

Abstract
Several lines of investigation have revealed the apparent interplay between the immune system of the host and many conventional, “standard-of-care” anticancer therapies, including chemotherapy and small molecule targeted therapeutics. In particular, preclinical and clinical studies have demonstrated the important role of regulatory T cells (Tregs) in inhibiting immune responses elicited by immunotherapeutic regimens such as those based on anticancer vaccines or checkpoint inhibitors. However, how the number and immunosuppressive function of Tregs change in cancer patients undergoing treatment with non-immune anticancer therapies remains to be precisely elucidated. To determine whether immunostimulatory therapies can be employed successfully in combination with conventional anticancer regimens, we have investigated both the number and function of Tregs obtained from the peripheral blood of carcinoma patients before the initiation and during the course of chemotherapeutic and targeted agent regimens. Our studies show that the treatment of breast cancer patients with tamoxifen plus leuprolide, a gonadotropin releasing hormone agonist, has minimal effects on Tregs, while sunitinib appears to exert differential effects on Tregs among patients with metastatic renal carcinoma. However, the administration of docetaxel to patients with metastatic prostate or breast cancer, as well as that of cisplatin plus vinorelbine to non-small cell lung cancer patients, appears to significantly increase the ratio between effector T cells and Tregs and to reduce the immunosuppressive activity of the latter in the majority of patients. These studies provide the rationale for the selective use of active immunotherapy regimens in combination with specific standard-of-care therapies to achieve the most beneficial clinical outcome among carcinoma patients.
Keywords: carcinoma, chemotherapy, drug therapy, T lymphocytes, Tregs
Introduction
Emerging evidence has revealed the complex interplay between the immune system of the host and many “standard-of-care” anticancer therapies such as chemotherapy, radiation therapy, hormonal therapy, and small molecule targeted therapeutics. For many cancer types, the extent and composition of the immune infiltrate into the primary lesion is a strong and independent predictor of response to subsequent therapies, hence significantly affecting disease outcome (reviewed in Refs. 1–4). Several classes of chemotherapeutics have previously been shown to induce the immunogenic demise of malignant cells, a cell death mode during which antigenic components from dying cancer cells are cross-presented to the immune system to generate a T cell-mediated tumor-specific response.2,5,6 Moreover, specific chemotherapeutic agents as well as radiation therapy have been shown to affect the phenotype of malignant cells that survive treatment (either because they are exposed to non-lethal doses of the therapeutic agent or because they are intrinsically resistant to it) such that they become susceptible to T cell-mediated lysis.7-9 Finally, it has been demonstrated in preclinical studies and some clinical trials that some chemotherapeutic agents and small molecule targeted therapeutics can have differential effects on specific components of the immune system, leading to enhanced or reduced antitumor effects.10-15 All of the abovementioned phenomena have important implications for the use of immunotherapeutic agents, such as anticancer vaccines, in combination with standard-of-care treatments.
Several different therapeutic anticancer vaccine platforms are currently being evaluated in multi-center Phase II and Phase III trials enrolling patients with a large range of carcinomas (reviewed in ref. 16). While some of these vaccines are providing evidence of clinical benefit, to date sipuleucel-T remains the only FDA-approved agent of this class, significantly increasing the survival of metastatic prostate cancer patients.17 Several reasons have been purported to account for the perceived failure of various anticancer vaccines. These include: (1) the lower potency of early generation vaccines relative to those currently available; (2) the clinical endpoint of choice (e.g., the use of strict RECIST criteria or time-to-progression vs. survival); and (3) the exposure of patients to numerous prior chemotherapy/radiotherapy regimens. Clinical studies have indeed shown that both the frequency of prior therapeutic regimens and a short interval since the last chemotherapy course can have a negative influence on the responses of cancer patients to vaccination.18,19 The presence of immunosuppressive cells and soluble factors can also inhibit vaccine-elicited immune responses.
Immunosuppressive cells and molecules are thus a major issue potentially limiting the efficacy of anticancer vaccines. These inhibitory factors are abundant in the tumor microenvironment as well as in the circulation of cancer patients.20-22 Preclinical and clinical studies have implicated regulatory T cells (Tregs) as one such immunosuppressive entity (reviewed in Refs. 23, 24). Both the abundance and immunosuppressive activity of Tregs can play a pivotal role in this process.25-33 For example, murine Tregs within the tumor microenvironment were more proliferative than those found in the periphery, whereas peripheral Tregs were more immunosuppressive than tumor-infiltrating ones.29 Unfortunately, obtaining biopsy material from most metastatic carcinoma lesions is difficult, and when tumor tissue is available its amount is usually insufficient for the analysis of Treg immunosuppressive functions. A comprehensive study has been performed on the Tregs found in the periphery of renal carcinomas treated with the tyrosine kinase inhibitor (TKI) sunitinib.10,12-14 A trend toward reduced numbers of Tregs was noted upon treatment, although not reaching statistical significance.12 Treg number and function were also evaluated in the peripheral blood of healthy individuals vs. patients with metastatic prostate cancer.34,35 In that study, a slight elevation in the number of Tregs was detected among cancer patients as compared with normal subjects. However, the most prominent difference between these 2 groups was the superior immunosuppressive activity of Tregs in the peripheral blood of prostate cancer patients. In another clinical evaluation of anticancer vaccination in patients with metastatic prostate cancer, the baseline levels of Tregs among peripheral blood mononuclear cells (PBMCs) did not impact overall survival.34 There was also no difference in the number of Tregs pre- vs. post-vaccination in patients who survived more than predicted or less than predicted using the prognostic Halabi nomogram.34-36 There were, however, strong trends linking vaccination-induced changes in the immunosuppressive function of Tregs to patient outcome. In response to vaccination therapy, patients who survived longer than predicted exhibited attenuated Treg functions, whereas patients whose survival was shorter than predicted exhibited enhanced Treg-dependent immunosuppression. Thus, it appears that changes in both the number and the immunosuppressive activity of Tregs in response to therapy should be evaluated.
Preclinical findings now suggest that anticancer vaccines can be additive or synergistic in terms of antitumor responses when used just prior to, concomitantly, or immediately following standard-of-care therapies. In this study, we have analyzed the PBMCs of carcinoma patients both before the initiation and during the course of several conventional therapies to evaluate the effect, if any, of the treatment on both the number and function of Tregs. Such standard-of-care treatments included: the administration of docetaxel to metastatic prostate and breast carcinoma patients; the use of cisplatin plus vinorelbine as an adjuvant intervention among non-small cell lung cancer (NSCLC) patients; the administration of sunitinib (Sutent®, Pfizer) to subjects with metastatic renal cell carcinoma; and the use of tamoxifen plus gonadotropin releasing hormone (GnRH) agonists as an adjuvant intervention in women with breast carcinoma.
