

Introduction
Calcifying Fibrous Tumors (CFT) are rare benign lesions. They are believed to be true neoplasms with a tendency for nondestructive local recurrence. We report such a case of a large retroperitoneal CFT which was treated successfully at our center.
Case report
A 64-year-old postmenopausal female patient, with no co-morbidities and no history of previous abdominal surgery, presented to our OPD with complaints of a lump in the left side of the abdomen for last eight years. The lump had slowly increased in size and was painless except for the previous three months when she started experiencing local pain which was dull aching in nature, non-colicky, non-radiating and with no relation to intake of meals. There was no history of any sudden increase in the size of the lump or severe abdominal pain or any constitutional symptoms. Physical examination revealed an elderly, thinly built lady with a 20 × 15 cm intra-abdominal, firm lump. It was non-tender and occupied the left lumbar and the umbilical regions and extended into the lower half of the left hypochondrium and the epigastrium. The fingers could be insinuated between the lump and the left hypochondrium. It had well defined margins and a smooth surface. Liver and spleen were not palpable and there was no free fluid in the abdomen. USG abdomen revealed a well-defined hetero-echoic mass lesion occupying the entire left half of the abdomen. It was pushing the left kidney medially and was not fixed to the spleen, the left ovary or the pancreas. CECT abdomen was suggestive of a large left sided retroperitoneal neoplastic mass (Fig. 1). CA 125 was not raised. Colonoscopy was essentially normal. Other routine investigations including chest radiograph were normal.
Fig. 1.
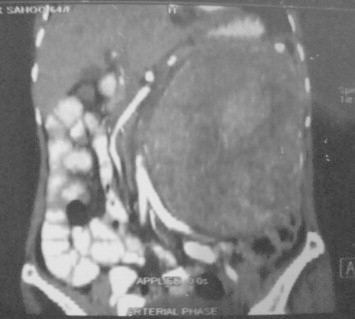
CT Scan showing large retroperitoneal tumor.
The patient was taken up for exploratory laparotomy and the abdomen was entered through a vertical midline incision. A large tumor abutting on to the upper pole of left kidney was encountered which occupied the entire left side of the abdomen (Fig. 2). It was carefully mobilized and complete excision was carried out. The gross weight of the tumor was 10 kg with dimensions of 20 × 14 × 15 cm (the max cranio-caudal, transverse and the antero-posterior measurements). Postoperatively the patient had a smooth recovery and was discharged on 5th postoperative day. Histopathological examination of the tumor showed that it was circumscribed, but not encapsulated and was composed of paucicellular, hyalinized fibrosclerotic tissue with psammomatous calcification scattered throughout. Immunohistochemistry studies showed Vimentin positivity and focal positivity for Actin in the blood vessels consistent with the diagnosis of CFT (Figs. 3 and 4). The patient is presently well and recurrence free after one year of follow-up.
Fig. 2.
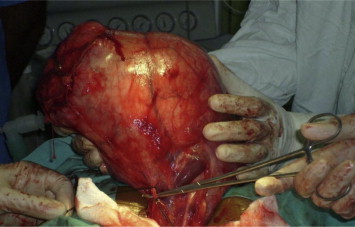
Operative picture of tumor.
Fig. 3.
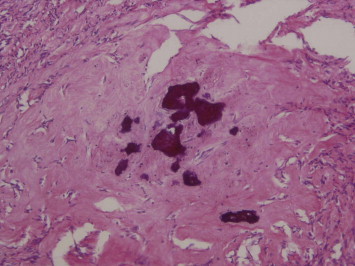
Hyalinized fibrosclerotic tissue with foci of psammomatous calcification (H & E ×200).
Fig. 4.
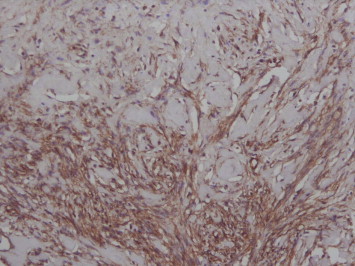
IHC showing Vimentin positivity (×200).
