended to determine how HbF chemical inducers upregulate HbF expression in erythroid cells13 and has resulted in reams of scholarly work on the subject. In contrast, very little is known about how high levels of HbF expression are sustained in particular erythroid cells. Using primary erythroid cells from cord blood and the mononuclear cells (MNCs) of patients with β-thalassemia, we discovered that these erythroid cells express high levels of HbF. We then investigated the phosphorylation status of protein kinases involved in intracellular signaling pathways that play a role in regulating HbF expression, and found that the cAMP signaling pathway is consistently activated in the erythroid cells of cord blood and β-thalassemia, while some MAP kinases such as extracellular signal-regulated protein kinases are phosphorylated. These results indicate that cAMP signaling is an important pathway for sustaining high-level HbF in erythroid cells.
Materials and methods
Materials
Bone marrow MNCs from normal subjects were obtained from Poietics (Walkersville, MD, USA). Human CD34+ cells were provided by the National Heart, Lung and Blood Institute Programs of Excellence in Gene Therapy Hematopoietic Cell Processing Core (Fred Hutchinson Cancer Research Center, Seattle, WA, USA). Cord blood was from the National Disease Research Interchange (Philadelphia, PA, USA). MNCs from patients with β-thalassemia intermedia (at least 10 patients or more) were as described6,14 and the patients had not been transfused for at least 3 months before the blood draw. 8-CTP-cAMP and 8-CTP-cGMP were purchased from Enzo (Ann Arbor, MI, USA). Antibodies were from Cell Signaling Technology (Beverly, MA, USA) unless stated otherwise. Epo was kindly provided by Amgen (Thousand Oaks, CA, USA); other cytokines were from Peprotech (Rocky Hill, NJ, USA). Informed consent was obtained from all patients and normal subjects according to the Declaration of Helsinki of 1975 and as revised in 2000. This study was approved by the Georgia Regents University institutional review board.
Isolation of primary erythroid cells from cord blood and patients with β-thalassemia
MNCs from cord blood were isolated by density gradient centrifugation with Histopaque®-1077 (Sigma-Aldrich, St Louis, MO, USA). Primary erythroid cells from cord blood were prepared as described.11 Peripheral blood was obtained from patients with β-thalassemia intermedia because a high proportion of circulating nucleated erythroblasts, representing between 70% and 90% of MNCs, is typical in this form of thalassemia.14 MNCs were isolated from peripheral blood and nucleated erythroblast-rich fractions were obtained by removing nonerythroid cells either with cell panning using anti-human CD45 antibody or with magnetic-activated cell sorting LD columns using a MidiMACS separator and CD45 microbeads (Miltenyi Biotec Inc, Auburn, CA, USA) according to the protocol supplied by the manufacturer, as described previously11 These cell preparations had at least 80% or more nucleated erythroblasts by morphology after Wright’s staining and benzidine staining using 0.05% 2,7-diaminofluorene (Sigma-Aldrich).15
Isolation of whole cell extracts from erythroblasts and immunoblotting
Whole cellular extracts were prepared from nucleated erythroblasts of β-thalassemic patients as described.14 Briefly, nucleated erythroblasts (5–10 × 106 cells) were suspended with 1× lysis buffer (Cell Signaling Technology) supplemented with 1 mM phenyl-methyl sulfonyl fluoride and 0.1% sodium dodecyl sulfate. Whole cellular extracts were obtained by centrifugation at 14,000 × g for 15 minutes. Immunoblotting was performed as previously described.16 Approximately 20–30 μg of cellular extracts were separated on 12% sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membranes (Invitrogen). All antibodies used for immunoblotting analyses were purchased from Cell Signaling Technology unless otherwise stated. Protein bands were visualized by the Phototope®-HRP Western blot detection system (Cell Signaling Technology) according to the protocol provided by the supplier.
Analysis of globin mRNA by primer extension
Total RNA was extracted from cells as previously described.17 Ten micrograms of total RNA were subjected to primer extension analysis as described.18 Oligonucleotides used as primers were 5′-end-labeled with T4 polynucleotide kinase. Total RNAs were annealed with P32-labeled oligonucleotides in 30 mL of hybridization buffer (80% formamide/40 mM Pipes, pH 6.4, 1 mM ethylenediaminetetraacetate, pH 8.0, 0.4 M NaCl) at 30°C. The extension reaction was performed as described.18 The extension products were analyzed on 8% urea-polyacrylamide gels, and the gels were exposed to Kodak X-AR film. The DNA sequences of oligonucleotides used as primers were as described.18
Globin chain analysis by reverse-phase HPLC
Nucleated erythroblasts were isolated from semisolid cultures as described above and washed three times with phosphate-buffered saline. Red blood cells were isolated by density gradient centrifugation with Histopaque-1077. Red blood cells were lysed by adding 5 mM ethylenediaminetetraacetate solution and incubating at 37°C for 30 minutes. The cell suspensions were centrifuged at 14,000× g for 15 minutes and the supernatants were recovered. Impurities were removed from the supernatant using a 0.45 μM filter before high-performance liquid chromatography (HPLC) analysis. Reverse-phase HPLC was performed using an LC-VP series system (Shimadzu, Kyoto, Japan) and a Vydac C4 column (250 × 4.6 mm) according to the method previously described.11
Measurement of intracellular levels of cyclic nucleotides in erythroid cells
Intracellular levels of cyclic nucleotides (cAMP and cGMP) in red blood cells from cord blood and β-thalassemic patients were determined as described elsewhere.6,19 Briefly, washed red blood cells (1 × 108 cells) or nucleated erythroblasts (2 × 106 cells) were incubated with 1 mM 3-isobutyl-1-methylxanthine for 30 minutes at room temperature. Cyclic nucleotides were extracted by suspending the cells in 0.5 M perchloric acid. The supernatant was removed and the pH neutralized by addition of 8 M KOH. Intracellular cAMP levels were determined by a nonacetylation method using a cAMP enzyme-linked immunosorbent assay kit (Cayman Laboratories, Ann Arbor, MI, USA). To measure intracellular cGMP levels, the extracted cyclic nucleotide samples were acetylated and assayed using a cGMP enzyme-linked immunosorbent assay kit (Cayman Laboratories). Results were expressed as pmol/mg protein.