Results
Docetaxel is a standard-of-care therapy for men with hormone-refractory malignancies of the prostate, in other words, metastatic castration-resistant prostate cancer (mCRPC). In our study, patients underwent docetaxel monotherapy (35 mg/m2) weekly for 3 weeks followed by 1 week of rest, making up 4-week therapy cycles. PBMCs were collected from each of 15 patients prior to the start of therapy (baseline), immediately after the first cycle of therapy (i.e., prior to the initiation of the week of rest) and just prior to the beginning of the second cycle. Cytofluorometric analyses of PBMCs revealed no statistical differences between the PBMCs collected at baseline and post-docetaxel in terms of PBMC amount, total number of CD4+ T cells and percent of CD4+ cells relative to total PBMCs (Fig. 1A and B; Table 1). There was, however, a significant decrease in the percent of CD4+CD25highCD127-FoxP3+ Tregs among CD4+ lymphocytes between samples collected at baseline and before the second cycle of chemotherapy (P < 0.001; Fig. 1C; Table 1). We also observed an increase in the ratio of CD4+ effector T cells (Teffs) to Tregs (CD4+ Teff:Treg ratio) from baseline to post-cycle I (P < 0.01; Table 1) and post-cycle II samples (P < 0.001; Table 1). The relative change in the CD4+ Teff:Treg ratio upon therapy relative to baseline is shown in Figure 2A for individual patients. A ≥ 20% increase in this parameter was detected in 11/15 (73%) patients, while only 2 of them exhibited decreased CD4+ Teff:Treg ratio upon therapy.

Figure 1. Circulating immune cell subsets in patients with prostate cancer or non-small cell lung cancer before and during the course of chemotherapy. (A–C) Patients (n = 15) with hormone refractory metastatic prostate cancer were treated with docetaxel and evaluated before and during therapy for the number of peripheral blood mononuclear cells (PBMCs) (A), the frequency of CD4+ T cells among PBMCs (B), and the frequency of regulatory T cells (Tregs) among CD4+ T cells (C). Peripheral blood samples were collected at the indicated time points. Statistical analyses were performed using Friedman and Dunn’s multiple comparison tests and revealed a significant decrease in the frequency of Tregs at post-cycle I (**P < 0.01) and pre-cycle II (***P < 0.001). (D–F) Patients (n = 14) with stage IB, II, and IIIA non-small cell lung cancer (NSCLC) were evaluated before and during therapy with cisplatin plus vinorelbine for the number of PBMCs (D), the frequency of CD4+ T cells among PBMCs (E), and the frequency of Tregs among CD4+ T cells (F). Peripheral blood samples were collected at the indicated time points. Statistical analyses were performed using Friedman and Dunn’s multiple comparison tests; * P < 0.05, *** P < 0.001. In all dot plots, the median and interquartile range are shown.
Table 1. Evaluation of prostate cancer patients prior to and in the course of standard-of-care anticancer therapy.
| Baseline | Post-cycle I | Pre-cycle II | n | |
|---|---|---|---|---|
| No. of PBMCs ( × 103/µL) |
1.4 (0.9–2.2) |
0.9 (0.6–1.6) |
1.7 (0.7–2.4) |
15 |
| No. of CD4+ T cells ( × 103/µL) |
0.43 (0.19–0.55) |
0.23 (0.14–0.45) |
0.42 (0.24–0.64) |
15 |
| CD4+ T cells (%) |
32 (22–35) |
25 (21–38) |
28 (19–38) |
15 |
| Tregs among CD4+ T cells (%) |
4.5 (2.6–5.2) |
3.5 (2.0–4.6)** |
2.9 (1.6–5.2)*** |
15 |
| No. of Tregs (#/µL) |
14 (10–24) |
7 (6–16) |
8 (5–20) |
15 |
| CD4+ Teff:Treg ratio | 15.0 (11.0–28.0) | 19.0 (13.0–30.0)** | 23.0 (14.0–55.0)*** | 15 |
Values are the median (interquartile range). Friedman and Dunn’s multiple comparison tests between baseline and all other time points; ** P < 0.01, and *** P < 0.001 compared with baseline. PBMC, peripheral blood mononuclear cell; Teff, effector T cell; Treg, regulatory T cell.
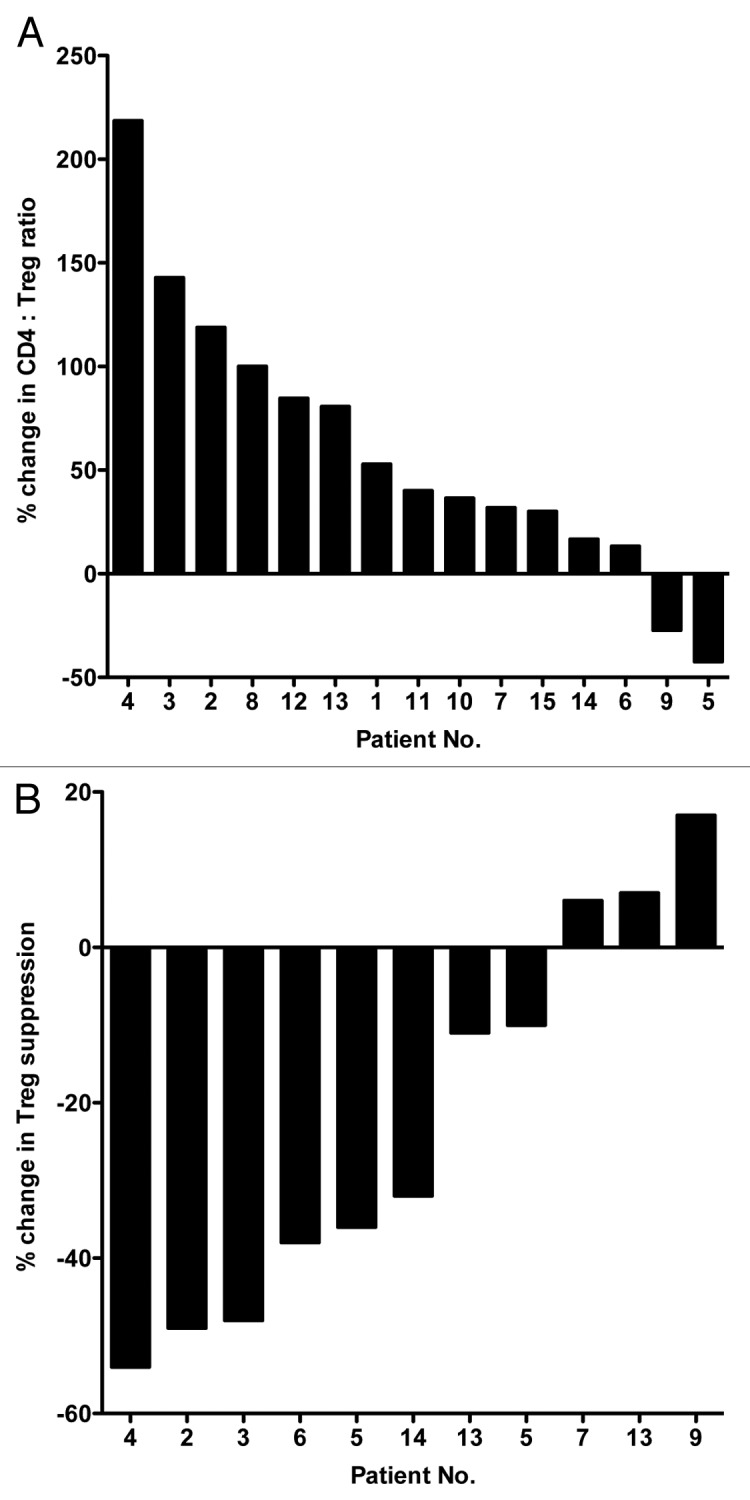
Figure 2. Docetaxel-induced changes in the ratio of effector to regulatory T cells and in the immunosuppressive activity of the latter in hormone-refractory prostate cancer patients. (A) Waterfall plot of the change in the ratio between effector T cells and regulatory T cells (Teff:Treg ratio) in the course of docetaxel therapy in patients with hormone-refractory prostate cancer (n = 15). Patients were treated with docetaxel weekly for 3 weeks followed by 1 week of rest; this comprised a 4-week cycle of therapy. Peripheral blood samples were collected prior to therapy (baseline, day 0) and at the end of the week of rest before starting cycle II. (B) Waterfall plot of the change in suppressive activity of Tregs in the course of docetaxel therapy in patients with hormone-refractory prostate cancer (n = 11).