Discussion
Calcifying Fibrous Tumors are rare, benign soft tissue neoplasms. These lesions are characterized by hypocellular, densely hyalinized collagenization with variably prominent mononuclear inflammatory infiltrate and scattered psammomatous and/or dystrophic calcification.1 These rare benign fibrous lesions were originally described as “childhood fibrous pseudotumor with psammoma bodies” in 2 and 11 year old girls by Rosenthal and Abdul-Karim2 in 1988, from Case Western University, Ohio, USA. The term calcifying fibrous pseudotumor was coined by Fetsch et al3 in 1993. This name was adopted to reflect their belief that the underlying process was most likely fibro-inflammatory and reactive. However, later studies suggested a true neoplasm with a tendency for nondestructive local recurrence.4 The word “childhood” was dropped from the name because the lesion involved a broader age range than initially known, as also evident in our case. The adjective “calcifying” was used as a substitute for “psammoma bodies” because the calcification was sometimes of the dystrophic type. This lesion has recently been renamed as ‘calcifying fibrous tumor’ in the newly published World Health Organization classification of tumors of soft tissue and bone.1
The etiology of CFT is unknown. A few were thought to evolve from inflammatory myofibroblastic tumor (IMT), but larger studies failed to confirm this and ultra structural studies revealed fibroblastic features.4,5 A genetic susceptibility theory was also floated but has not been substantiated.6 These lesions were originally described as arising in the subcutaneous and deep soft tissues, mostly in the extremities and the neck areas3 but more recently CFT have been found to be ubiquitous. Most lesions are solitary but multifocal lesions are also reported specially arising from pleura and peritoneum.4,6 However, very few cases arising from the retroperitoneum have been reported and none of them were as large as the one described by us.4 The treatment modality of choice for CFT is surgical excision. Recurrence is rare and malignant transformation has not been reported.7
The histopathological differential diagnosis is broad. Fibromatoses are usually more cellular lesions and they generally express nuclear beta-catenin. Inflammatory myofibroblastic tumor (IMT) has been related by some authors to CFT, and they have considered this latter entity as the final collagenous stage of the former. However, these tumors show a different immunohistochemical profile and a different biological behavior.5,6 Retroperitoneal fibrosis can also be considered as a differential diagnosis for the tumor in retroperitoneal location specially when associated with other fibrosing conditions, such as inflammatory pseudotumor or retractile mesenteritis. But in these lesions, there are no psammomatous calcifications, as seen in our case. More recently, there has been a proposal to consider all these fibrosis-inducing tumors as part of the so-called IgG4-positive multiorgan lymphoproliferative syndrome. This entity is clinically characterized by the presence of elevated serum IgG4 levels and a dense infiltration of different organs and tissues by IgG4-positive plasma cells. The patients with IgG4-positive multiorgan lymphoproliferative syndrome (also called in the literature IgG4-related sclerosing disease) can have involvement of several target organs or just localized disease in one of them.8
In summary, Calcifying Fibrous Tumors are rare soft tissue neoplasms and a huge lesion arising from the retroperitoneum as described above is extremely uncommon. Complete surgical excision is the treatment of choice and local recurrence is rare. These lesions may be identified more often in the future due to increased awareness and technical advances in diagnosis.
Conflicts of interest
All authors have none to declare.
References
- 1.Montgomery E. Calcifying fibrous tumour. In: Fletcher C.D.M., Unni K.K., Mertens F., editors. Pathology and Genetics of Tumours of Soft Tissue and Bone (World Health Organization Classification of Tumours) IARC Press; Lyon, France: 2002. pp. 77–78. [Google Scholar]
- 2.Rosenthal N.S., Abdul-Karim F.W. Childhood fibrous tumor with psammoma bodies. Clinicopathologic features in two cases. Arch Pathol Lab Med. 1988;112:798–800. [PubMed] [Google Scholar]
- 3.Fetsch J.F., Montgomery E.A., Meis J.M. Calcifying fibrous pseudotumor. Am J Surg Pathol. 1993;17:502–508. doi: 10.1097/00000478-199305000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Nascimento A.F., Ruiz R., Hornick J.L., Fletcher C.D. Calcifying fibrous “pseudotumor”: clinicopathologic study of 15 cases and analysis of its relationship to inflammatory myofibroblastic tumor. Int J Surg Pathol. 2002;10:189–196. doi: 10.1177/106689690201000304. [DOI] [PubMed] [Google Scholar]
- 5.Sigel J.E., Smith T.A., Reith J.D., Goldblum J.R. Immunohistochemical analysis of anaplastic lymphoma kinase expression in deep soft tissue calcifying pseudotumor: evidence of a late sclerosing stage of inflammatory myofibroblastic tumor? Ann Diagn Pathol. 2001;5:10–14. doi: 10.1053/adpa.2001.21474. [DOI] [PubMed] [Google Scholar]
- 6.Chen K.T.K. Familial peritoneal multifocal calcifying fibrous tumor. Am J Clin Pathol. 2003;119:811–815. doi: 10.1309/MXC6-TWEL-UUH4-20W0. [DOI] [PubMed] [Google Scholar]
- 7.Farah R.B., Dimet S., Bidault A.T. Multiple peritoneal calcifying fibrous tumors revealed by ischemic colitis. Ann Diagn Pathol. 2007;11:460–463. doi: 10.1016/j.anndiagpath.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Kamisawa T., Okamoto A. IgG4-related sclerosing disease. World J Gastroenterol. 2008;14:3948–3955. doi: 10.3748/wjg.14.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]