PKA assays using cellular extracts of primary erythroid cells
Measurements of cAMP-dependent protein kinase (PKA) activity in cellular extracts were performed using a SignaTECT PKA assay system (Promega, Madison, WI, USA), in which biotinylated Kemptide (LRRASLG) was used as the substrate for PKA. Briefly, cellular extracts of primary erythroblasts were prepared by suspending the cells in extraction buffer (25 mM Tris-HCl (pH 7.4), 0.5 mM ethylenediaminetetraacetate, 0.5 mM EGTA, 10 mM β-mercaptoethanol, 1 μg/mL leupeptin, 1 μg/mL aprotinin). To measure PKA activity in the cellular extracts, the following reaction was set up: 5 μL of PKA buffer, 200 mM Tris-HCl, pH 7.4, 100 mM MgCl2, 0.5 mg/mL bovine serum albumin, 25 μM cAMP, 0.5 mM biotinylated Kemptide, 5 μL of [γ-32P]ATP (3,000 Ci/mmol) 10 μCi/μL diluted with 0.5 mM ATP, and 5 μL of cellular extracts. The reaction was performed at 30°C for 5 minutes, then terminated by adding 12.5 μL of termination buffer (7.5 M guanidine hydrochloride), and spotted onto SAM2® Biotin capture membranes (Promega). After washing, radioactivity was determined in a liquid scintillation counter and PKA activity was calculated according to the instructions provided by the supplier.
Microarray analysis for gene expression in primary erythroid cells
Microarray analysis was performed as described previously20 Briefly, total RNAs (~10 μg) were isolated from primary erythroid cells using an Absolutely RNA Miniprep kit (Qiagen, Venlo, the Netherlands). Double-stranded cDNA was synthesized from total RNA and in vitro transcription reactions were then performed in the presence of biotinylated UTP and CTP to produce biotin-labeled cRNA from the double-stranded cDNA using one-cycle target labeling and control reagents (Affymetrix, Santa Clara, CA, USA). The labeled cRNAs were hybridized to Human Genome U133 Plus 2.0 Array (Affymetrix) for 16 hours at 45°C. The images were analyzed using Microarray Suite software and the comparison analysis was performed according to the instructions provided by Affymetrix. Expression levels of genes were adjusted by using β-actin as an internal control. Assays were performed three times.
Statistical analysis
Statistical analysis was performed using the two-samples Student’s t-test. P-values<0.05 were considered to be statistically significant.
Results
Both cAMP and cGMP induced HbF expression in primary erythroid cells
We previously showed that chemicals stimulating adenylate cyclase activity such as forskolin and prostaglandin E2 induce β-globin mRNA expression in adult erythroid cells.20 To confirm that the cAMP and cGMP signaling pathways indeed exert positive regulatory effects on HbF expression at the protein level, we first performed immunoblotting, as described previously,16 to determine whether treatment of cyclic nucleotides increased HbF expression levels in primary erythroid cells. 8-CPT-cAMP and 8-CPT-cGMP are membrane-permeable, resistant to the activities of phosphodiesterase, and more potent than 8-Bromo-cAMP and 8-Bromo-cGMP.21 HbF expression levels in primary erythroid cells treated with 8-CPT-cGMP (Figure 1, lanes 2–5) or 8-CPT-cAMP (Figure 1, lanes 6–9) were increased in a dose-dependent manner. Interestingly, erythroid cells had higher HbF levels when treated with cAMP than with cGMP, suggesting that cAMP more strongly enhances HbF activity. This is consistent with previously reported mRNA studies.11
Figure 1.

Induction of fetal hemoglobin expression by cyclic nucleotides in adult erythroid cells.
Notes: Adult erythroid cells prepared from bone marrow mononuclear cells were treated with various concentrations (1 μM to 1 mM) of 8-CPT-cGMP or 8-CPT-cAMP for 4 days. GAPDH was used as an internal control. The results summary is at the bottom of the figure. *P<0.05 and **P<0.01 versus untreated control cells (lane 1).