The assay measuring the ability of Tregs to suppress the proliferation of Teffs requires a considerable amount of PBMCs, which was not always available from all patients. Sufficient PBMCs were available to conduct immunosuppression assays for 11/15 patients. As shown in Figure 2B, comparing the samples obtained before the second cycle of therapy with those collected at baseline reveals a ≥ 20% decrease in the immunosuppressive function of circulating Tregs in 6 patients, whereas 3 individuals exhibited a slight (≤ 20%) increase in Treg immunosuppressive activity.
Although the primary objective of this study was to evaluate the effect of chemotherapy on Teffs and Tregs, we also sought to correlate these immunophenotypes with patient outcome. Thus, we obtained the circulating levels of the prostate cancer biomarker prostate-specific antigen (PSA) for each patient at baseline and before the initiation of the second docetaxel cycle. PSA values decreased ≥ 50% in 8/15 patients, and slightly increased in 2/15 patients (#5 and #9). Of particular interest, these 2 patients (#5 and #9; Fig. 2A) were the only subjects whose CD4+ Teff:Treg ratio decreased upon therapy.
A limited number of PBMC samples were able to be obtained from a separate trial in which patients with metastatic breast carcinoma were treated with docetaxel. The dosages and schedule of the therapeutic regimen were similar to those used for prostate cancer patients, but in this case 3 cycles (rather than 2) were administered, and PBMCs were collected at approximately day 91 during the second week of the third cycle of docetaxel-based chemotherapy. PBMC preparations were analyzed by flow cytometry using 40 markers that delineate specific immune cell subsets (Table S1A and B). In comparison to baseline levels (pre-treatment), both CD4+ and CD8+ T cells decreased in the PBMCs obtained from docetaxel-treated patients (Fig. 3A and B). For the majority of individuals, however, more substantial reductions were observed in the relative amounts of CD4+CD25highCD127-FoxP3+ Tregs among PBMCs (Fig. 3C), resulting in increased CD4+ Teff:Treg and CD8+ Teff:Treg ratios post-treatment (Fig. 3D and E). The relative abundance of natural killer (NK) cells also increased in most patients upon therapy (Fig. 3F). Thus, in these 2 docetaxel-based clinical trials, 16/23 patients (70%) manifested > 20% increase in the CD4+ Teff:Treg ratio upon treatment, while 2/23 subjects (9%) had a > 20% decrease.

Figure 3. Docetaxel treatment of metastatic breast cancer patients increased the ratio of CD4+ or CD8+ T lymphocytes vs. regulatory T cells. (A–F) Cytofluorometric analysis of peripheral blood mononuclear cells (PBMCs) from patients (n = 8) with metastatic breast cancer treated with docetaxel. PBMCs were analyzed before treatment (baseline) and at the time of first re-staging near day 91 (at the second week of cycle III). (A) Percentage of CD4+ T lymphocytes among PBMCs. (B) Percentage of CD8+ T lymphocytes among PBMCs. (C) Percentage of CD4+CD25highCD127–FoxP3+ regulatory T cells (Tregs) among PBMCs. (D) Ratio of CD4+ effector T cells to Tregs (CD4+ Teff:Treg ratio). (E) Ratio of CD8+ effector T cells to Tregs (CD8+ Teff:Treg ratio). (F) Percentage of CD3–CD56+ natural killer (NK) cells among PBMCs. Medians at baseline and cycle III are shown. Statistical analyses were performed by Wilcoxon matched-pairs signed rank test and P < 0.05 was considered statistically significant.
The impact of a second chemotherapeutic regimen, i.e., the administration of cisplatin plus vinorelbine as an adjuvant intervention in patients with Stage IB, II, and IIIA NSCLC on Tregs was also evaluated. Upon surgical tumor resection, patients received cisplatin plus vinorelbine at day 1 and vinorelbine at day 8, followed by 14 d of rest, making up a 3-week treatment cycle. PBMCs were collected and analyzed from 14 patients prior to therapy (baseline), at day 4 (to evaluate early responses to cisplatin), and at day 8 (prior to the administration of vinorelbine alone) to evaluate delayed cisplatin effects. PBMCs were also collected and evaluated after the first (day 21) and third (day 63) cycles of treatment. There were no significant changes in the absolute amount of PBMCs, the number of CD4+ T cells, and percent CD4+ T cells among total PBMCs between samples collected at any time point post-therapy and specimens obtained at baseline (Table 2; Fig. 1D and E). There was, however, a significant difference in the absolute number of Tregs and in the relative abundance of Tregs among CD4+ cells between the samples collected after the third cycle of therapy and at baseline (P < 0.001; Fig. 1F; Table 2). The percentage of Tregs among total CD4+ cells decreased by ≥ 20% after the second cycle of chemotherapy in 10/14 patients (71%), while only 2/14 patients exhibited an increase in this parameter of ≥ 20%.
Table 2. Evaluation of non-small cell lung cancer patients prior to and in the course of standard-of-care anticancer therapy.
| Baseline | Post-4 d | Post-8 d | Post-cycle I | Post-cycle III | n | |
|---|---|---|---|---|---|---|
| No. of PBMCs ( × 103/µL) |
2.6 (2.1–3.0) |
2.3 (1.7–3.1) |
2.1 (1.9–3.4) |
2.7 (1.7–3.2) |
2.6 (1.8–3.6) |
14 |
| No. of CD4+ T cells ( × 103/µL) |
1.1 (0.8–1.5) |
1.1 (0.7–1.4) |
1.0 (0.7–1.8) |
1.1 (0.7–1.6) |
0.9 (0.7–1.5) |
14 |
| CD4+ T cells (%) |
43 (37–55) |
44 (35–51) |
47 (37–57) |
41 (33–54) |
45 (31–51) |
14 |
| Tregs among CD4+ T cells (%) |
4.8 (3.9–6.0) |
4.5 (3.1–6.4)* |
3.6 (2.5–5.1)*** |
2.9 (2.1–5.8)*** |
3.1 (2.5–5.6)*** |
14 |
| No. of Tregs (#/µL) |
56 (41–70) |
50 (33–63)** |
48 (17–68)*** |
45 (13–66)*** |
36 (19–64)*** |
14 |
| CD4+ Teff:Treg ratio | 15.5 (13.0–19.0) | 19.5 (12.5–23.2) | 21.0 (14.5–30.0)** | 27.5 (15.0–40.0)*** | 27.0 (15.8–34.8)*** | 14 |
Values are the median (interquartile range). Friedman and Dunn’s multiple comparison tests between baseline and all other time points; * P < 0.05, ** P < 0.01, and *** P < 0.001 compared with baseline. PBMC, peripheral blood mononuclear cell; Teff, effector T cell; Treg, regulatory T cell.