Abbreviation: GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
HbF is highly expressed in cord blood and β-thalassemia erythroid cells
Our previous study found that HbF is highly expressed in erythroid cells isolated from some types of β-thalassemia intermedia.14 To examine the expression levels of β-globin in primary erythroid cells prepared from cord blood and β-thalassemia, we first examined globin mRNA levels by primer extension. The ratio of β-globin mRNA to non-β-globin mRNA in cord blood erythroid cells ranged from 60% to 70% (Figure 2, lane 1), while those of β-thalassemia intermedia erythroid cells were from 50% to 60% (lane 3); β-globin mRNA was not expressed in erythroid cells isolated from adult bone marrow MNCs (lane 2). This demonstrated that the β-globin mRNA expression levels of erythroid cells derived from cord blood and β-thalassemia intermedia are comparable. To confirm that both erythroid cell types express similar levels of HbF, we performed reverse-phase HPLC. The elution patterns of both erythroid cell types were quite similar to each other, suggesting that HbF expression of cord blood and β-thalassemia erythroid cells are comparable (Figure 2B and C).
Figure 2.
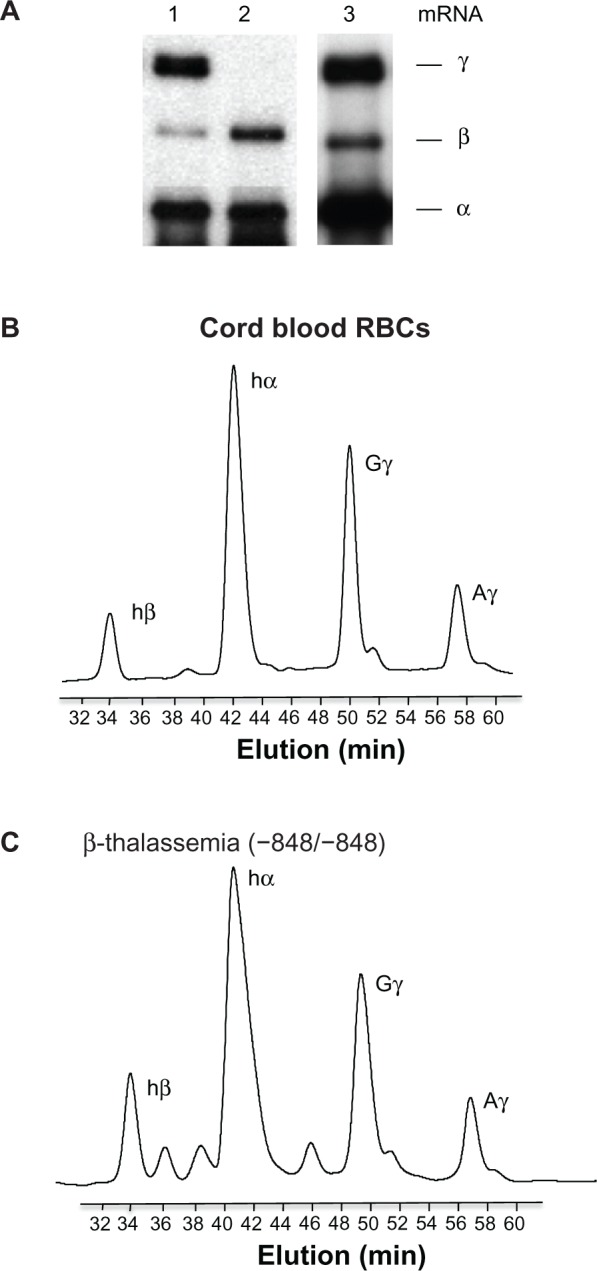
Fetal hemoglobin expression in cord blood and β-thalassemia erythroid cells.
Notes: (A) Expression of human globin genes analyzed by primer extension. Positions of the extension products for individual globin genes are shown to the right of the figure. Lanes: 1, erythroid cells isolated from cord blood; 2, erythroid cells isolated from adult bone marrow mononuclear cells; 3, erythroid cells from β-thalassemia (genotype of the mutation −848/−848). Globin chain analysis in RBCs isolated from cord blood (B) and from β-thalassemia (C) by reverse-phase high-pressure liquid chromatography. Note that the elution profiles of globin chains in both preparations are similar to each other, suggesting that the fetal hemoglobin levels are comparable among cell types.
Abbreviations: RBCs, red blood cells; mRNA, messenger ribonucleic acid; min, minutes.