The CD4+ Teff:Treg ratio increased post-therapy by ≥ 20% in 11/14 patients (78%) (P < 0.001; Fig. 4A) while it slightly decreased in 2 individuals. Sufficient PBMCs to conduct immunosuppression assays were collected at baseline and after the third cycle of therapy from 10 patients. A marked decrease in the immunosuppressive activity of circulating Tregs was observed in all patients. Such a reduction was ≥ 20% in 7/10 patients (Fig. 4B).
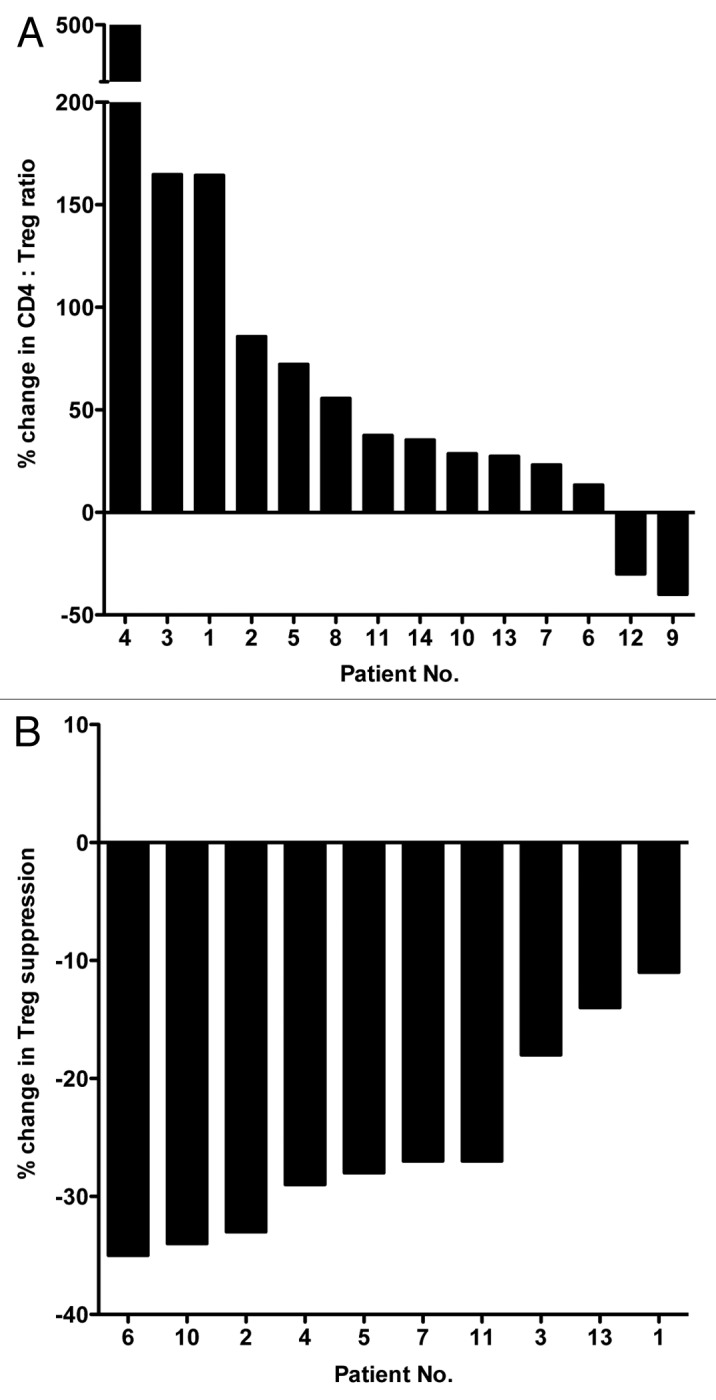
Figure 4. Cisplatin plus vinorelbine-induced changes in the ratio of effector T cells to regulatory T cells and in the immunosuppressive activity of the latter in non-small cell lung cancer patients. (A) Waterfall plot of the change in the ratio between CD4+ effector T cells and regulatory T cells (Teff:Treg ratio) in non-small cell lung cancer (NSCLC) patients before and during therapy with cisplatin plus vinorelbine (n = 14). Patients with NSCLC (stages IB, II, and IIIA) were treated in the adjuvant setting, post-surgery, with cisplatin plus vinorelbine at day 1 and vinorelbine at day 8. This was followed by 14 days of rest, which comprised the 3-week cycle of therapy. PBMC were collected from peripheral blood at baseline and post-cycle III. (B) Waterfall plot of the change in immunosuppressive activity of Tregs from NSCLC patients before and during therapy with cisplatin plus vinorelbine (n = 10). The Treg suppressive activity was evaluated baseline and post-cycle III.
In addition to the studies involving conventional chemotherapeutics, we were also able to evaluate how 2 different anticancer regimens involving small molecule targeted agents affected Tregs in patients. The first of these treatments involved the daily use of 20 mg tamoxifen along with the GnRH agonist leuprolide acetate (11.25 mg/mL) every 3 mo in premenopausal (< 50 y) breast cancer patients. PBMCs were collected from 8 patients prior to therapy (baseline), at days 15 and 30 after treatment, as well as at the end of the third month of therapy, prior to the administration of the GnRH agonist. There were no significant changes in the total number of PBMCs, the total amount of CD4+ cells, the percent of CD4+ cells among total PBMCs, and the relative abundance of Tregs among CD4+ cells (Fig. 5A-C; Table 3) in this setting. The CD4+ Teff:Treg ratio increased upon therapy by ≥ 20% in only 3/8 patients and decreased ≥ 20% in 1 patient (Fig. 6A). Sufficient PBMCs to analyze the immunosuppressive functions of Tregs were available from 6 patients. Mixed results were obtained, with an increase of ≥ 20% in the immunosuppressive function of circulating Tregs upon therapy observed in 2/8 patients, and a decrease ≥ 20% recorded in 2/8 patients (Fig. 6B).

Figure 5. Circulating immune cell subsets in patients with breast cancer and metastatic renal cell carcinoma before and during therapy. (A–C) Premenopausal (< 50 y) breast cancer patients (n = 8) were treated upon surgery with tamoxifen plus a gonadotropin-releasing hormone (GnRH) agonist, and evaluated before and during therapy for the number of circulating peripheral blood mononuclear cells (PBMCs; A), the frequency of CD4+ T cells among PBMCs (B), and the frequency of regulatory T cells (Tregs) among CD4+ T cells (C). Peripheral blood samples were collected at the indicated time points. (D–F) Metastatic renal cell carcinoma patients on sunitinib monotherapy (n = 10) were evaluated before and during treatment for the frequency of CD4+ T cells among PBMCs (D), the frequency of Tregs among CD4+ T cells (E), and the absolute number of Tregs (F). Peripheral blood was collected at the indicated time points. Statistical analyses were performed using Friedman and Dunn’s multiple comparison test; * P < 0.05, ** P < 0.01, *** P < 0.001. In all dot plots, the median and interquartile ranges are shown.
Table 3. Evaluation of breast cancer patients prior to and in the course of standard-of-care anticancer therapy.
| Baseline | Post-15 d | Post-30 d | 3 mo | n | |
|---|---|---|---|---|---|
| No. of PBMCs ( × 103/µL) |
2.4 (2.0–2.7) |
2.0 (1.8–2.5) |
1.8 (1.5–2.4) |
2.0 (1.8–2.6) |
8 |
| No. of CD4+ T cells ( × 103/µL) |
0.8 (0.6–0.9) |
0.7 (0.5–0.9) |
0.6 (0.4–0.8) |
0.6 (0.4–0.9) |
8 |
| CD4+ T cells (%) |
32 (29–37) |
35 (27–40) |
31 (22–38) |
29 (24–37) |
8 |
| Tregs among CD4+ T cells (%) |
3.7 (2.9–4.4) |
3.2 (2.3–4.2) |
3.7 (2.7–4.3) |
3.6 (2.7–4.6) |
8 |
| No. of Tregs (#/µL) |
30 (16–39) |
24 (12–34) |
20 (14–35) |
19 (13–42) |
8 |
| CD4+ Teff:Treg ratio | 19.0 (16.0–25.2) | 24.0 (18.0–35.8) | 19.5 (16.5–27.5) | 19.0 (16.5–30.0) | 8 |
Values are the median (interquartile range). Friedman and Dunn’s multiple comparison tests between baseline and all other time points showed no statistical differences. PBMC, peripheral blood mononuclear cell; Teff, effector T cell; Treg, regulatory T cell.

Figure 6. Effects of targeted anticancer therapeutics on the ratio between effector and regulatory T cells and on the immunosuppressive activity of the latter in patients with breast cancer or metastatic renal cell carcinoma. (A–B) Waterfall plots of the change in the ratio between CD4+ effector T cells and regulatory T cells (Teff:Treg ratio) (A), and in the immunosuppressive activity of Tregs (B), in breast cancer patients receiving tamoxifen plus a gonadotropin-releasing hormone (GnRH) agonist upon surgery (n = 8). Patients of premenopausal status were treated in the adjuvant setting post-surgery with tamoxifen daily and GnRH agonist every 3 months. Peripheral blood samples were collected prior to therapy (baseline, day 0) and at post 3 months. (C–D) Waterfall plots of the change in CD4+ Teff:Treg ratio (C), and Treg-mediated immunosuppression (D), in metastatic renal cell carcinoma patients receiving sunitinib monotherapy (n = 10). Patients with metastatic renal cell carcinoma were treated with sunitinib monotherapy daily for 4 weeks followed by 2 weeks of rest; this comprised a 6-week cycle of therapy. Peripheral blood was collected prior to therapy, and at the end of the second round of chemotherapy in cycle II.
The targeted therapeutic agent sunitinib is widely used for the treatment of metastatic renal cell carcinoma (mRCC), with differing dosages and dosing schedules under evaluation for use as monotherapy or in combinatorial therapies. In this study, 10 patients with mRCC received 50 mg sunitinib monotherapy daily for 4 weeks followed by 2 weeks of rest, making up a 6-week treatment cycle. PBMCs were collected and analyzed prior to treatment (baseline), at the end of 4 weeks of drug, at the end of the first (6 weeks) and second (12 weeks) cycles of therapy. Cytofluorometric analyses revealed no significant changes in the number of total PBMCs, the total amount of CD4+ cells, the relative abundance of CD4+ cells among PBMC, or the percentage of Tregs among CD4+ cells at any time point (Fig. 5D and E; Table 4). There was, however, a clear decrease in the absolute number of Tregs detected after the first (P < 0.001) and second (P < 0.01) cycle of sunitinib-based therapy (Fig. 5F). The analysis of PBMCs collected after the second cycle of treatment and at baseline revealed an increase of ≥ 20% in the CD4+ Teff:Treg ratios in 6/10 patients and a decrease of a similar extent in 2/10 patients (Fig. 6C). Among patients with sufficient PBMCs for this type of assay, 4/9 individuals exhibited a decrease of ≥ 20% in the immunosuppressive functions of Tregs after the second cycle of therapy (as compared with baseline), while a substantial increase in this parameter was evident in 3/9 subjects (Fig. 6D). Potential explanations for the immunophenotypic variations observed between patients are discussed below.
Table 4. Evaluation of renal cell carcinoma patients prior to and in the course of standard-of-care anticancer therapy.
| Baseline | Post-28 d | Pre-cycle II | Post-cycle II | n | |
|---|---|---|---|---|---|
| No. of PBMC ( × 103/µL) |
2.1 (1.9–2.6) |
1.7 (1.1–2.2) |
1.8 (1.4–2.2) |
1.6 (1.2–2.1) |
10 |
| No. of CD4+ T cells ( × 103/µL) |
0.8 (0.6–1.2) |
0.7 (0.4–0.9) |
0.8 (0.5–1.1) |
0.6 (0.3–1.0) |
10 |
| CD4+ T cells (%) |
41 (25–57) |
39 (26–55) |
48 (36–56) |
36 (22–54) |
10 |
| Tregs among CD4+ T cells (%) |
4.4 (3.1–5.6) |
2.7 (2.4–4.4) |
3.6 (3.0–4.4) |
3.6 (3.0–4.6) |
10 |
| No. of Tregs (#/µL) |
45 (28–52) |
24 (15–36)*** |
30 (20–47)* |
20 (12–28)** |
10 |
| CD4+ Teff:Treg ratio | 16.0 (11.8–25.5) | 22.5 (16.2–33.2) | 18.0 (15.5–26.0) | 19.0 (12.2–29.5) | 10 |
Values are the median (interquartile range). Friedman and Dunn’s multiple comparison tests between baseline and all other time points showed no statistical differences, except for the absolute number of Tregs, which were lower after treatment; * P < 0.05, ** P < 0.01, and *** P < 0.001 compared with baseline. PBMC, peripheral blood mononuclear cell; Teff, effector T cell; Treg, regulatory T cell.
Discussion
The findings reported herein provide further rationale to pursue more comprehensive clinical studies with immunostimulatory agents (such as vaccines and/or checkpoint inhibitors) in combination with “standard-of-care” anticancer therapies. Specifically, our results relative to 2 chemotherapeutic regimens—docetaxel and cisplatin plus vinorelbine—support the initiation of clinical studies in which these agents may be used concomitantly with active immunotherapy. Alternatively, these chemotherapeutics could be used immediately prior to vaccination, so as to reduce the number and function of Tregs. The use of tamoxifen in breast carcinoma patients did not appear to have any profound effect on the number of effector CD4+ cells or Tregs, providing evidence that this therapy does not boost Tregs, albeit it does not inhibit them either. Thus, a trial employing activating immunotherapy in combination with tamoxifen plus a GnRH agonist could be contemplated without concerns that this regimen may potentiate Tregs and compromise the efficacy of immunotherapy.