cAMP signaling pathway is activated in erythroid cells prepared from cord blood and β-thalassemia
In the cAMP signaling pathway, adenylate cyclase, a membrane protein, converts ATP to cAMP (Figure 3). After cAMP binds to the regulatory subunits of PKA, PKA releases the catalytic subunits. The catalytic subunits move to the nucleus and phosphorylate the cAMP-response element binding (CREB) protein.22 Our finding that cAMP is more potent than cGMP in inducing HbF activity (Figure 1) led us to investigate the status of the cAMP signaling pathway. First, we determined the intracellular cAMP levels in cord blood-derived and β-thalassemic erythroid cells (Figure 4A). The intracellular cAMP levels in erythroid cells isolated from cord blood and β-thalassemia were much higher than those prepared from bone marrow MNCs (Figure 4A), suggesting that adenylate cyclase is activated at higher levels in erythroid cells from cord blood and β-thalassemia. We then studied the PKA activity of these erythroid cells. Although the baseline activity of PKA in cord blood and β-thalassemia erythroid cells was a little higher than that of adult erythroid cells, the addition of cAMP to the reaction mixture raised PKA activity by more than two-fold (Figure 4B), which is consistent with the results in Figure 4A. CREB is a transcription factor and a typical substrate of PKA.22 Whereas the phosphorylation level of CREB was low in adult erythroid cells (Figure 4C, lane 1), it was high in the cord blood and β-thalassemia erythroid cells (lanes 2 and 3). These results indicate that the cAMP signaling pathway is highly activated in erythroid cells prepared from both cord blood and β-thalassemia.
Figure 3.
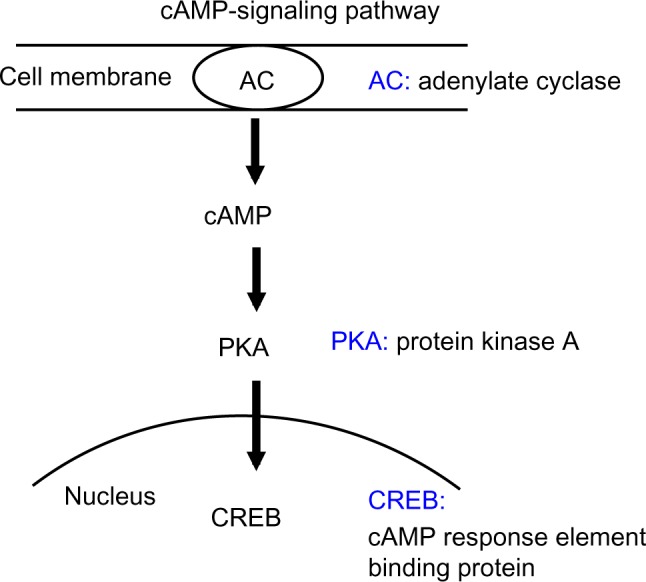
cAMP signaling pathway.
Notes: Adenylate cyclase, which is a key enzyme located in the cell membrane, generates cAMP. When cAMP binds to regulatory subunits of PKA, catalytic subunits are released. Catalytic subunits move to the nucleus and phosphorylate the CREB protein.
Abbreviations: AC, adenylate cyclase; PKA, protein kinase; CREB, cAMP-response element binding.
Figure 4.
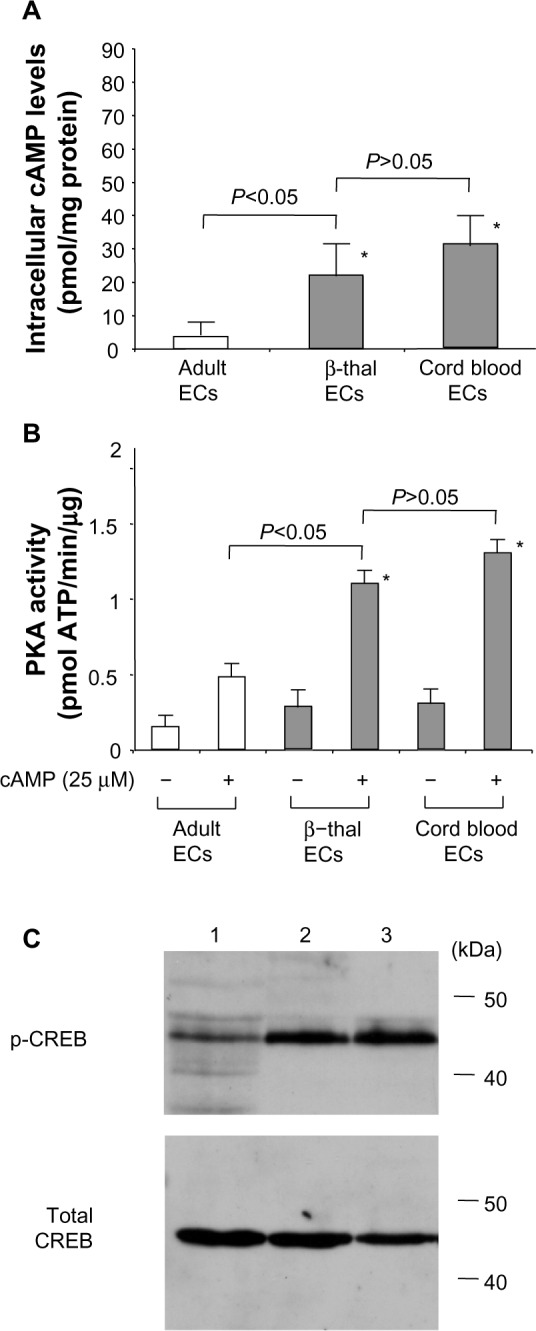
Activation of the cAMP signaling pathway in cord blood and β-thalassemia erythroid cells.
Notes: (A) Intracellular cAMP concentrations as measured by enzyme-linked immunosorbent assay and expressed as pmol/mg cellular protein. *P<0.05, compared with adult mononuclear cells. (B) PKA assays in erythroid cells. The PKA activity of cellular extracts were determined as described in the Materials and methods section. *P<0.01 compared with adult mononuclear cells. (C) CREB phosphorylation in erythroid cells. Cellular extracts were isolated from erythroid cells and CREB phosphorylation was studied by immunoblotting. Lanes: 1, erythroid cells isolated from adult bone marrow mononuclear cells; 2, erythroid cells from β-thalassemia (genotype of the mutation −848/−848); 3, erythroid cells isolated from cord blood.
Abbreviations: ECs, erythroid cells; PKA, protein kinase; CREB, cAMP-response element binding.
Status of MAP kinase pathways in erythroid cells with high HbF expression
Several studies have shown that MAP kinase pathways are involved in the induction of HbF expression by growth factors and chemicals.7,8,23,24 We examined the phosphorylation status of MAP kinase pathways including extracellular signal-regulated protein kinases, p38, and c-JUN NH2-terminal protein kinase/stress-activated protein kinase (JNK/SAPK) by investigating the phosphorylation levels of the kinases. These MAP kinases were expressed at variable levels in the cord blood and β-thalassemia erythroid cells. Phosphorylation levels were likewise inconsistent with HbF levels (Figure 5). This suggests that MAP kinases are not likely to be involved in high-level expression of HbF in cord blood and β-thalassemia erythroid cells.
Figure 5.

Status of MAP kinase pathways in erythroid cells from cord blood and from β-thalassemia.
Note: Phosphorylation of MAP kinases in cellular extracts was assayed by immunoblotting. β-actin was used as loading control.
Abbreviations: CB, cord blood; ERKs, extracellular signal-regulated protein kinases; JNK/SAPK, c-JUN NH2-terminal protein kinase/stress-activated protein kinase; MAP, mitogen-activated protein.
Effect of cAMP signaling on BCL11A expression
Recent studies have shown that BCL11A silences β-globin gene expression both in vivo and in vitro.25,26 We examined whether BCL11A mRNA expression in primary erythroid cells was affected by activation of the cAMP signaling pathway. Primary erythroid cells prepared from adult bone marrow were treated with 10 μM forskolin, which activates adenylate cyclase.27 This brought about a three-fold induction of β-globin mRNA expression, but suppressed β-globin mRNA expression by ~30%, demonstrating that activating cAMP signaling will reactivate the β-globin gene (Figure 6A). This result is consistent with our previous study.11 By performing microarray analyses using mRNAs from adult erythroid cells, we next determined the genes for which expression was affected by forskolin treatment. Interestingly, the mRNA level of BCL11A in forskolin-treated adult erythroid cells was reduced by more than 55% compared with untreated control cells (Figure 6B), indicating that the cAMP signaling pathway may negatively regulate BCL11A expression.
Figure 6.

Effects of cAMP signaling on BCL11A expression in erythroid cells. (A) Expression of human globin genes in adult erythroid cells was analyzed by primer extension. Adult erythroid cells were treated with 10 μM forskolin for 3 days. (B) Results of microarray study. Poly (A)+ RNAs were isolated from erythroid cells cultured in the absence or presence of 10 μM forskolin for 3 days and were subjected to microarray assays. Microarray analysis was repeated three times.
Abbreviations: M, molecular marker; mRNA, messenger ribonucleic acid.
Discussion
The regulation of HbF expression has been extensively studied because HbF ameliorates the clinical severity of β-globin disorders. Several studies have identified transcription factors involved in upregulating β-globin gene transcription, among them KLF128 and BCL11A.29 However, transcription factors do not have catalytic domains and are therefore not ideal molecular targets around which to develop therapeutics.30
We hypothesized that HbF chemicals take advantage of the intracellular signaling pathway to induce HbF in erythroid cells.9 Our previous studies attempted to determine which signaling mechanisms are needed for HbF-inducing chemicals, such as hydroxyurea, to induce HbF. Such knowledge would propel the development of efficacious HbF inducers.6 We subsequently found that both cAMP and cGMP signaling pathways are involved in upregulation of HbF expression in primary erythroid cells.11,20
In this study, we showed that high HbF expression levels activate cAMP signaling in erythroid cells. First, we showed that, in addition to adenylate cyclase activators such as forskolin and prostaglandin E2,11 the membrane-permeable cAMP analog 8-PCT-cAMP upregulated HbF expression in adult erythroid cells by more than five-fold (Figure 1). This suggests that activating cAMP signaling induces high HbF expression levels in adult erythroid cells; such cells have low-level HbF expression. Second, using HPLC analysis, we showed that HbF is highly expressed in erythroid cells isolated from cord blood and β-thalassemia (Figure 2). More importantly, the cAMP signaling pathway, which includes adenylate cyclase, PKA, and CREB (Figure 3), was activated in both erythroid cell types (Figure 4). More specifically, the intracellular cAMP levels were elevated (Figure 4A), and the PKA activity (Figure 4B) and CREB phosphorylation levels were much higher compared with those of adult erythroid cells with low-level HbF. On the other hand, MAP kinases, such as extracellular signal-regulated protein kinases, were phosphorylated in cord blood erythroid cells, while the phosphorylation levels of other MAP kinases, including p38 kinases and JNKs/SAPKs, were highly variable. These results indicate that MAP kinases are unlikely to contribute to high-level HbF expression in these erythroid cells. Furthermore, we showed that using forskolin to activate the cAMP signaling pathway downregulates BCL11A gene expression (Figure 6); this gene silences the β-globin gene.25 Thus, it is likely that cAMP signaling upregulates HbF expression in adult erythroid cells in part by inhibiting BCL11A expression.