Our data obtained in metastatic renal carcinoma patients treated with sunitinib are confounding, but extend previously reported findings.10,12-14 In these studies, sunitinib inhibited Tregs in some patients, although the effect was not statically significant. Furthermore, sunitinib has been previously shown to deplete myeloid-derived suppressor cells (MDSC), a phenomenon correlating with reduced numbers of Tregs but not with changes in their biological activity or in tumor burden. Here we report a range of responses—including increases as well as decreases in the number and immunosuppressive activity of Tregs—among patients on sunitinib therapy. Preclinical murine studies have recently demonstrated that while Tregs are decreased in the course of sunitinib treatment, upon therapy cessation, the amount of Tregs rebounds to levels that are even higher than those seen prior to therapy.37 This may be one causal factor driving the rapid tumor progression seen in some renal cell carcinoma patients upon termination of sunitinib therapy.
It is interesting to point out that both docetaxel and cisplatin plus vinorelbine reduced the Teff:Treg ratio and the immunosuppressive activity of Tregs. A previous study analyzed the expression of more than 38,000 genes in Tregs from both healthy individuals and metastatic prostate cancer patients.38 This study demonstrated an upregulation of both proliferative and migratory genes in the Tregs of metastatic prostate cancer patients as compared with those from healthy subjects. Considering that docetaxel and vinorelbine are both microtubule inhibitors, such agents should preferentially inhibit the proliferation and function of rapidly dividing cells, including Tregs. In a separate preclinical study, mice treated with a combination of cisplatin plus vinorelbine exhibited reductions in both CD4+ Teffs and Tregs. However, the recovery time of Teffs was shorter than that of Tregs, resulting in increased Teff:Treg ratios at later time points.39 It is presently unclear whether these chemotherapeutics were acting on natural or induced Tregs. As previously reported,40 one marker expressed on natural CD4+CD25highCD127–FoxP3+ Tregs is inducible T-cell co-stimulator (ICOS). In the breast cancer patients included in this study, we observed that the level of both ICOS+ and ICOS– Tregs was similarly reduced by docetaxel (−72.2% and −81.8%, respectively, p = 0.4331). However, there have been contradictory reports41 as to whether ICOS is a valid marker discriminating natural from inducible Tregs.
Noteworthy, in the studies reported here the activation status of the Teffs utilized in immunosuppression assays was evaluated both prior to and during the course of chemotherapy. For example, in both lung cancer patients treated with cisplatin plus vinorelbine and prostate cancer patients treated with docetaxel, the proliferation of the patient-derived CD4+CD25− Teffs was measured upon stimulation with anti-CD3 antibodies. We observed no changes in the proliferative ability of the CD4+CD25− Teffs upon the administration of cisplatin plus vinorelbine or docetaxel to NSCLC or prostate cancer patients, respectively (data not shown). Similarly, in breast cancer patients treated with docetaxel, the percentage of CD4+CD25− Teffs expressing the activation marker ICOS was 4.5% at baseline and 2.7% before the initiation of the second cycle of treatment (p = 0.1484). However, in marked contrast, the percentage of CD4+CD25+CD49d− Tregs expressing the activation marker tumor necrosis factor receptor superfamily, member 18 (TNFRSF18, better known as GITR) was 18.9% at baseline and 0.19% at cycle III of docetaxel treatment (p = 0.0039).
The relevance of host immune responses in the success or failure of conventional anticancer treatments has become increasingly apparent from multiple perspectives. Galon and Fridman1,3 demonstrated that a detailed analysis of specific lymphocyte populations in the primary lesions of colorectal cancer patients can be employed as a strong prognostic indicator, a phenomenon that has been confirmed by numerous groups in colorectal cancer as well as other carcinomas (reviewed in Refs. 2, 4). Zitvogel, Kroemer and colleagues have elegantly shown that certain chemotherapeutic agents can induce the immunogenic death of cancer cells, resulting in the cross-priming of T cells specific for tumor-associated antigens.2,5,6 Other studies have demonstrated an alternative, but not necessarily mutually exclusive, phenomenon of immunogenic modulation of tumor cells. In this scenario, non-lethal amounts of several chemotherapeutic agents and radiation therapy may alter the phenotype of human carcinoma cells, thereby rendering them susceptible to T cell-mediated lysis.7-9
The results reported herein represent only a fragment of the complex manifestation of host immune responses to conventional anticancer agents. Nonetheless, these results suggest that not all chemotherapeutic regimens are likely to have a negative impact on the immune system of cancer patients, whereas others have either no effect or may actively repress immunosuppressive pathways, such as those mediated by Tregs. Furthermore, with these immunological responses in mind, our results imply that some specific conventional anticancer therapies could potentially be combined with active immunotherapy to achieve synergistic anticancer responses. Thus, our results may inform clinicians on the rational design of future trials implementing conventional, standard-of-care therapies (such as those investigated here) together with cutting-edge immunostimulatory interventions toward an ever more effective treatment of cancer patients.
Materials and Methods
Patients and anticancer regimens
Five distinct groups of cancer patients undergoing different anticancer treatments were enrolled in the study. (1) Patients with hormone-refractory metastatic prostate cancer (n = 15) underwent 35 mg/m2 docetaxel monotherapy weekly for 3 weeks followed by 1 week of rest, making up a 4-week cycle of therapy. PBMC samples were collected prior to the start of cycle I, prior to the start of the week of rest, and at the end of the week of rest, prior to the start of cycle II. (2) Patients at the NIH Clinical Center with evidence of metastatic breast carcinoma and a life expectancy of at least 4 mo were enrolled in a randomized Phase II study of docetaxel alone or in combination with an anticancer vaccine (NCI-6977).42 Their PBMCs were collected before treatment (baseline) and at the time of first re-staging near day 91 (at the second week of cycle III). Patients received weekly 35 mg/m2 docetaxel i.v. over 30 min plus 4 mg dexamethasone orally 12 h before, 1 h before, and 12 h after docetaxel for cycles of 3 weeks followed by 1 week of rest. (3) Patients with any histological subtype of stage IB, II and IIIA NSCLC (n = 14) were treated upon surgical tumor resection with 80 mg/m2 cisplatin plus 25 mg/m2 vinorelbine at day 1 and vinorelbine at day 8, followed by 14 d of rest, making up a 3-week cycle of therapy. PBMC samples were collected at day 1 (prior to the start of treatment), at day 4, at day 8 (prior to the vinorelbine treatment), at day 21 (prior to the start of cycle II), and at the first day of cycle IV, prior to the start of treatment. (4) Patients with breast cancer in premenopausal (< 50 y) status (n = 8) were treated upon surgical tumor resection with 20 mg tamoxifen daily and 11.25 mg/mL leuprolide acetate every 3 mo. PBMC samples were collected prior to the start of the treatment, at day 15, at day 30, and at the end of the third month prior to the administration of leuprolide acetate. (5) Patients with mRCC (n = 10) were treated with 50 mg sunitinib daily for 4 weeks followed by 2 weeks of rest, making up a 6-week therapeutic cycle. PBMC samples were collected prior to the start of cycle I, at the end of cycle I, prior to the start of cycle II and at the end of cycle II.