It is important to clarify how the cAMP signaling pathway is activated in both cord blood and β-thalassemia erythroid cells. It is known that cord blood contains elevated levels of cytokines including erythropoietin.31 Galanello et al reported that plasma erythropoietin levels correlate with HbF levels in β-thalassemia intermedia.32 Our previous study also found increases in cytokines including erythropoietin, stem cell factor, and tumor growth factor-β in β-thalassemic patients;11 both stem cell factor and tumor growth factor-β strongly enhance HbF activity in primary erythroid cells.33 However, the erythropoietin levels in cord blood are much lower than those in β-thalassemia intermedia. Thus, it is likely that the HbF expression levels in cord blood erythroid cells may be sustained by other cytokines or mechanisms. For instance, interleukin-3, which we have shown to stimulate HbF expression,19 may contribute in part to high-level expression of HbF in cord blood erythroid cells.
An important objective in investigating the signaling mechanisms by which HbF is expressed at high levels in erythroid cells is to determine whether cAMP-elevating agents could be used to treat β-globin disorders. Although previous studies had shown that the above cytokines enhance HbF, a clinical trial of erythropoietin in patients with sickle cell disease was not successful, and the clinical application of other cytokines such as stem cell factor and tumor growth factor-β may not be feasible because of undesirable adverse effects.33 Another possible approach to reactivating the cAMP signaling pathway may involve cAMP-dependent phosphodiesterases (PDEs). We found that PDE3, a cAMP-dependent PDE, is expressed at a significantly high level in primary erythroid cells.34 Nitric oxide may also be capable of inducing HbF expression in erythroid cells. Our recent study found that nitric oxide, a potent soluble guanylate cyclase activator, demonstrates strong HbF-inducing activity in adult erythroid cells in vitro (Ikuta, unpublished observation). Nitric oxide activates the cGMP signaling pathway35 and thereby increases cGMP levels in erythroid cells;6 however, this activation may be occurring through either cross-talk between the cGMP and cAMP signaling pathways or activation of cyclo-oxygenases, which are linked to G protein-coupled receptors.36,37
In regard to the signaling effects of cyclic nucleotides on HbF expression, an important question is: Does hydroxyurea utilize the cAMP signaling pathway to induce HbF? Hydroxyurea is the only therapeutic approved to treat sickle cell disease and its HbF-enhancing effects have been tested in clinical trials of β-thalassemia.38,39 Although Keefer et al reported that hydroxyurea-induced HbF is downregulated by an adenylate cyclase inhibitor,40 it is unclear whether hydroxyurea activates cAMP-dependent pathways in vivo. Further studies are required to clarify this.
Although the current study has shown that activation of the cAMP signaling pathway is associated with a significant reduction of BCL11A, which is a strong silencer of β-globin gene expression,41 it is not certain whether BCL11A inhibitors, if available, are capable of inducing HbF expression in patients with the β-globin disorders. A recent study showed that BCL11A is essential for the development of lymphoid cells42 and it is possible that administering BCL11A inhibitors as HbF inducers to patients with β-globin disorders may impair the functioning of T-cells and B-cells.
With regard to the ability of cyclic nucleotides to enhance HbF activity, we initially demonstrated that activating the cGMP signaling pathway leads to elevated HbF expression in erythroid cells.6 Our subsequent studies found that both cGMP and cAMP likely exert positive regulatory effects on HbF expression.11 It has been shown that erythroid and megakaryocytic cells share a number of cellular and molecular characteristics.43,44 For instance, in megakaryocytes, both cAMP and cGMP inhibit platelet activation.45 Thus, it would be expected that both cAMP and cGMP have similar molecular roles in HbF expression.
In conclusion, the current study has shown that the cAMP signaling pathway is commonly activated in primary erythroid cells with high-level HbF expression, which are isolated from cord blood and β-thalassemic patients. Activation of the cAMP signaling pathway may prove to be an important signaling mechanism with which to reactivate HbF expression in erythroid cells. If so, this will open up a novel avenue to develop therapeutics for the β-globin disorders. Recently, Lowrey et al showed that HbF expression can be regulated by post-transcriptional mechanisms.46 It would be interesting to study whether the cAMP signaling pathway induces HbF expression through such mechanisms.
Acknowledgments
This study was supported by grants from the National Institutes of Health (DK61806, HL73452, and P20 MD003383 to TI).
Author contributions
TI designed, performed the research, analyzed the data, wrote the paper, and approved the final version. YK and SL obtained research data, assisted with writing, and approved the final version. NO analyzed the data, assisted, edited the paper, and approved the final version.
Disclosure
The authors report no conflict of interest in this work.
References
- 1.Miller BA, Olivieri N, Salameh M, et al. Molecular analysis of the high-hemoglobin-F phenotype in Saudi Arabian sickle cell anemia. N Engl J Med. 1987;316(5):244–250. doi: 10.1056/NEJM198701293160504. [DOI] [PubMed] [Google Scholar]
- 2.Atweh GF, Schechter AN. Pharmacologic induction of fetal hemoglobin: raising the therapeutic bar in sickle cell disease. Curr Opin Hematol. 2001;8(2):123–130. doi: 10.1097/00062752-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanna J, Wernig M, Markoulaki S, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318(5858):1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 5.Eggleston B, Patience M, Edwards S, et al. Effect of myeloablative bone marrow transplantation on growth in children with sickle cell anaemia: results of the multicenter study of haematopoietic cell transplantation for sickle cell anaemia. Br J Haematol. 2003;136(4):673. doi: 10.1111/j.1365-2141.2006.06486.x. [DOI] [PubMed] [Google Scholar]
- 6.Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cyclic gmp-dependent protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98(4):1847–1852. doi: 10.1073/pnas.041599798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pace BS, Qian XH, Sangerman J, et al. p38 MAP kinase activation mediates gamma-globin gene induction in erythroid progenitors. Exp Hematol. 2003;31(11):1089–1096. doi: 10.1016/s0301-472x(03)00235-2. [DOI] [PubMed] [Google Scholar]
- 8.Bhanu NV, Trice TA, Lee YT, Miller JL. A signaling mechanism for growth-related expression of fetal hemoglobin. Blood. 2004;103(5):1929–1933. doi: 10.1182/blood-2003-05-1624. [DOI] [PubMed] [Google Scholar]
- 9.Ikuta T, Cappellini MD. A novel mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cyclic GMP pathway [Abstract] Blood. 1999;94:615a. doi: 10.1073/pnas.041599798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cokic VP, Smith RD, Beleslin-Cokic BB, et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Invest. 2003;111(2):231–239. doi: 10.1172/JCI16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey L, Kuroyanagi Y, Franco-Penteado CF, et al. Expression of the γ-globin gene is sustained by the cAMP-dependent pathway in β-thalassaemia. Br J Haematol. 2007;138(3):382–395. doi: 10.1111/j.1365-2141.2007.06673.x. [DOI] [PubMed] [Google Scholar]
- 12.Ikuta T, Adekile AD, Gutsaeva DR, et al. The proinflammatory cytokine GM-CSF downregulates fetal hemoglobin expression by attenuating the cAMP-dependent pathway in sickle cell disease. Blood Cells Mol Dis. 2011;47(4):235–242. doi: 10.1016/j.bcmd.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrine SP, Castaneda SA, Boosalis MS, White GL, Jones BM, Bohacek R. Induction of fetal globin in beta-thalassemia: cellular obstacles and molecular progress. Ann N Y Acad Sci. 2005;1054:257–265. doi: 10.1196/annals.1345.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikuta T, Kan YW, Swerdlow PS, Faller DV, Perrine SP. Alterations in protein-DNA interactions in the γ-globin gene promoter in response to butyrate therapy. Blood. 1998;92(8):2924–2933. [PubMed] [Google Scholar]
- 15.Worthington RE, Bossie-Codreanu J, Van Zant G. Quantitation of erythroid differentiation in vitro using a sensitive colorimetric assay for hemoglobin. Exp Hematol. 1987;15(1):85–92. [PubMed] [Google Scholar]
- 16.Suzuki Y, Takeda Y, Ikuta T. Immunoblotting conditions for human hemoglobin chains. Anal Biochem. 2008;378(2):218–220. doi: 10.1016/j.ab.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Ikuta T, Papayannopoulou T, Stamatoyannopoulos G, Kan YW. Globin gene switching. In vivo protein-DNA interactions of the human beta-globin locus in erythroid cells expressing the fetal or the adult globin gene program. J Biol Chem. 1996;271(24):14082–14091. doi: 10.1074/jbc.271.24.14082. [DOI] [PubMed] [Google Scholar]
- 19.Conran N, Saad STO, Costa FF, Ikuta T. Leukocyte numbers correlate with plasma levels of granulocyte-macrophage colony-stimulating factor in sickle cell disease. Ann Hematol. 2007;86(4):255–261. doi: 10.1007/s00277-006-0246-6. [DOI] [PubMed] [Google Scholar]
- 20.Kuroyanagi Y, Kaneko Y, Muta K, et al. cAMP differentially regulates γ-globin gene expression in erythroleukemic cells and primary erythroblasts through c-Myb expression. Biochem Biophys Res Commun. 2006;344(3):1038–1047. doi: 10.1016/j.bbrc.2006.03.203. [DOI] [PubMed] [Google Scholar]
- 21.Connolly BJ, Willits PB, Warrington BH, Murray KJ. 8-(4-Chlorophenyl) thio-cyclic AMP is a potent inhibitor of the cyclic GMP-specific phosphodiesterase (PDE VA) Biochem Pharmacol. 1992;44(12):2303–2306. doi: 10.1016/0006-2952(92)90673-7. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto KK, Gonzalez GA, Biggs WHD, Montminy MR. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature. 1988;334(6182):494–498. doi: 10.1038/334494a0. [DOI] [PubMed] [Google Scholar]