Patients involved in the study did not receive any radiation therapy or chemotherapy within 6 mo prior to PBMC collections. All patients were informed and the procedures were conducted in accordance with the Helsinki Declaration of 1975 and signed an informed consent form.
Collection of PBMCs
PBMCs were isolated by Ficoll (MP Biomedicals) density gradient separation, washed twice and cryopreserved in 90% heated-inactivated human AB serum and stored in 10% dimethylsulfoxide (DMSO) in liquid nitrogen at a concentration of 1 × 107 cells/mL until assayed.
Flow cytometry studies
Cryopreserved PBMCs were analyzed by 4-color flow cytometry for the phenotypic characterization of Tregs, as described by Vergati et al.34 Cells were resuspended in staining buffer (PBS containing 3% fetal bovine serum) and stained for 30 min at 4°C with fluorescein isothiocyanate (FITC)-conjugated anti-CD4 (BD Pharmingen, BD Biosciences), phycoerythrin-(PE) conjugated anti-CD25 (BD Pharmingen) and PerCP Cy5.5-conjugated anti-CD127 (eBioscience) antibodies. FoxP3 intracellular staining was performed on cells pre-stained as indicated above with anti-CD4, anti-CD25, and anti-CD127 antibodies. Cells were fixed and permeabilized using a Fix/Perm buffer (eBioscience) according to the manufacturer’s instructions, then labeled with allophycocyanin (APC)-conjugated anti-FoxP3 (eBioscience) antibodies or an isotype-matched antibody as negative control. Unless otherwise indicated, samples were run on a FACSCalibur flow cytometer (BD Biosciences) with 5 × 104 events acquired and data analyzed using the CellQuest software (BD Biosciences). To determine the percentage of Tregs, lymphocytes were gated by plotting forward (FSC) vs. side scatter (SSC). The CD4+ population was gated first, followed by the CD25+CD127- population, and—lastly—the FoxP3+ population was gated in the CD4+CD25+CD127− cell subset. Tregs are thus defined as the CD4+CD25highCD127-FoxP3+ population. The cytofluorometric analysis of PBMCs from metastatic breast cancer patients receiving docetaxel was performed on a BD LSR-II flow cytometer (BD Biosciences) equipped with a 488 nm blue, a 405 nm violet, a 355 nm UV, and a 633 nm red laser. Live cells were discriminated by a LIVE/DEAD® Fixable Blue Dead Cell Stain Kit for UV excitation (Molecular Probe, Life Technologies, Grand Island, NY), following the manufacturer’s protocol. The following antibodies were purchased from BD Biosciences: anti-human PeCy7 CD8 clone RPA-T8, BD HorizonTM V500 CD3 clone UCHT1, APC-Cy7 CD25 clone M-A251, PerCP-Cy5.5 CD16 clone 3G8, and Alexa Fluor 700 CD33 clone WM53. The following antibodies were purchased from BioLegend (San Diego, CA): AlexaFluor 700 CD4 clone RPA-T4, FITC CD127 (IL-7Rα) clone A019D5, FITC CD19 clone HIB19, FITC CD20 clone 2H7, Pe-Cy7 CD56 clone MEM-188, and Brilliant Violet 605TM HLA-DR clone L243. The anti-human Pe-Cy7 FoxP3 antibody (clone 236A/E7) was purchased from eBioscience. The following immunophenotypic markers were used to define immune cell subsets: CD4+ T lymphocytes were CD3+CD4+; CD8+ T lymphocytes were CD3+CD8+; NK cells were CD3-CD56+; Tregs were CD3+CD4+CD25highCD127-FoxP3+ and MDSCs were CD3-CD16−CD19−CD20−CD56−HLA-DRlowCD33+.
CD4+CD25high T-cell enrichment
CD4+CD25high T cells were enriched using a CD4+CD25+ Treg isolation kit (Miltenyi Biotec), with modifications to the manufacturer’s instructions. CD4+ T cells were negatively enriched and positive selection for CD25high T cells was subsequently done on the negatively selected CD4+ T cells. In order to achieve a consistently high CD25high purity rate, the amount of CD25 antibody-microbeads was decreased by 70% and the incubation time was decreased by 15%. The CD25high cell fraction was collected by eluting twice the cells through a magnetic separation (LS) column to further enrich for CD4+CD25high T cells.
Immunosuppression assays
T cells were cultured in RPMI 1640 medium supplemented with 10% AB serum, 100 units/mL penicillin, 100 μg/mL of streptomycin (Mediatech), and 2 mM L-glutamine (Mediatech). Responder CD4+CD25– T cells were labeled with 2 μM carboxyfluorescein succinimidyl ester (CFSE; Sigma). In suppression assays, to assess the suppressive capacity of CD4+CD25high T cells, 1 × 104 CFSE-labeled responder CD4+CD25– T cells were cultured alone or together with 1 × 104 CD4+CD25high T cells in the presence of mitomycin-treated T-depleted PBMCs as antigen presenting cells. The immune cell culture mixture was stimulated with 0.5 μg/mL plate-bound anti-CD3 antibody (OKT3; eBioscience) in 96-well round-bottom plates.43 The proliferation of CFSE-labeled cells was assessed by flow cytometry after 4 d of culture. Relative suppression was calculated using the following formula: [(percent proliferating CD4+CD25– cells cultured alone - percent proliferating CFSE-diluting CD4+CD25– cells cultured in the presence of Tregs at a 1:1 ratio)/percent proliferating CD4+CD25– cells cultured alone] × 100.
Statistical analysis
Statistical analyses were performed using non-parametric Wilcoxon or Friedman tests with Dunn’s multiple comparison for paired samples. P < 0.05 was considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. This work has been partially performed within the Ph.D. program in “Oncology rehabilitation” XXV Ciclo. The authors thank Debra Weingarten for her editorial assistance in the preparation of the manuscript.