- 23.Aerbajinai W, Zhu J, Gao Z, Chin K, Rodgers GP. Thalidomide induces γ-globin gene expression through increased reactive oxygen species mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood. 2007;110(8):2864–2871. doi: 10.1182/blood-2007-01-065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mabaera R, West RJ, Conine SJ, et al. A cell stress signaling model of fetal hemoglobin induction: what doesn’t kill red blood cells may make them stronger. Exp Hematol. 2008;36(9):1057–1072. doi: 10.1016/j.exphem.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 26.Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. KLF1 regulates BCL11A expression and [gamma]- to [beta]-globin gene switching. Nat Genet. 2010;42(9):742–744. doi: 10.1038/ng.637. [DOI] [PubMed] [Google Scholar]
- 27.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci U S A. 1981;78(6):3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol Cell Biol. 1993;13(5):2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc Natl Acad Sci U S A. 2008;105(5):1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koehler AN. A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol. 2010;14(3):331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maier RF, Bohme K, Dudenhausen JW, Obladen M. Cord blood erythropoietin in relation to different markers of fetal hypoxia. Obstet Gynecol. 1993;81(4):575–580. [PubMed] [Google Scholar]
- 32.Galanello R, Barella S, Turco MP, et al. Serum erythropoietin and erythropoiesis in high- and low-fetal hemoglobin beta-thalassemia intermedia patients. Blood. 1994;83(2):561–565. [PubMed] [Google Scholar]
- 33.Bhanu NV, Trice TA, Lee YT, et al. A sustained and pancellular reversal of gamma-globin gene silencing in adult human erythroid precursor cells. Blood. 2005;105(1):387–393. doi: 10.1182/blood-2004-04-1599. [DOI] [PubMed] [Google Scholar]
- 34.Ikuta T, Gutsaeva DR, Parkerson JB, Yerigenahally SD, Head CA. Hydroxyurea induces fetal hemoglobin expression by activating camp signaling pathways in a cAMP- and cGMP-dependent manner; new hypothesis to account for a role of non-erythroid cells in fetal hemoglobin induction. Blood. 2010;116:3455. [Google Scholar]
- 35.Hobbs AJ, Ignarro LJ. Nitric oxide-cyclic GMP signal transduction system. Methods Enzymol. 1996;269:134–148. doi: 10.1016/s0076-6879(96)69016-8. [DOI] [PubMed] [Google Scholar]
- 36.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci U S A. 1993;90(15):7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tetsuka T, Daphna-Iken D, Srivastava SK, Baier LD, DuMaine J, Morrison AR. Cross-talk between cyclooxygenase and nitric oxide pathways: prostaglandin E2 negatively modulates induction of nitric oxide synthase by interleukin 1. Proc Natl Acad Sci U S A. 1994;91(25):12168–12172. doi: 10.1073/pnas.91.25.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fucharoen S, Siritanaratkul N, Winichagoon P, et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood. 1996;87(3):887–892. [PubMed] [Google Scholar]
- 39.Bradai M, Abad MT, Pissard S, Lamraoui F, Skopinski L, de Montalembert M. Hydroxyurea can eliminate transfusion requirements in children with severe β-thalassemia. Blood. 2003;102(4):1529–1530. doi: 10.1182/blood-2003-01-0117. [DOI] [PubMed] [Google Scholar]
- 40.Keefer JR, Schneidereith TA, Mays A, Purvis SH, Dover GJ, Smith KD. Role of cyclic nucleotides in fetal hemoglobin induction in cultured CD34+ Cells. Exp Hematol. 2006;34(9):1150–1160. doi: 10.1016/j.exphem.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 41.Sankaran VG, Xu J, Ragoczy T, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460(7259):1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu P, Keller JR, Ortiz M, et al. Bcl1 1a is essential for normal lymphoid development. Nat Immunol. 2003;4(6):525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 43.Martin DI, Zon LI, Mutter G, Orkin SH. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature. 1990;344(6265):444–447. doi: 10.1038/344444a0. [DOI] [PubMed] [Google Scholar]
- 44.Komatsu N, Kirito K, Shimizu R, et al. In vitro development of erythroid and megakaryocytic cells from a UT-7 subline, UT-7/GM. Blood. 1997;89(11):4021–4033. [PubMed] [Google Scholar]
- 45.Waldmann R, Nieberding M, Ulrich W. Vasodilator-stimulated protein phosphorylation in platelets is mediated by cAMP- and cGMP-dependent protein kinases. Eur J Biochem. 1987;167(3):441–448. doi: 10.1111/j.1432-1033.1987.tb13357.x. [DOI] [PubMed] [Google Scholar]
- 46.Hahn CK, Lowrey CH. Eukaryotic initiation factor 2α phosphorylation mediates fetal hemoglobin induction through a post-transcriptional mechanism. Blood. 2013;122(4):477–485. doi: 10.1182/blood-2013-03-491043. [DOI] [PMC free article] [PubMed] [Google Scholar]