Glossary
Abbreviations:
- GnRH
gonadotropin releasing hormone
- ICOS
inducible T-cell co-stimulator
- mCRPC
metastatic castration-resistant prostate cancer
- MDSC
myeloid-derived suppressor cell
- mRCC
metastatic renal cell carcinoma
- NK
natural killer
- NSCLC
non-small cell lung cancer
- PBMC
peripheral blood mononuclear cell
- PSA
prostate-specific antigen
- Teff
effector T cell
- Treg
regulatory T cell
Citation: Roselli M, Cereda V, Giovanna di Bari M, Formica V, Spila A, Jochems C, Farsaci B, Donahue R, Gulley JL, Schlom J, et al. Effects of conventional therapeutic interventions on the number and function of regulatory T cells. OncoImmunology 2013; 2:e26440; 10.4161/onci.26440
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/27025
References
- 1.Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 3.Galon J, Pagès F, Marincola FM, Thurin M, Trinchieri G, Fox BA, Gajewski TF, Ascierto PA. The immune score as a new possible approach for the classification of cancer. J Transl Med. 2012;10:1. doi: 10.1186/1479-5876-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jochems C, Schlom J. Tumor-infiltrating immune cells and prognosis: the potential link between conventional cancer therapy and immunity. Exp Biol Med (Maywood) 2011;236:567–79. doi: 10.1258/ebm.2011.011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 6.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–91. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 7.Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64:7985–94. doi: 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 8.Hodge JW, Ardiani A, Farsaci B, Kwilas AR, Gameiro SR. The tipping point for combination therapy: cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin Oncol. 2012;39:323–39. doi: 10.1053/j.seminoncol.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–71. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adotevi O, Pere H, Ravel P, Haicheur N, Badoual C, Merillon N, Medioni J, Peyrard S, Roncelin S, Verkarre V, et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J Immunother. 2010;33:991–8. doi: 10.1097/CJI.0b013e3181f4c208. [DOI] [PubMed] [Google Scholar]
- 11.Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, Levi J, Daphtary MM, Biedrzycki B, Wolff AC, et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol. 2009;27:5911–8. doi: 10.1200/JCO.2009.23.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–82. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 13.Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, Rini B, Finke JH, Cohen PA. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res. 2010;70:3526–36. doi: 10.1158/0008-5472.CAN-09-3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 15.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–51. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlom J. Therapeutic cancer vaccines: current status and moving forward. J Natl Cancer Inst. 2012;104:599–613. doi: 10.1093/jnci/djs033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. IMPACT Study Investigators Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 18.von Mehren M, Arlen P, Gulley J, Rogatko A, Cooper HS, Meropol NJ, Alpaugh RK, Davey M, McLaughlin S, Beard MT, et al. The influence of granulocyte macrophage colony-stimulating factor and prior chemotherapy on the immunological response to a vaccine (ALVAC-CEA B7.1) in patients with metastatic carcinoma. Clin Cancer Res. 2001;7:1181–91. [PubMed] [Google Scholar]
- 19.von Mehren M, Arlen P, Tsang KY, Rogatko A, Meropol N, Cooper HS, Davey M, McLaughlin S, Schlom J, Weiner LM. Pilot study of a dual gene recombinant avipox vaccine containing both carcinoembryonic antigen (CEA) and B7.1 transgenes in patients with recurrent CEA-expressing adenocarcinomas. Clin Cancer Res. 2000;6:2219–28. [PubMed] [Google Scholar]
- 20.Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, Harlin H. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–45. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 21.Gajewski TF, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29:233–40. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 22.Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343–51. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
- 23.deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18:3022–9. doi: 10.1158/1078-0432.CCR-11-3216. [DOI] [PubMed] [Google Scholar]
- 24.Ménétrier-Caux C, Curiel T, Faget J, Manuel M, Caux C, Zou W. Targeting regulatory T cells. Target Oncol 2012; 7:15-28;; 10.1007/s11523-012-0208-y [DOI] [PubMed]
- 25.Cesana GC, DeRaffele G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, Cheung K, Hesdorffer C, Kim-Schulze S, Kaufman HL. Characterization of CD4+CD25+ regulatory T cells in patients treated with high-dose interleukin-2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol. 2006;24:1169–77. doi: 10.1200/JCO.2005.03.6830. [DOI] [PubMed] [Google Scholar]
- 26.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007;13:5256–61. doi: 10.1158/1078-0432.CCR-07-0892. [DOI] [PubMed] [Google Scholar]
- 28.Lindenberg JJ, Fehres CM, van Cruijsen H, Oosterhoff D, de Gruijl TD. Cross-talk between tumor and myeloid cells: how to tip the balance in favor of antitumor immunity. Immunotherapy 2011; 3:77-96;; 10.2217/imt.10.95 [DOI] [PubMed]
- 29.Lutsiak ME, Tagaya Y, Adams AJ, Schlom J, Sabzevari H. Tumor-induced impairment of TCR signaling results in compromised functionality of tumor-infiltrating regulatory T cells. J Immunol. 2008;180:5871–81. doi: 10.4049/jimmunol.180.9.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mocellin S, Wang E, Marincola FM. Cytokines and immune response in the tumor microenvironment. J Immunother. 2001;24:392–407. doi: 10.1097/00002371-200109000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–35. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184:3106–16. doi: 10.4049/jimmunol.0902661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shevach EM. The resurrection of T cell-mediated suppression. J Immunol. 2011;186:3805–7. doi: 10.4049/jimmunol.1100364. [DOI] [PubMed] [Google Scholar]
- 34.Vergati M, Cereda V, Madan RA, Gulley JL, Huen NY, Rogers CJ, Hance KW, Arlen PM, Schlom J, Tsang KY. Analysis of circulating regulatory T cells in patients with metastatic prostate cancer pre- versus post-vaccination. Cancer Immunol Immunother. 2011;60:197–206. doi: 10.1007/s00262-010-0927-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gulley JL, Arlen PM, Madan RA, Tsang KY, Pazdur MP, Skarupa L, Jones JL, Poole DJ, Higgins JP, Hodge JW, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother. 2010;59:663–74. doi: 10.1007/s00262-009-0782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halabi S, Small EJ, Kantoff PW, Kattan MW, Kaplan EB, Dawson NA, Levine EG, Blumenstein BA, Vogelzang NJ. Prognostic model for predicting survival in men with hormone-refractory metastatic prostate cancer. J Clin Oncol. 2003;21:1232–7. doi: 10.1200/JCO.2003.06.100. [DOI] [PubMed] [Google Scholar]
- 37.Farsaci B, Higgins JP, Hodge JW. Consequence of dose scheduling of sunitinib on host immune response elements and vaccine combination therapy. Int J Cancer. 2012;130:1948–59. doi: 10.1002/ijc.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huen NY, Pang AL, Tucker JA, Lee TL, Vergati M, Jochems C, Intrivici C, Cereda V, Chan WY, Rennert OM, et al. Up-regulation of proliferative and migratory genes in regulatory T cells from patients with metastatic castration-resistant prostate cancer. Int J Cancer. 2013;133:373–82. doi: 10.1002/ijc.28026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gameiro SR, Caballero JA, Higgins JP, Apelian D, Hodge JW. Exploitation of differential homeostatic proliferation of T-cell subsets following chemotherapy to enhance the efficacy of vaccine-mediated antitumor responses. Cancer Immunol Immunother. 2011;60:1227–42. doi: 10.1007/s00262-011-1020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, Qin FX, Gilliet M, Liu YJ. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity. 2008;28:870–80. doi: 10.1016/j.immuni.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J. 2012;26:2253–76. doi: 10.1096/fj.11-193672. [DOI] [PubMed] [Google Scholar]
- 42.Docetaxel Alone or in Combination With Vaccine to Treat Breast Cancer [NCI-6977]. http://www.cancer.gov/clinicaltrials/search/view?cdrid=442407&version=HealthProfessional
- 43.Yokokawa J, Cereda V, Remondo C, Gulley JL, Arlen PM, Schlom J, Tsang KY. Enhanced functionality of CD4+CD25(high)FoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clin Cancer Res. 2008;14:1032–40. doi: 10.1158/1078-0432.CCR-07-2056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.